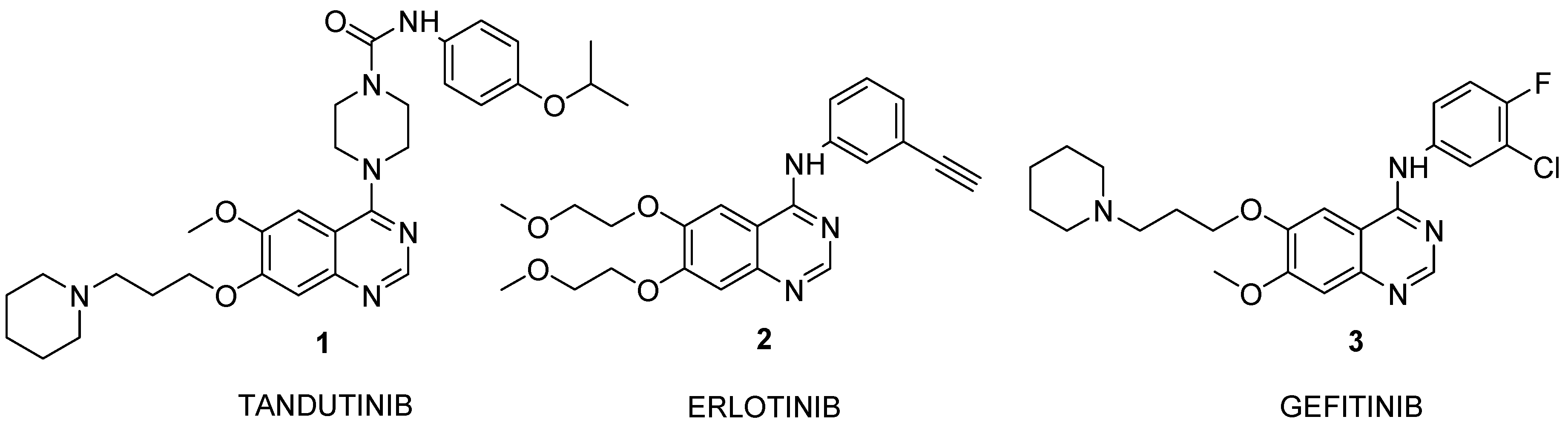
Experimental
General
Melting points were measured on a Büchi melting point apparatus B-545. 1H- and 13C-NMR spectra were recorded on a Bruker AC-200 (200 MHz) pulse Fourier-transform NMR spectrometer in CDCl3 or DMSO-d6. Thin layer chromatography (TLC) was performed on Merck TLC aluminum sheets silica 60 F254. Visualization was by UV light at 254 and 366 nm or spray reagents (molybdophosphoric acid and heating). Column chromatography was performed using silica gel (Baker 40-60 μm). MPLC (medium pressure liquid chromatography) was performed using a LC-8A pump (Shimadzu), a SPD-6AV UV-detector (Shimadzu) and Büchi preparative columns. HPLC was performed using a Waters 2695 instrument and Merck Chromolith RP18 columns and a gradient of 3 % to 60 % acetonitrile/water (0.1 % TFA) at a flow of 3.0 ml/min. The HPLC purity reported is the number generated for the peak area as calculated using the Waters Millennium Software with the Maxplot option for the UV maximum of the corresponding peak.
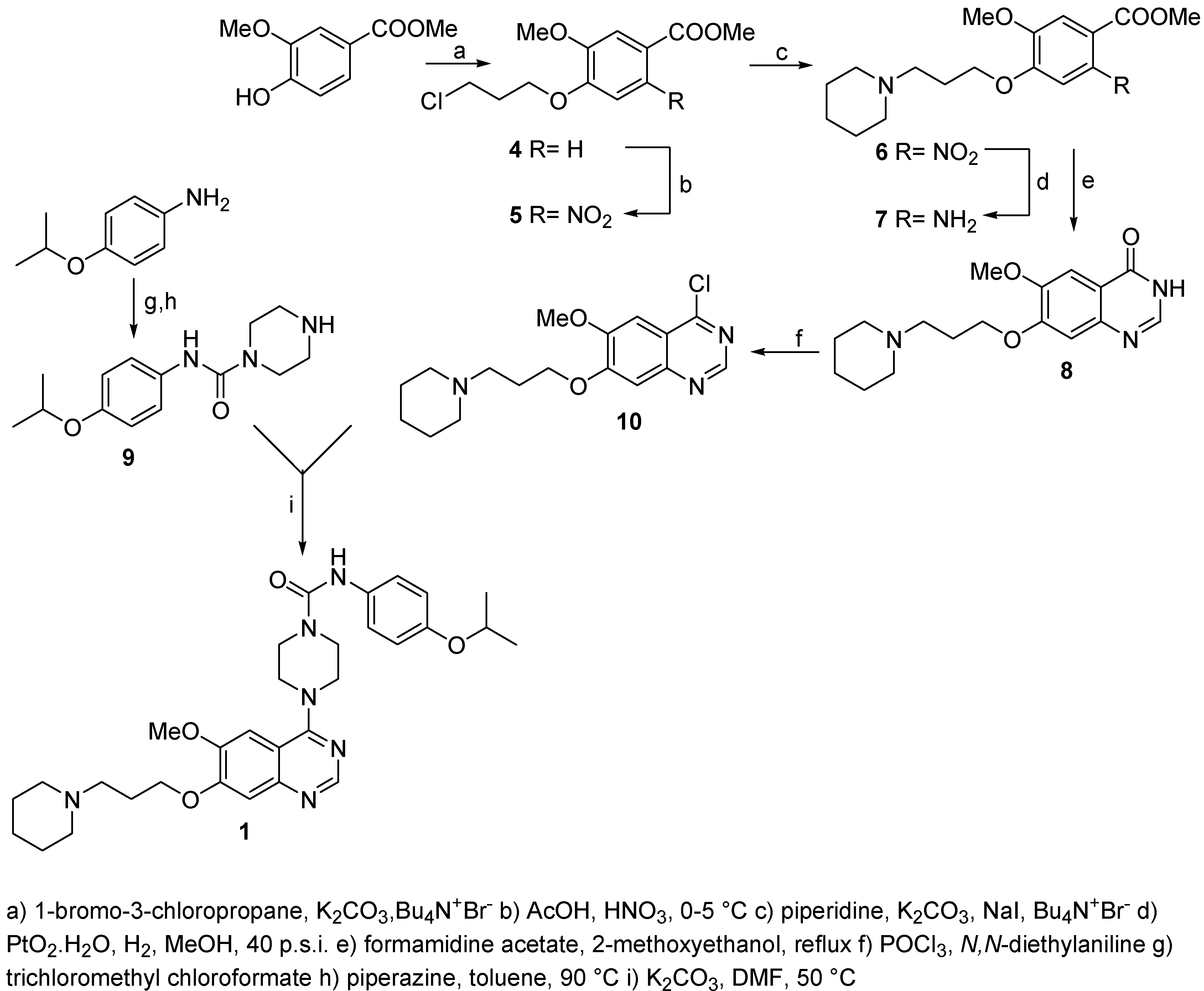
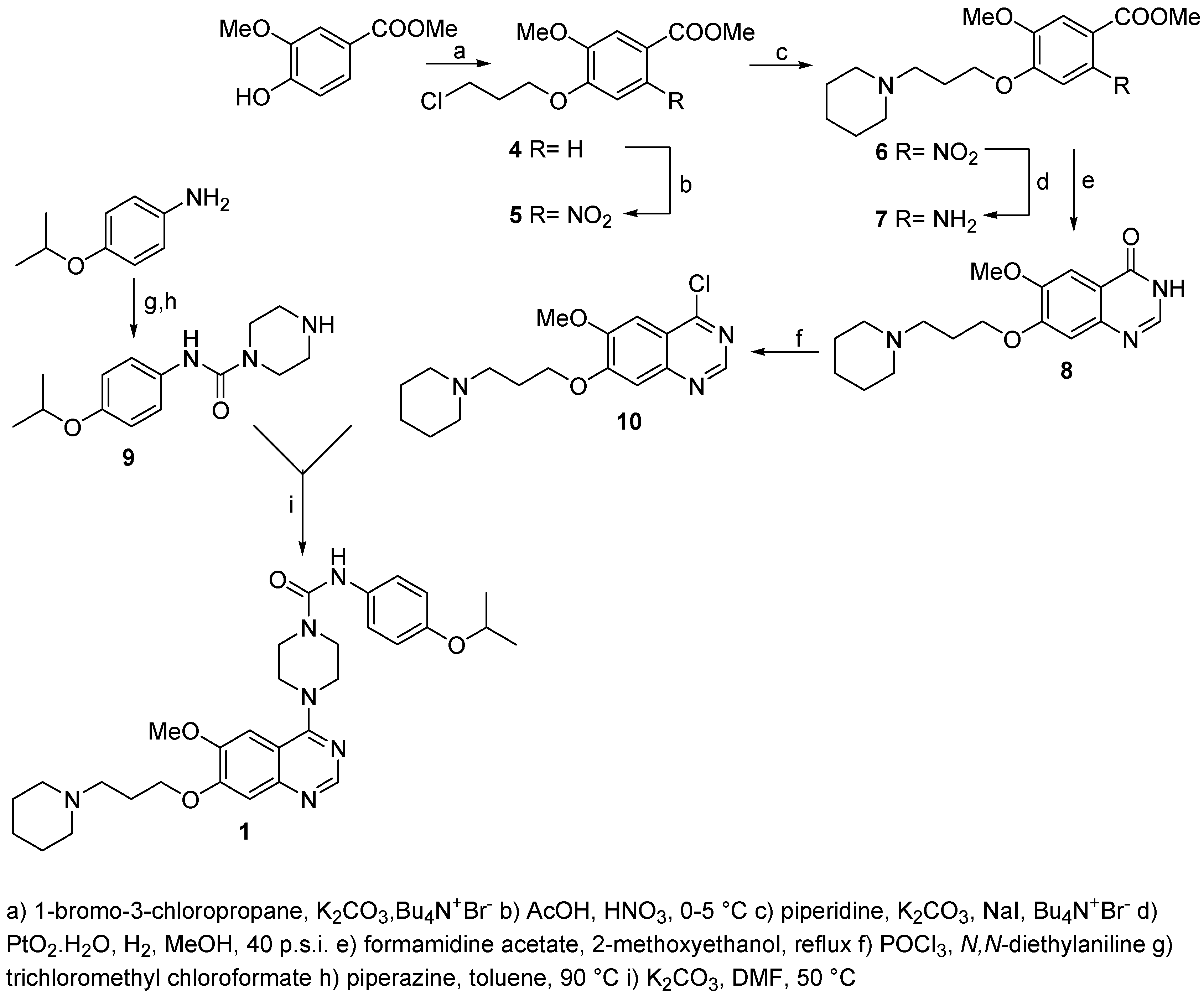
Methyl 4-(3-chloropropoxy)-3-methoxybenzoate (4)
