Abstract
This paper describes the fine-tuning of the acidichromic properties of a coumarin-containing colorant 1 by incorporation of electron-donating and electron-withdrawing substituents on the coumarin moiety. Colorant 1 can undergo two distinct and reversible color changes under both strongly acidic and basic conditions, but not in the presence of gaseous ammonia. The results indicated that the bromo-substituted compound 5b changes from red to yellow when exposed to gaseous ammonia, both in solution and on polycarbonate film, suggesting that an electron-withdrawing group at the 7-position of the coumarin moiety made the enolic hydrogen on 5b more susceptible to deprotonation by a base than in the unsubstituted compound 1.
Introduction
The term acidichromic colorants refers to compounds exhibiting reversible color changes depending on either the pH in the solution or on alternating exposure to HCl and NH3 in films [1,2,3,4,5,6,7,8,9]. These compounds are highly promising for use in acid-base sensors [2], photo- and chemical-switching systems [10] and gas-controlled reversible color-change devices [11]. Given that most reported acidichromic colorants contain only one pH-sensitive functional group in their molecules, their color changes are limited to a narrow pH range. In an effort to develop acidichromic colorants that can change their color under both extremely high basicity and high acidity, we have previously reported [12,13] the rational design and chemical synthesis of a novel acidichromic colorant 1 by the combination of an acid and base-sensitive triketone-containing functional group with an aniline-containing molecule. The resulting compound underwent two distinct and reversible color changes under strong acidic and basic conditions (Scheme 1). When exposed to ammonia gas, however, compound 1 failed to exhibit a distinct red to yellow color change. The UV-vis spectra also indicated that two thirds of enolic hydrogen atoms remained intact after bubbling with gaseous ammonia [12]. A simple explanation of this observation is that the basicity of ammonia is not sufficiently strong to completely deprotonate the intramolecular hydrogen-bonded hydrogen in compound 1. Presumably, the relative strength of the intramolecular hydrogen bond in 1 can be further adjusted by incorporating different substitutents on the benzene ring of the coumarin moiety. Since hydrochloric acid and ammonia are two of the most extensively used acidic and basic gases in industry, design of an organic sensor which can detect or respond to both gases is highly desired for safety reasons [14,15,16,17]. In an effort to tune finely the acidichromic properties of 1, electron-donating and electron-withdrawing substituents were introduced to the 7-position of the coumarin moiety. The synthesis and the evaluation of their acidichromic properties are reported.
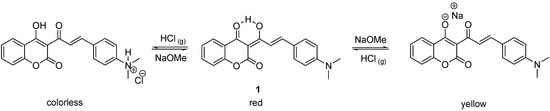
Scheme 1.
The acidichromic switch of 1 and the corresponding colors in acidic, neutral and basic conditions.
Scheme 1.
The acidichromic switch of 1 and the corresponding colors in acidic, neutral and basic conditions.
Results and Discussion
Scheme 2 shows the preparation of the target molecules 5a-b. The syntheses started with the esterification of 7-substituted-4-hydroxycoumarins 2a-b with acetyl chloride in methylene chloride using triethylamine as a base, followed by a potassium cyanide-catalyzed isomerization to 3-acetyl-4-hydroxycoumarins 4a-b [17]. The subsequent piperidine-catalyzed aldol condensation of 4a-b with 4-N,N-dimethylaminobenzaldehyde in benzene using the Dean-Stark trap afforded 7-methoxy-substituted 5a and 7-bromo-substituted 5b with overall yields of 61 and 57 %, respectively.
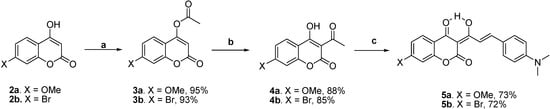
Scheme 2.
Synthesis of 5a-b.
Scheme 2.
Synthesis of 5a-b.
