Introduction
The behavior of 2,6-dichloro-9-thiabicyclo[3.3.1]nonane (
1) is a good example of the power of anchimeric assistance in organic chemistry [
1]. Historically, the electrophilic addition of SCl
2 to certain cyclic and acyclic dienes provides an inexpensive route to
anti-dichlorosulfide structures of the mustard class [1a, 2]. Competing polymerization pathways often dominate, but the reaction parameters are nearly optimal for the addition of SCl
2 to a few cyclic polyenes and above all for1,5-cyclooctadiene.
Compound
1 has interesting applications as an electrophilic connector and chiral scaffold. Its sulfur center, poised with C
2-symmetry behind two C-Cl bonds, is able to form episulfonium intermediates that can be efficiently captured by a wide variety of nucleophiles [1b-c]. As examples of this facile and clean substitution chemistry we report here the synthesis of the bis(azide)
2 and dicyano derivative
3, in addition to a practical improvement in the large-scale synthesis of
1. The preparation of
2 is improved from our previous published method [1b], in that azide substitution is performed at room temperature in acetonitrile/water rather than at reflux in water only. Compounds
2 and
3 may be further derivatized in a number of ways, including by 1,3-dipolar cycloaddition of
2 with alkynes. Indeed, the original motivation for the synthesis of
2 was its potential incorporation into functional molecules and polymers by virtue of this “
click chemistry” cycloaddition connection [
3].
Results and Discussion
The transannular addition of SCl
2 to 1,5-cyclooctadiene [
4] gives 2,6-dichloro-9-thiabicyclo[3.3.1]nonane (
1) in excellent yield at a variety of concentrations and scales, but the reaction was successful (at least in our hands) only when the hazardous reagent SCl
2 was purified by careful double distillation just before use [
5]. As an alternative, we have found that the sequential treatment of diene with sulfur monochloride (S
2Cl
2) followed by sulfuryl chloride (SO
2Cl
2) provides high yields of
1 in a robust and convenient manner (
Scheme 1) [
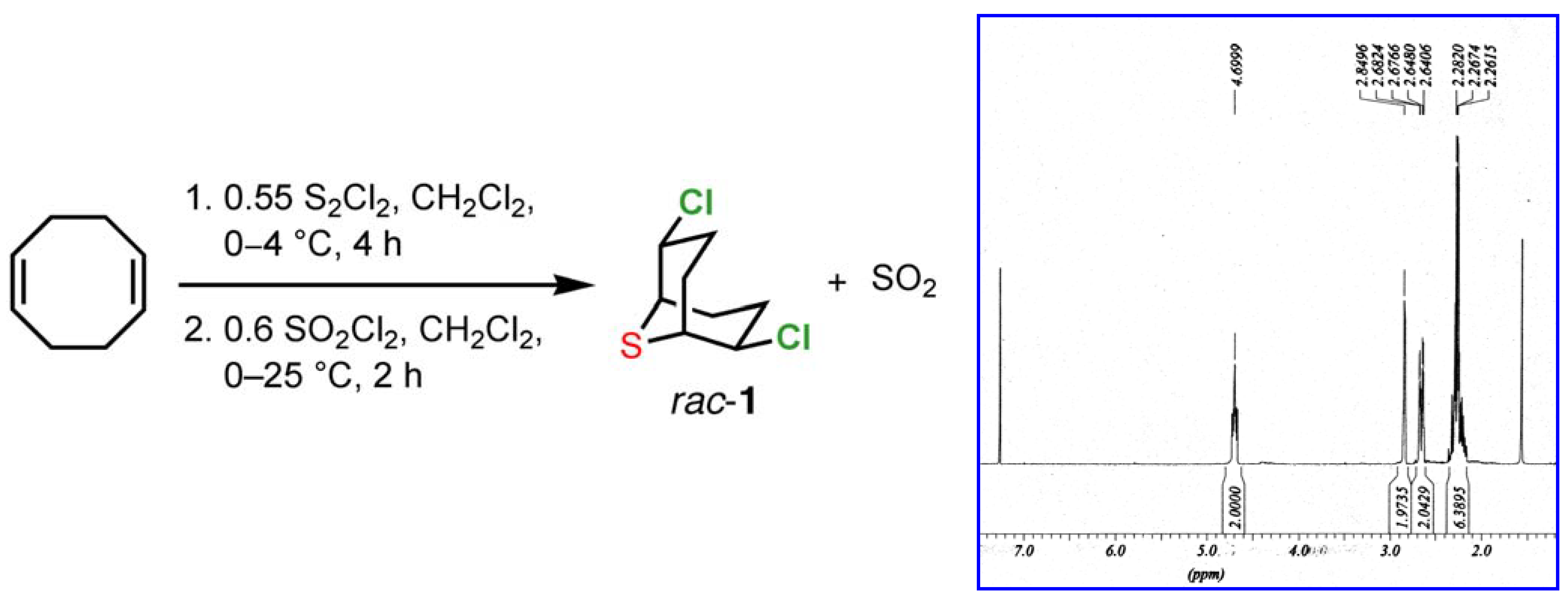
1b]. The crude material obtained from this process displays a very clean
1H-NMR spectrum, showing only trace quantities of contaminants (
Scheme 1).