A solution of methyl 3-methoxy-4-hydroxybenzoate (1, 87.0 g, 478 mmol), 1-bromo-3-chloro-propane (200.0 g, 1.270 mmol), potassium carbonate (350.0 g, 2.533 mol) and tetrabutylammonium iodide (9.0 g, 27 mol) in acetone (900 mL) was heated to reflux for 2 h. Reaction progress was monitored by HPLC and the reaction was found to be complete after this time. The reaction mixture was cooled to room temperature, solid material was filtered and washed with acetone. The combined filtrates and washings were evaporated under reduced pressure to give a yellow, solid product (110.6 g) which was crystallized from acetonitrile (500 mL) to afford the title compound (m.p.: 111-113 °C, 93.5 g, 76%, 94% HPLC purity). 1H-NMR (CDCl3): 2.25-2.42 (tt, 2H, -CH2CH2CH2-, 3Ja= 6.26 Hz, 3Jb= 6.06 Hz), 3.74-3.80 (t, 2H, CH2Cl, 3Ja= 6.26 Hz), 3.89 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 4.19-4.25 (t, 2H, CH2O, 3Jb=6.06 Hz), 6.89-6.93 (d, 1H, HAr, 3J= 8.41 Hz), 7.54-7.55 (d, 1H, HAr, 4J= 1.96 Hz), 7.63-7.68 (dd, 1H, HAr, 3J= 8.41 Hz, 4J=1.96 H); 13C-NMR (CDCl3): 32.52, 40.99, 50.06, 68.73, 116.13, 116.05, 123.23, 123.12, 148.63, 149.25.
Methyl 4-(3-chloropropoxy)-5-methoxy-2-nitrobenzoate (5)
A solution of methyl 4-(3-chloropropoxy)-3-methoxybenzoate (4, 93.0 g, 128 mmol) in acetic acid (350 mL) was added dropwise to nitric acid (84.5 mL, 66%) at 0-5 °C and this mixture was stirred at room temperature for 1 h and than for 2 h at 50 °C. According to HPLC the reaction was complete. The reaction mixture was poured on ice/water (1300 mL) and extracted with ethyl acetate (6 x 200 mL). The combined organic phases were collected, washed with saturated sodium bicarbonate (2 x 150 mL), brine (500 mL), dried (Na2SO4) and decolorized (charcoal). Ethyl acetate was then removed under reduced pressure to give a yellow oil (130.3 g) which was crystallized from ethyl acetate/petroleum ether to afford the product as light yellow crystals (m.p.: 54-56 °C, 81.3 g, 75%, 97% HPLC purity). 1H-NMR (CDCl3): 2.26-2.38 (tt, 2H, -CH2CH2CH2-, 3Ja= 6.16 Hz, 3Jb= 5.87 Hz), 3.74-3.80 (t, 2H, CH2Cl, 3Ja= 6.16 Hz), 3.90 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 4.22-4.28 (t, 2H, CH2O, 3Jb=5.87 Hz), 7.07 (s, 1H, HAr), 7.48 (s, 1H, HAr); 13C-NMR (CDCl3): 32.95, 42.33, 50.03, 58.54, 70.02, 112.22, 115.62, 120.16, 143.15, 150.05, 154.87, 169.87.
Methyl 5-methoxy-2-nitro-4-(3-piperidin-1-yl-propoxy)benzoate (6)
A solution of methyl 4-(3-chloropropoxy)-5-methoxy-2-nitrobenzoate (5, 110.0 g, 0.362 mol), potassium carbonate (200.0 g, 1.447 mol), sodium iodide (110 g, 0.734 mol) and tetrabutylammonium iodide (5.7 g, 10.7 mmol) in acetonitrile (900 mL) was stirred for 5-10 min at room temperature. Piperidine (95 g, 1.116 mol) was added and this mixture heated to reflux for 3 h. The reaction was found to be complete after this time (HPLC). Solid material was removed by filtration and washed with acetone. The combined filtrates were evaporated and the dark product obtained dissolved in dichloromethane (650 mL) and extracted with water (4 x 250 mL). The organic phase was dried (Na2SO4), decolorized (charcoal), filtered and evaporated to afford the product (110.0 g, 86%, 98% HPLC purity) as an amber oil. 1H-NMR (CDCl3): 1.41-1.67 (m, 6H, 3 x CH2pip.), 2.26-2.38 (m, 2H, -CH2CH2CH2) 2.48-2.62 (m, 6H, 3 x CH2N), 3.95 (s, 3H, OCH3), 4.22 (s, 3H, OCH3), 4.28-4.39 (t, 2H, CH2O), 7.13 (s, 1H, HAr), 7.62 (s, 1H, HAr); 13C-NMR (CDCl3): 24.32, 26.25, 32.33, 49.95, 51.15, 53.03, 71.23, 111.12, 116.26, 117.87, 141.15, 150.07, 153.02, 170.08.
Methyl 2-amino-5-methoxy-4-(3-piperidin-1-yl-propoxy)benzoate (7)
A solution of methyl 5-methoxy-2-nitro-4-(3-piperidin-1-yl-propoxy)benzoate (6, 90.0 g, 0.25 mol) in methanol (700 mL) was hydrogenated with PtO2·H2O (1.0 g) at room temperature and an initial pressure of 40 p.s.i. for 48 h until no further hydrogen uptake was noted. The catalyst was filtered, washed with methanol and volatiles evaporated to give a dark oil, which was dissolved in dichloromethane (400 mL), extracted with a saturated solution of sodium carbonate (1 x 100 mL) and brine (1 x 100 mL), dried (Na2SO4) and decolorized (charcoal). Dichloromethane was removed on the rotavapor and the brown, semisolid residue triturated with diisopropyl ether to afford the product as a light brown powder (m.p.: 96-98 °C, 63.4 g, 77%, 97% HPLC purity). 1H-NMR (CDCl3): 1.35-1.47 (m, 2H, CH2pip.), 1.52-1.63 (m, 4H, 2 x CH2pip.), 1.94-2.08 (tt, 2H, -CH2CH2CH2-, 3J= 6.85 Hz), 2.35-2.48 (m, 6H, 3 x CH2N), 3.79 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 4.00-4.06 (t, 2H, CH2O, 3J=6.85 Hz), 5.56 (w, 2H, NH2), 6.16 (s, 1H, Hard), 7.29 (s, 1H, Hard); 13C-NMR (CDCl3): 25.07, 27.06, 30.99, 50.25, 51.65, 53.80, 73.06, 101.65, 110.20, 118.85, 141.18, 136.65, 149.07, 153.02, 167.88.
6-Methoxy-7-(3-piperidin-1-yl-propoxy)quinazoline-4(3H)-one (8)
A solution of methyl 2-amino-5-methoxy-4-(3-piperidin-1-yl-propoxy)benzoate (7, 25.0 g, 78 mol) and formamidine acetate (10.4 g, 100 mmol) in 2-methoxyethanol (250 mL) was heated to reflux for 4 h. To complete the reaction another portion of formamidine acetate (2.00 g, 19.2 mmol) was added and heating continued for further 2 h. The reaction progress was monitored by HPLC and found to be complete after this time. Volatiles were removed under reduced pressure and the orange, sticky residue obtained triturated with diethyl ether (4 x 150 mL) to afford the product as a white powder which was collected by filtration and air dried (m.p.: 218-219 °C, 20.9 g, 85%, 99% HPLC purity). 1H-NMR (CDCl3): 1.32-1.40 (m, 2H, CH2pip.), 1.43-1.53 (m, 4H, 2 x CH2pip.), 1.83-1.97 (tt, 2H, -CH2CH2CH2-, 3J= 6.26 Hz), 2.29-2.41 (m, 6H, 3 x CH2N), 3.86 (s, 3H, OCH3), 4.09-4.15 (t, 2H, CH2O, 3J=6.26 Hz), 7.10 (s, 1H, HAr), 7.43 (s, 1H, HAr), 7.97 (s, 1H, HAr); 13C-NMR (CDCl3): 27.02, 29.34, 31.37, 51.25, 53.78, 55.23, 71.19, 109.78, 115.52, 120.57, 142.07, 146.36, 150.15, 165.28, 176.32.