Scheme 1.
Synthesis and 1H NMR spectrum of 2,6-dichloro-9-thiabicyclo[3.3.1]nonane (1) in CDCl3.
Scheme 1.
Synthesis and 1H NMR spectrum of 2,6-dichloro-9-thiabicyclo[3.3.1]nonane (1) in CDCl3.
The reagents S
2Cl
2 and SO
2Cl
2 employed here may be used as received from commercial suppliers or even after extended storage, requiring no additional purification prior to use. In accord with previous observations of sulfur monochloride reactivity [
6], we suggest that the immediate addition product of S
2Cl
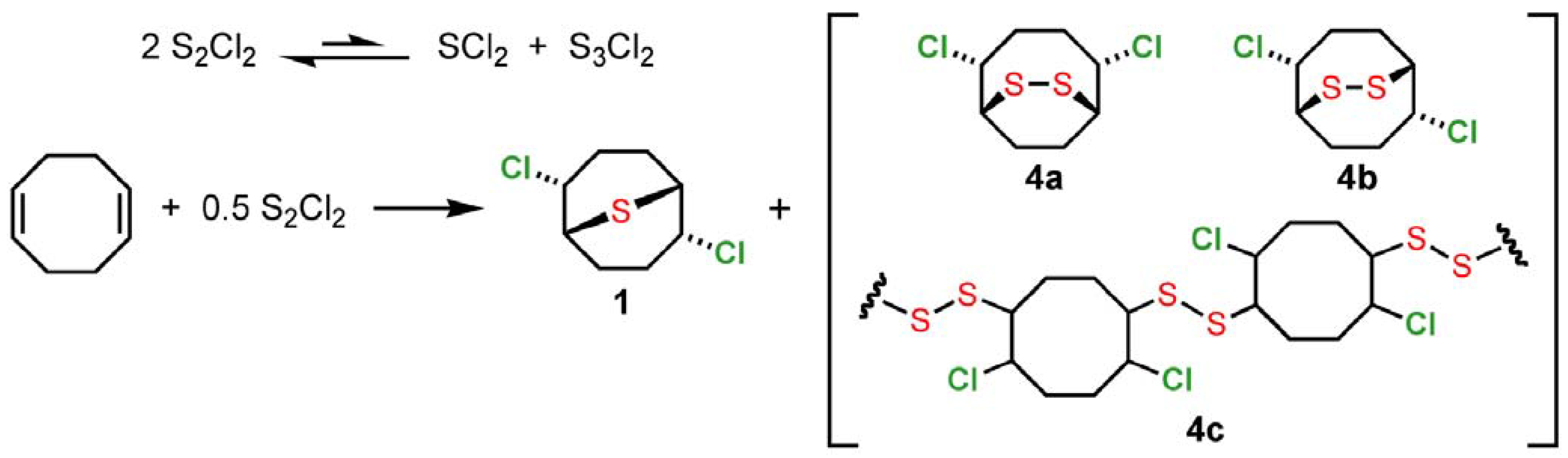
2 and 1,5-cyclooctadiene is likely to be a mixture of monomeric (
1,
4a,
4b) and oligomeric (
4c) adducts (
Scheme 2). Reaction of SCl
2 with the diolefin is faster than reaction of the higher members of the family and shifts the disproportionation equilibrium of S
nCl
2 to the right. In the second step, the addition of sulfuryl chloride initiates a cascade of reactions that has the effect of providing an additional equivalent of chlorine (shown in
Scheme 3 for
4a as an example). The production of
1 is the result of reversible elimination-addition of SCl
2 and the high thermodynamic stability of
1 relative to other monomeric and oligomeric sulfur dichloride addition products.
Scheme 2.