4-Chloro-6-methoxy-7-(3-piperidin-1-yl-propoxy)quinazoline (10)
Phosphoryl chloride (80 mL) was added to N,N-diethylaniline (6.2 mL) with magnetic stirring followed by addition of 6-methoxy-7-(3-piperidin-1-ylpropoxy)quinazoline-4(3H)-one (8, 10.0 g, 31.51 mmol) and the reaction flask was immersed in a preheated oil bath (70 °C).The temperature was increased to 90 °C over a period of 10 min. and kept at 80-90 °C for another 30 min. Most of the excess of phosphoryl chloride was then removed under reduced pressure and the resulting dark oil triturated with toluene (3 x 40 mL). The residue was dissolved in water (20 mL) and extracted with ethyl acetate (2 x 15 mL). The pH of the aqueous phase was adjusted to 9-10 with sodium bicarbonate and it was extracted with dichloromethane (3x150 mL), dried (Na2SO4), decolorized (charcoal) and the organic solvent was removed to afford the product as a yellowish oil (9.1 g, yield 86%, 99% HPLC purity). 1H-NMR (CDCl3): 1.30-1.39 (m, 2H, CH2pip.), 1.41-1.50 (m, 4H, 2 x CH2pip.), 1.82-1.96 (tt, 2H, -CH2CH2CH2-), 2.16-2.35 (m, 6H, 3 x CH2N), 3.80 (s, 3H, OCH3), 4.00-4.09 (t, 2H, CH2O), 7.41 (s, 1H, HAr), 7.45 (s, 1H, HAr), 8.76 (s, 1H, HAr); 13C-NMR (CDCl3): 26.22, 27.94, 30.27, 51.45, 52.97, 56.54, 70.03, 101.88, 108.52, 121.57, 147.07, 155.26, 160.95, 157.28, 159.32.
N-(4-Isopropoxyphenyl)-4-[6-methoxy-7-(3-piperidin-1-yl-propoxy)quinazolin-4-yl]piperazine-1-carboxamide (1)
A suspension of N-(4-isopropoxyphenyl)piperazine-1-carboxamide (9, 6.90 g, 88%, 23 mmol) and potassium carbonate (12.7 g, 96.2 mmol) in dry DMF (60 mL) was stirred at room temperature for 20 min, then 4-chloro-6-methoxy-7-(3-piperidin-1-yl-propoxy)quinazoline (10, 7.83 g, 23 mmol, 98.8%) was added and this mixture was heated at 50 °C for 18 h. The reaction was monitored by HPLC and found to be complete. The reaction mixture was poured on water (500 mL) and extracted with dichloromethane (4 x 150 mL). The combined phases were washed with brine (2 x 80 mL), dried (Na2SO4), decolorized (charcoal) and the solvent was removed under reduced pressure to dryness to give a crude yellow, sticky material (12.4 g, yield 96%, 95% HPLC purity) which was crystallized from ethyl acetate/petroleum ether to afford the product >99% HPLC purity. 1H-NMR (CDCl3): 1.29-1.32 (d, 6H, 2 x CH3, 3J=6.06 Hz) 1.39-1.50 (m, 2H, CH2pip.), 1.53-1.64 (m, 4H, 2 x CH2pip.), 2.03-2.17 (tt, 2H, -CH2CH2CH2-, 3Ja= 6.84 Hz, 3Jb= 6.65 Hz), 2.38-2.43 (m, 4H, 2 x CH2N), 2.47-2.54 (t, 2H, CH2N, 3Ja= 6.84 Hz), 3.71 (w, 8H, 4 x CH2N), 3.96 (s, 3H, OCH3), 4.19-4.25 (t, 2H, CH2O, 3Jb=6.65 Hz), 4.38-4.56 (m, 1H, (CH3)2CH), 6.50 (s, 1H, NH), 6.81-6.86 (m, 2H, HAr) 7.09 (s, 1H, HAr), 7.22-7.27 (m, 3H, HAr), 8.67 (s, 1H, HAr); 13C-NMR (CDCl3): 23.02, 25.65, 28.82, 31.11, 49.89, 52.20, 54.70, 56.76, 58.89, 72.31, 73.76, 103.11, 108.99, 114.45, 118.25, 122.43, 131.32, 148.87, 150.12, 155.67, 158.87, 160.76, 161.73, 165.75, 180.11.
N-(4-Isopropoxyphenyl)piperazine-1-carboxamide (9)
A solution of 4-isopropoxyphenylamine (1.0 g, 6.61 mmol) in dry toluene (10 mL) was added drop- wise to a solution of trichloromethyl chloroformate (13.0 g, 9.94 mmol) in dry toluene (10 mL) at 0-5 °C and the mixture was stirred at the same temperature for 20 min. The reaction mixture was then stirred and heated to 100 °C over 1h and kept at this temperature for 1 h. Reaction progress was monitored by HPLC and found to be complete by this time. The reaction mixture was cooled to room temperature and extracted with saturated solution of sodium bicarbonate (1 x 10 mL). The toluene solution was separated and aqueous phase extracted with toluene (5 mL). The combined toluene layers were added dropwise to a solution of piperazine (2.80 g, 32.5 mmol) in toluene (10 mL) and this mixture was heated at 90 °C for 3 h, cooled and washed with water (2 x 15). The toluene layer was dried (Na2SO4) and evaporated to afford the product (0.91 g, 52%, 95% HPLC purity). 1H-NMR (DMSO): 1.20-1.23 (d, 6H, 2 x CH3, 3J= 6.06), 2.73-2.78 (m, 4H, 2 x CH2N), 3.15 (w, 1H, NH), 3.39-3.48 (m, 4H, 2 x CH2N), 4.41-4.53 (s, 1H, 3J=6.06), 6.75-6.80 (d, 2H, HAr, 3J=8.9), 7.31-7.36 (d, 2H, HAr, 3J=8.9), 8.37 (s, 1H, NH); 13C-NMR (DMSO): 20.28, 49.05, 55.63, 68.82, 111.33, 120.20, 126.35, 155.05, 160.01.
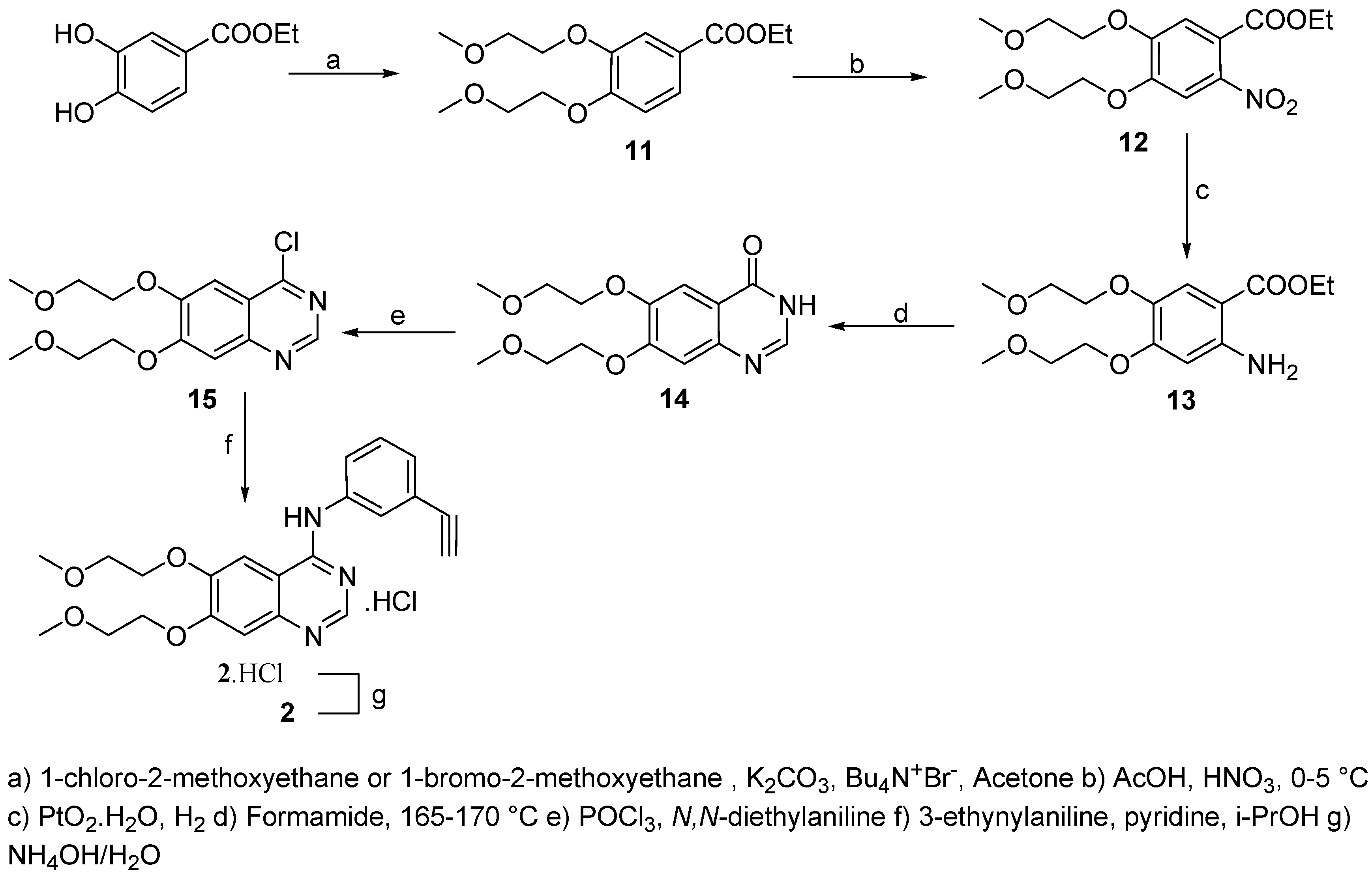
Ethyl 3,4-bis(2-methoxyethoxy)benzoate (11)
A suspension of ethyl 3,4-dihydroxybenzoate (40.0 g, 220 mmol), potassium carbonate (70.0 g, 507 mmol) and tetrabutylammonium iodide (2.80 g, 7.6 mmol) in acetone (300 mL) was stirred for 20 min at room temperature. 1-Chloro-2-methoxyethane (158.0 g, 1.671 mol) was added and the solution stirred and heated to reflux for 5 days. Reaction progress was monitored by HPLC and the reaction was found to be complete after this time.The reaction mixture was cooled to room temperature and diethyl ether (250 mL) was added. Inorganic salts were filtered off and washed with diethyl ether (2 x 250 mL). The combined organic layers were dried (Na2SO4), decolorized (charcoal) and volatiles evaporated under reduced pressure to afford the product as light oil (72.5 g) which was dissolved in petrol ether (250 mL) and cooled to 0-5 °C. The resulting white precipitate was collected by filtration and dried on the air (m.p.: 55-58 °C, 61.8 g, yield: 94 %, 97% HPLC purity). 1H-NMR (CDCl3): 1.27-1.34 (t, 3H, CH3CH2, 3J= 7.10 Hz), 3.38 (s, 6H, 2 x OCH3), 3.69-3.74 (m, 4H, 2 x CH2O), 4.11-4.15 (m, 4H, 2 x CH2O), 4.21-4.32 (q, 2H, CH2, 3J=7.10 Hz), 6.82-6.86 (d, 1H, HAr, 3J=8.41 Hz), 7.51-7.52 (d, 1H, HAr, 4J=1.96 Hz), 7.57-7.62 (dd, 1H, HAr, 3J=8.41 Hz, 4J=1.96 Hz); 13C-NMR (CDCl3): 15.08, 50.25, 61.62, 70.76, 73.89, 111.54, 116.27, 121.83, 124.35, 139.12, 152.55, 165.13.