Monomeric and oligomeric adducts intermediates from S2Cl2 and 1,5-cyclooctadiene.
Scheme 2.
Monomeric and oligomeric adducts intermediates from S2Cl2 and 1,5-cyclooctadiene.
Scheme 3.
Evolution of monomeric intermediate 4a by addition of sulfuryl chloride.
Scheme 3.
Evolution of monomeric intermediate 4a by addition of sulfuryl chloride.
2,6-Dichloro-9-thiabicyclo[3.3.1]nonane (
1) may be used as connector and as a chiral scaffold through nucleophilic substitution of the chloride atoms [7, 1b-c]. Our interest was to optimize a multigram synthesis of
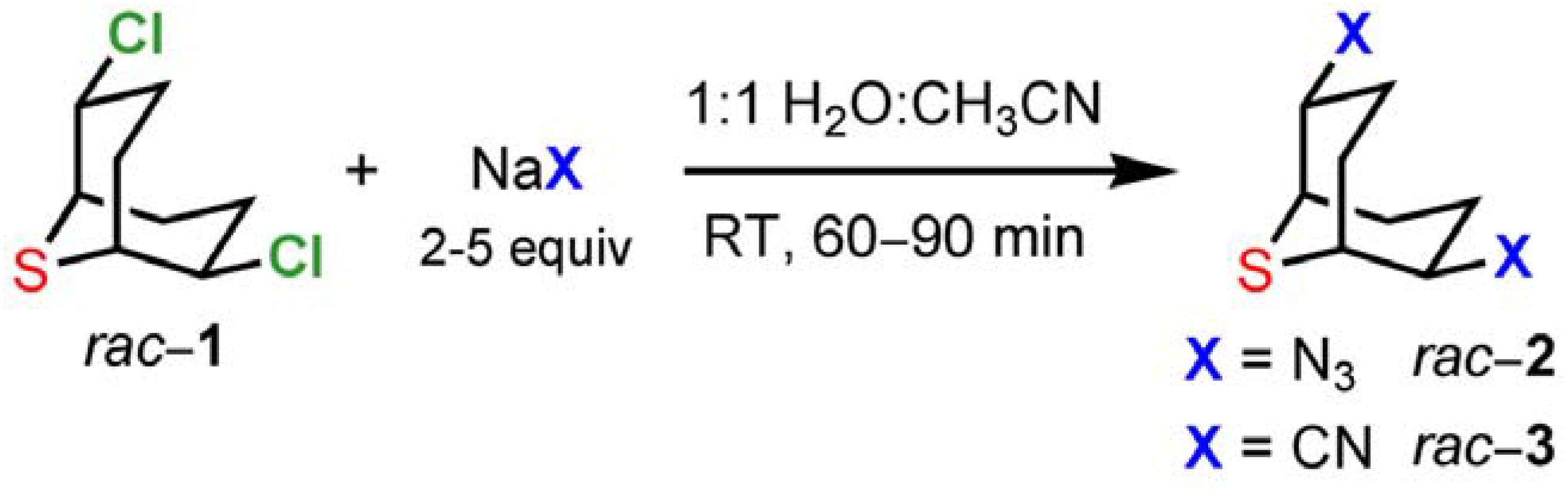
1 and substitution products using nucleophiles that allow the facile introduction of functionalities in subsequent steps. For this purpose we selected azides and cyanides as nucleophiles (
Scheme 4). The substitution reactions on
1 in aqueous solution (H
2O:CH
3CN 1:1) were completed within 60−90 min at room temperature affording the desired compounds
2 and
3 in 90−94% and 83−90% yields, respectively. Simple extraction and washing allowed the isolation of analytically pure samples.
Scheme 4.
Reactivity of 2,6-dichloro-9-thiabicyclo[3.3.1]nonane (1).
Scheme 4.
Reactivity of 2,6-dichloro-9-thiabicyclo[3.3.1]nonane (1).