Ethyl 4,5-bis(2-methoxyethoxy)-2-nitrobenzoate (12)
A solution of ethyl 3,4-bis(2-methoxyethoxy)benzoate (11, 67.0 g, 0.23 mol) in AcOH (230 mL) was added dropwise to nitric acid (60 mL, 65%) at 0-5 °C, stirred at the same temperature for 30 min and than for 24 h at room temperature. Reaction progress was monitored by HPLC and it was found to be complete after this time. The reaction mixture was poured on ice/water (1000 mL) followed by extraction with ethyl acetate (5 x 250 mL). The combined organic layers were washed with saturated NaHCO3 (3 x 250 mL), brine (3 x 250 mL), dried (Na2SO4), decolorized (charcoal) and evaporated to give the product (71.30 g, 92%, 95% HPLC purity) as an amber oil. 1H-NMR (CDCl3): 1.35-1.43 (t, 3H, CH3CH2, 3J= 7.20 Hz), 3.35 (s, 6H, 2 x OCH3), 3.72-3.85 (m, 4H, 2 x CH2O), 4.20-4.26 (m, 4H, 2 x CH2O), 4.31-4.42 (q, 2H, CH2, 3J=7.20 Hz), 7.52 (s, 1H, HAr,), 7.82 (s, 1H, HAr); 13C-NMR (CDCl3): 20.12, 51.55, 59.92, 71.13, 76.18, 115.54, 118.03, 120.16, 142.24, 149.86, 150.12, 167.23.
Ethyl 2-amino-4,5-bis(2-methoxyethoxy)benzoate (13)
A solution of ethyl 4,5-bis(2-methoxyethoxy)-2-nitrobenzoate (12, 38.0 g, 0.11 mmol) in methanol (250 mL) was hydrogenated in a Parr apparatus with PtO2·H2O (0.5 g) at room temperature and 50 p.s.i. pressure. Reaction development was monitored by HPLC and the reduction continued until no more hydrogen was consumed. The catalyst was filtered off and methanol removed to afford a brown slurry (39.2 g) which was triturated with diethyl ether (500 mL). The light brown precipitate was filtered trough a glass funnel and dried on the air (34.2 g, 99%, 94% HPLC purity). 1H-NMR (CDCl3): 1.30-1.37 (t, 3H, CH3CH2, 3J= 7.10 Hz), 3.30 (s, 6H, 2 x OCH3), 3.70-3.81 (m, 4H, 2 x CH2O), 4.16-4.21 (m, 4H, 2 x CH2O), 4.25-4.34 (q, 2H, CH2, 3J=7.10 Hz), 5.25 (br, 2H, NH2), 6.38 (s, 1H, HAr,), 7.42 (s, 1H, HAr); 13C-MR (CDCl3): 18.54, 52.45, 60.92, 74.13, 77.33, 101.24, 108.76, 116.53, 133.55, 141.20, 150.05, 167.23.
6,7-Bis-(2-methoxyethoxy)-quinazolin-4(3H )-one (14)
A solution of ethyl 2-amino-4,5-bis(2-methoxyethoxy)benzoate (13, 4.0 g, 109 mmol) in formamide (50 mL) was heated to 165-170 °C under N2 for 12 h., when HPLC indicated the absence of starting material. The reaction mixture was cooled and the amber sticky precipitate triturated with diethyl ether (250 mL), filtered and triturated in boiling acetonitrile (100 mL) for 30 min, than cooled to 5 °C and diethyl ether (300 mL) was added. The resulting white powder was filtered using a sintered glass frit and dried under reduced pressure. (m.p.: 183-184 °C, 26.8 g, 84%, 98% HPLC purity). 1H- NMR (DMSO): 3.32 (s, 6H, OCH3), 3.67-3.73 (m, 4H, 2 x OCH2), 4.16-4.26 (m, 4H, 2 x OCH2), 7.13 (s, 1H, HAr), 7.45 (s, 1H, HAr), 7.97 (s, 1H, HAr); 13C-NMR (DMSO): 52.26, 70.13, 73.15, 110.44, 115.23, 121.22, 139.79, 140.12, 143.56, 150.22, 165.56, 172.23.
4-Chloro-6,7-bis-(2-methoxyethoxy)-quinazoline (15)
Phosphoryl chloride (30 mL) was added to N,N-diethylaniline (5.8 g, 38.9 mmol) with magnetic stirring followed by addition of 6,7-bis-(2-methoxyethoxy)-quinazolin-4(3H)-one (14, 10.0 g, 34 mmol) and the reaction flask was immersed in oil bath preheated to 70 °C. The temperature was increased to 90 °C over a period of 10 min. and kept at 80-90 °C for additional 30 min. Most of the excess of phosphoryl chloride was removed under reduced pressure and the dark oil triturated with toluene (3 x 150 mL). Oil residue was poured on crushed ice/water mixture (200 mL) and extracted with ethyl acetate (4 x 150 mL). The combined organic layers were dried (Na2SO4) and decolorized (charcoal). The volatiles were evaporated under reduced pressure to give the product as yellowish crystals (m.p.:105-107 °C, 10.8 g, 89%, 96% HPLC purity). 1H-NMR (CDCl3): 3.49 (s, 3H, OCH3), 3.50 (s, 3H, OCH3), 3.87-3.91 (m, 4H, 2 x OCH2), 4.30-4.36 (m, 4H, 2 x OCH2), 7.33 (s, 1H, HAr), 7.42 (s, 1H, HAr), 8.85 (s, 1H, HAr); 13C-NMR (CDCl3): 50.34, 69.99, 71.46, 102.72, 110.33, 122.24, 146.98, 150.42, 143.56, 158.87, 160.30, 163.33.
N-(3-ethynylphenyl)-6,7-Bis-(2-methoxyethoxy)-quinazolin-4-amine hydrochloride (2·HCl) and free base 2
A solution of 4-chloro-6,7-bis-(2-methoxyethoxy)-quinazoline (15, 20.30 g, 65 mmol) in i-PrOH (100 mL) was added drop wise to a solution of pyridine (5.90 g, 75 mmol) and 4-ethynylphenylamine (8.80 g, 75 mmol) in i-PrOH (260 mL). This mixture was stirred and heated at reflux for 4 h under argon resulting in the precipitation of an orange solid. After stirring at room temperature overnight the precipitate was filtered, washed with hot i-PrOH and dried on the air to afford the crude 2·HCl product (26.4 g, 95%, 95% HPLC purity) which was crystallized from methanol to afford a yellowish powder (m.p.: 219-221 °C, > 99% HPLC purity). To isolate the free base 0.5 g of 2·HCl was dissolved in water (10 mL) and basified using conc. aq. ammonia (5 mL). Extraction with dichloromethane (4 x 25 mL), gave after drying and evaporation, 2 as yellow crystals. (m.p.: 159-160 °C, 425 mg, 92%, 99% HPLC purity). 1H-NMR (CDCl3): 3.08 (s, 1H, CH), 3.40 (s, 3H, OCH3), 3.73-3.78 (m, 4H, 2 x CH2O), 4.13-4.21 (m, 4H, 2 x CH2O), 7.12 (s, 1H, HAr), 7.42-7.36 (m, 3H, HAr), 7.70-7.76 (m, 1H, HAr), 7.85 (s, 1H, HAr), 7.96 (w, 1H, NH), 8.60 (s, 1H, HAr); 13C-NMR (CDCl3): 51.25, 73.52, 75.78, 80.63, 86.76, 100.97, 108.83, 114.15, 117.65, 122.34, 124.35, 125.76, 130.89, 142.31, 147.28, 151.20, 155.07, 159.11, 170.22.
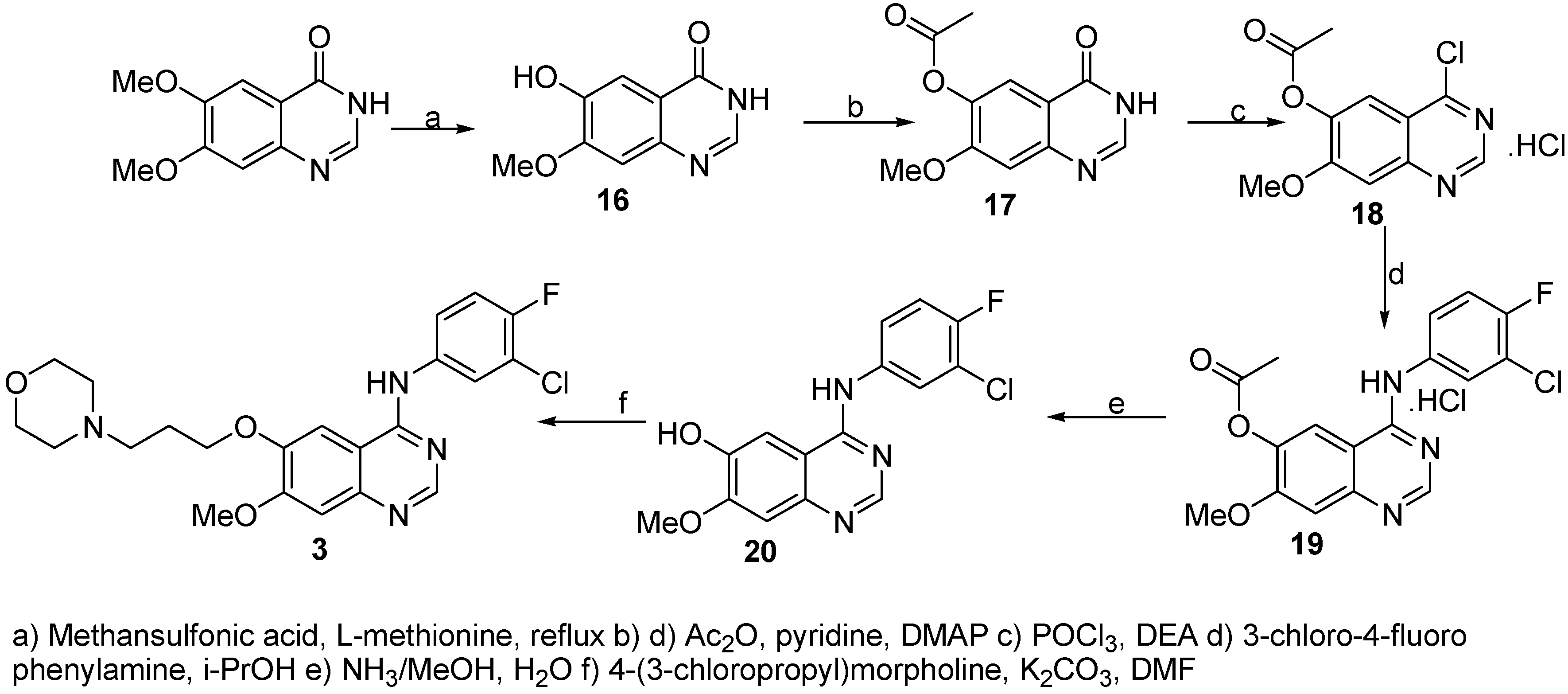
6-Hydroxy-7-methoxyquinazolin-4(3H)-one (16)
A mixture of 6,7-dimethoxyquinazolin-4(3H)-one (64.7 g, 0.31 mol) and L-methionine (53.7 g, 0.34 mol) was dissolved in methanesulfonic acid (425 mL) and heated to reflux for 12 h. Reaction was monitored by HPLC and no starting material was indicated after this time. Crushed ice/water mixture (200 mL) was added and the solution was cooled to 0 °C. Then NaOH (40% water solution) was added slowly (pH~7) resulting in precipitation of a white deposit. This product was collected by filtration using a sintered glass funnel, washed with water and dried at 50 °C and 23 mbar to afford the product (58.3 g, 98%, 90% HPLC purity) which was used without further purification.