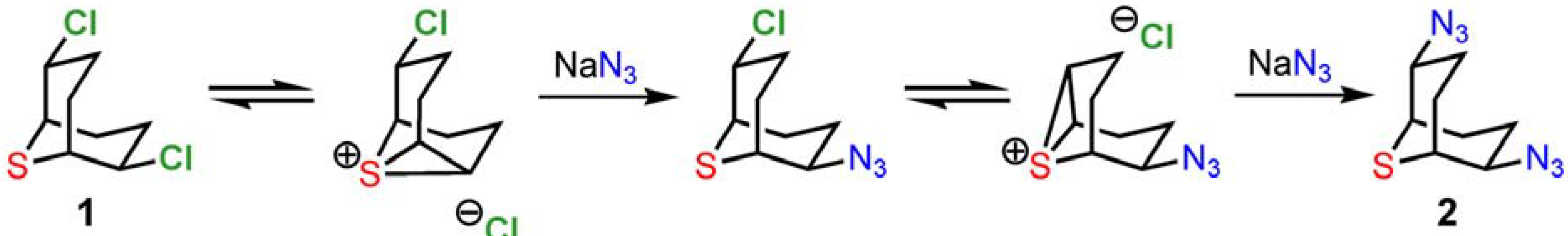
From a mechanistic point of view, the synthesis of
2 and
3 relies on the powerful anchimeric assistance of the neighboring sulfur atom [
8], as shown in
Scheme 5. The water-soluble and highly activated intermediate episulfonium ion allows facile reaction with a wide variety of nucleophiles on the β-chlorinated centers [1b-c]. The double inversion process preserves stereochemistry in substitution reactions with a broad collection of heteroatom nucleophiles. Optical resolution can be performed by chiral HPLC or by condensation with the chiral alkaloid brucine [1b]. Finally, the azido groups of
2 engage in useful 1,3-dipolar cycloaddition reactivity, providing for the facile introduction of heterocycles to the 9-thiabicyclo[3.3.1]nonane core.
Scheme 5.
Neighboring-group participation by the sulfur center of 1.
Scheme 5.
Neighboring-group participation by the sulfur center of 1.
Experimental Section
General
1H- and 13C-NMR spectra were obtained on a Bruker DRX-500 instrument. IR spectra were obtained on a MIDAC FTIR instrument using a horizontal attenuated total reflectance (HATR) accessory (Pike Instruments). Melting points were measured in a Thomas Hoover capillary melting point apparatus and are uncorrected. High-resolution mass spectral analyses were performed by the Scripps Center for Mass Spectrometry. TLC analysis was facilitated by the use of the following stains in addition to UV light with fluorescent-indicating plates (silica gel on aluminum, Sigma): phosphomolybdic acid, anisaldehyde/EtOH, or KMnO4/H2O. Dichloromethane and acetonitrile were purchased from Fisher (OPTIMA). Elemental analyses were performed by Midwest Microlabs, Inc. Sulfur monochloride (98%), 1,5-cyclooctadine (99%) and sodium cyanide (95%) were purchased from Aldrich. Sulfuryl chloride (98.5%) and sodium azide (99%) were purchased from Acros. Inhalation and toxicity hazards are associated with S2Cl2, SO2Cl2 and NaCN. All toxic materials were disposed of in accordance with "Prudent Practices in the Laboratory"; National Academy Press; Washington, DC, 1995.
2,6-Dichloro-9-thiabicyclo[3.3.1]nonane (1)
(Caution! Use a well ventilated hood.) A 4-L, round-bottomed flask equipped with a magnetic stirrer, dropping funnel, and capped with a drying tube was charged with a solution of 1,5-cyclo-octadiene (122 mL, 1.0 mol) in dichloromethane (2.5 L). The solution was cooled with an ice/water bath and sulfur monochloride (43 mL, 0.55 mol) was slowly added in dropwise fashion with vigorous stirring. The reaction mixture was allowed to stir for an additional 2.5 hours at 0–4°C and was then treated with sulfuryl chloride (48 mL, 0.6 mol) added through the dropping funnel over a period of 20 minutes. The cooling bath was removed and the reaction mixture was allowed to stir for 90 minutes while warming to room temperature. TLC analysis showed complete consumption of the starting diolefin. The mixture was washed with a saturated aqueous solution of Na
2S
2O
5 (2 x 200 mL), 0.5 N NaOH (2 x 200 mL), brine (1 x 200 mL), and the organic phase was dried over MgSO
4. Removal of the solvent by rotary evaporation gave a yellow solid (200.6 g, 95% crude yield), which was dissolved in the minimum amount of chloroform (400 mL). Twice the volume of hexane was added and the mixture was stirred with decolorizing charcoal. Following filtration through a pad of silica gel (8.5 cm diameter, 4 cm high) and removal of solvent, 152−169 g of off-white solid was obtained with a melting point of 93−94 ˚C (72−80% yield). The melting point has previously been reported to be 101−102 °C for material recrystallized from benzene [
5]. In our hands, recrystallization leads to poor mass recoveries. The above procedure represents a convenient alternative to the mole-scale synthesis of
1 by Bishop [
4].