7-Methoxy-4-oxo-3,4-dihydroquinazolin-6-yl acetate (17)
A suspension of 6-hydroxy-7-methoxyquinazolin-4(3H)-one (16, 36.7 g, 191 mmol) in acetic anhydride (300 mL), pyridine (41.2 mL) and N,N-dimethyl-4-aminopyridine (100 mg, 0.819 mmol) was stirred and heated to 100 °C under Ar atmosphere for 6 h. Reaction progress was monitored by HPLC. Crushed ice/water solution (300 mL) was added and the resulting white deposit filtered, washed with water and dried over P2O5 to afford the product (25.05 g, 56%, 95% HPLC purity). 1H-NMR (DMSO): 2.29 (s, 3H, CH3CO), 3.90 (s, 3H, OCH3), 7.26 (s, 1H, HAr.), 7.74 (s, 1H, HAr.), 8.07 (s, 1H, HAr.); 13C-NMR (DMSO): 21.22, 57.31, 110.12, 114.65, 120.82, 136.65, 144.99, 146.22, 164.23, 166.81, 170.25.
4-Chloro-7-methoxyquinazolin-6-yl acetate (18)
A solution of 7-methoxy-4-oxo-3,4-dihydroquinazolin-6-yl acetate (17, 30.0 g, 0.128 mmol) and N,N-diethylaniline (28.5 mL) in phosphoryl chloride (114 mL) was immersed in a preheated oil bath (100 °C) and at this temperature stirred for 30 min. The reaction mixture was cooled to 80 °C and stirred for further 30 min. Phosphoryl chloride was removed and the obtained crude material triturated with abs. toluene (3 x 150 mL). Crushed ice/water was added (200 mL), the light brown precipitate filtered and washed with ice water (500 mL). The brownish precipitate was dried over P2O5 to give the product (33.90 g, 92%, 99% HPLC purity). 1H-NMR (DMSO): 2.34 (s, 3H, CH3CO), 3.97 (s, 3H, OCH3), 7.48 (s, 1H, HAr.), 7.86 (s, 1H, HAr.), 8.76 (s, 1H, HAr.); 13C-NMR (DMSO): 20.29, 56.66, 114.53, 119.99, 108.82, 139.66, 148.09, 156.76, 158.42, 164.32, 168.39.
4-[(3-Chloro-4-fluorophenyl)amino]-7-methoxyquinazolin-6-yl acetate (19)
A solution of 4-chloro-7-methoxyquinazolin-6-yl acetate (18, 27.96 g, 0.111 mol) and 3-chloro-4-fluorophenylamine (16.12 g, 0.111 mol) in i-PrOH (500 mL) was stirred and heated to 90 °C under Ar atmosphere for 5 h. Reaction progress was monitored by HPLC and no starting materials were detected after this time. The reaction mixture was cooled to room temperature and the obtained precipitate was filtered trough a glass funnel, dried under reduced pressure (50.8 g) and crystallized from methanol to afford 19 (42.2 g, 96%, 96% HPLC purity) as a white powder. 1H-NMR (DMSO): 2.27 (s, 3H, CH3CO), 3.90 (s, 3H, OCH3), 7.38-7.46 (m, 1H, HAr.), 7.63-7.67 (m, 1H, HAr.), 7.92-7.97 (m, 1H, HAr.), 8.75 (s, 1H, HAr.), 8.84 (s, 1H, HAr.), 11.55 (w, 1H, NH); 13C-NMR (DMSO): 20.55, 57.11, 110.22, 112.25, 112.82, 114.46, 116.29, 118.52, 119.30, 122.34, 141.35, 143.77, 147.82, 153.76, 158.82, 161.01, 169.23, 170.11.
4-[(3-Chloro-4-fluorophenyl)amino]-7-methoxyquinazolin-6-ol (20)
A solution of 4-[(3-chloro-4-fluorophenyl)amino]-7-methoxyquinazolin-6-yl acetate (19, 35.0 g, 97 mmol) in NH3/MeOH (350 mL, 7 N solution)/H2O (200 mL) was stirred at room temperature for 24 h. No starting material was present after this time indicated by HPLC. The white precipitate was filtered off and washed with small amount of water. The product was dried at 50 °C to afford 20 as a white solid (27.8 g, 90%, 98.5% HPLC purity).
N-(3-Chloro-4-fluorophenyl)-7[methoxy-6-[(3-morpholin-4-yl)propoxy]-quinazolin-4-yl]amine (3)
A solution of 4-[(3-chloro-4-fluorophenyl)amino]-7-methoxyquinazolin-6-ol (20, 27.8 g, 87 mmol) and potassium carbonate (24.1 g, 175 mmol) in DMF (500 mL) was stirred at 40 °C for 20 min and than 4-(3-chloropropyl)morpholine was added. This mixture was stirred and heated at 80 °C for 12 h under an Ar atmosphere. Reaction development was monitored by HPLC and reaction found to be complete after this time.The reaction mixture was poured on crushed ice/water mixture and extracted with ethyl acetate (4 x 150 mL). Organic layers were combined, extracted with brine (2 x 100 mL), dried (Na2SO4) and solvent was removed. The crude material was crystallized from toluene to afford the product (29.5 g, 76%, > 99% HPLC purity). 1H-NMR (DMSO): 1.93-2.00 (m, 2H, CH2CH2CH2), 2.34-2.51 (m, 6H, N(CH3)3), 3.54-3.59 (m, 4H, O(CH2)2), 3.92 (s, 3H, OCH3), 4.12-4.18 (m, 2H, ArOCH2), 7.16 (s, 1H, HAr.), 7.35-7.45 (m, 1H, HAr.),7.45-7.82 (m, 2H, HAr.), 8.08-8.13 (m, 1H, HAr.), 8.48 (s, 1H, HAr.), 9.51 (s, 1H, HAr.); 13C-NMR (DMSO): 25.84, 38.20, 38.62, 39.04, 39.46, 39.87, 40.29, 40.71, 53.35, 54.90, 55.69, 66.11, 67.10, 102.53, 107.11, 108.74, 116.02, 116.45, 118.45, 118.81, 122.02, 122.16, 123.29, 136.78, 136.84, 146.86, 148.22, 150.58, 152.42, 154.36, 155.40, 155.87.

{kind=link}
{kind=link}
{kind=link}
{kind=link}