2,6-Diazido-9-thiabicyclo[3.3.1]nonane (2)
(Caution! Sodium azide will evolve the toxic and explosive hydrazoic acid gas in the presence of acid! Use safety glasses and nitrile gloves under a well-ventilated hood.) A 2-liter round-bottomed flask was charged with 730 mL of a 5 M aqueous solution of sodium azide and 730 mL of acetonitrile. 2,6-Dichloro-9-thiabicyclo[3.3.1]nonane (1, 154.1 g, 0.73 mol) was added, and the mixture (containing some undissolved material) was stirred at room temperature. After 1h the reaction was complete as judged by TLC, NMR, and by the disappearance of the solid. The product was extracted with ethyl acetate (3 x 200 mL, and the combined organic phases were dried with Na2SO4; removal of the solvent provided rac-2 as a yellow oil (155−160 g, 95−98% yield). If the yellow coloration is too dark or the NMR shows baseline noise, purity can be improved by dissolving the oil in a 6:4 hexane/ethyl acetate mixture and treating the solution with activated carbon, which diminishes the yield to 90−94%. This further treatment is more likely to be necessary if old samples of sulfur-containing starting materials were employed for the synthesis of the starting dichloride. 1H-NMR (CDCl3) δ 1.85 (m, 2H), 2.02 (m, 2H), 2.13 (m, 2H), 2.40 (m, 2H), 2.73 (bs, 2H), 4.12 (m, 2H); 13C-NMR (CDCl3) δ 26.7, 27.3, 34.6, 62.7; HRMS (FAB+) calcd. for C8H13N6S [(M+H)+]: 225.0922, found: 225.0920. Small-molecule azides such as 2 must always be treated with caution due to the potential for explosive decomposition. However, we found 2 to be quite stable in this regard, in the following three tests: (1) The material was inert to the standard “hammer test” for shock sensitivity. (2) Compound 2 was dropped into conc. H2SO4 with no visual or thermal evidence of decomposition. (3) The material was heated in a capillary tube in a melting point apparatus. It began to boil at a bath temperature of approximately 197 °C, started turning yellow at 210 °C, and slowly decomposed through a temperature range up to 240 °C. At that point, the material was brown and smelled unpleasant. No evidence of rapid decomposition was observed on any of several repeated trials. In spite of these comforting observations, however, compound 2 should not be distilled or stored in the absence of solvent.
2,6-Dicarbonitrile-9-thiabicyclo[3.3.1]nonane (3)
(Caution! Avoid mixing NaCN with acids; hydrogen cyanide gas will be created. Use safety glasses and nitrile gloves under a well-ventilated hood.) A 250-mL round-bottomed flask equipped with a magnetic stirring bar and a vacuum-take off adapter was charged with H2O (50 mL) and sodium cyanide (10.8 g, 210 mmol) at room temperature. After the NaCN was dissolved, CH3CN (50 mL) was added. 2,6-Dichloro-9-thiabicyclo[3.3.1]nonane (1, 21.2 g, 100 mmol) was then added through an addition funnel to the turbid solution and the heterogeneous mixture was stirred vigorously for 1.5 h. The solid gradually dissolved, producing a cloudy solution and then a white precipitate after 50−60 min, at which time TLC showed complete consumption of the starting material. The reaction mixture was diluted with EtOAc (100 mL) and extracted with brine (2 x 75 mL). The aqueous layer was washed twice with EtOAc (2 x 75 mL) and the combined organic extracts were dried over MgSO4. The solvent was removed under reduced pressure to yield rac-3 as a white solid (16−17 g, 83−90%). If necessary, colored impurities may be removed by washing rapidly with a small amount of cold EtOAc/hexanes (7:3) or by recrystallization from EtOAc/hexanes. For the non-recrystallized product, 1H-NMR (CDCl3) δ 2.23−2.36 (m, 6H), 2.52−2.54 (m, 2H), 3.03 (m, 2H), 3.41−3.44 (m, 2H); 13C-NMR (CDCl3) δ 25.0, 26.9, 32.3, 33.0, 120.4; IR (thin film, cm−1) 2979, 2870, 2234, 1184, 894. Anal. Calcd. for C10H12N2S: C, 62.46; H, 6.29, N, 14.57; S, 16.68. Found: C, 62.45; H, 6.37, N, 14.36; S, 16.60; mp 158−160 ºC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}