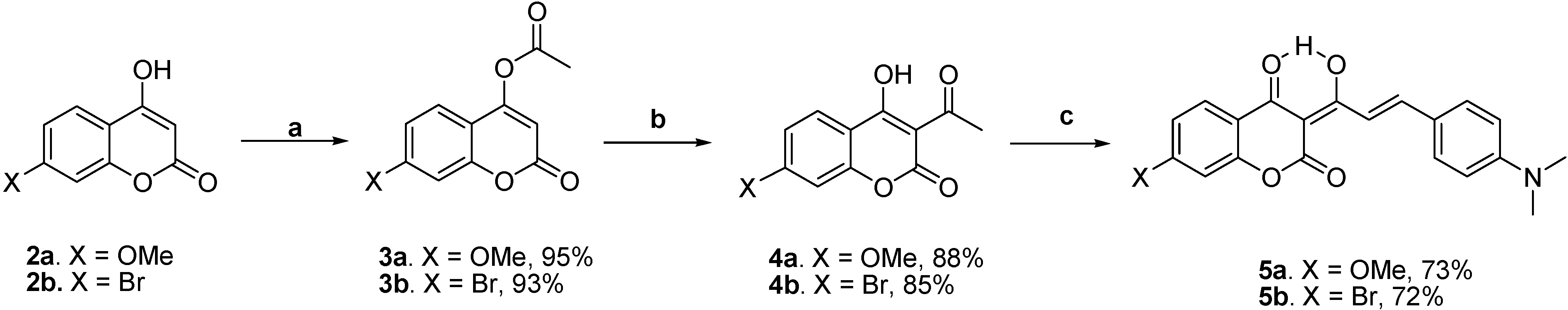
Reagents and conditions: (a) AcCl, Et3N, CH2Cl2, 0 oC, 0.5 h; (b) KCN, Et3N, 18-crown-6, CH2Cl2, rt, 72 h; (c) 4-N,N-dimethylaminobenzaldehyde, piperidine, benzene, Dean-Stark trap,3 h.
With the availability of 5a, the acidichromic behavior in both gaseous hydrochloric acid and ammonia was then determined. Under neutral conditions in methylene chloride, compound 5a was red because of extended conjugation with a UV-vis absorption λmax value of 490 nm (ε = 49,509 M-1 cm-1). When gaseous hydrochloric acid was bubbled through the solution, it turned colorless instantly, because of protonation of the nitrogen atom. When treated with gaseous ammonia, however, compound 5a failed to turn yellow, suggesting that the enolic hydrogen is too stable to be deprotonated by ammonia. Figure 1 shows the UV-vis spectra of 5a under neutral, basic (ammonia) and acidic (hydrochloric acid) conditions in methylene chloride. Before bubbled with ammonia, the UV absorbance at 490 nm for compound 5a under neutral conditions was 1.35 (Fig. 1). After bubbled with ammonia, the UV absorbance at that wavelength decreased to about 1.0, indicating that approximately one fifth of the enolic hydrogens of 5a were deprotonated by ammonia only, while the rest of them remained intact. This result demonstrated that 7-methoxy-substituted 5a displayed stronger intramolecular hydrogen bonding than the corresponding unsubstituted compound 1.
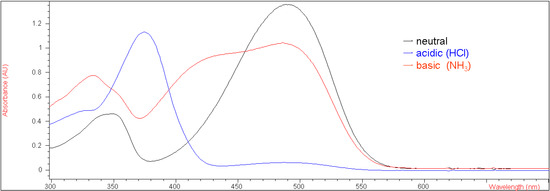
Figure 1.
UV-vis spectra of 5a (2.74 x 10-5 M in CH2Cl2) under neutral, basic and acidic conditions.
Figure 1.
UV-vis spectra of 5a (2.74 x 10-5 M in CH2Cl2) under neutral, basic and acidic conditions.
Under neutral pH conditions in methylene chloride compound 5b was red, similar to 5a, because of extended conjugation, with a UV-vis absorption λmax value of 515 nm (ε = 50,623 M-1 cm-1). It also turned colorless instantly, when bubbled with gaseous hydrochloric acid. However, compound 5b exhibited different acidichromic behavior from 5a when treated with ammonia. Upon bubbling of gaseous ammonia, compound 5b did change color from red to yellow, as shown in Scheme 3.
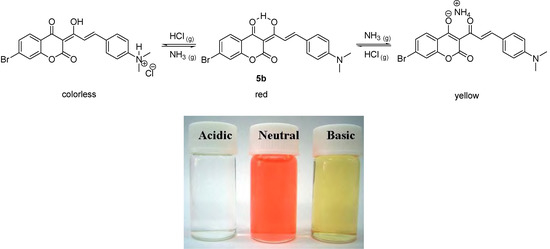
Scheme 3.
The acidichromic switch of 5b and the corresponding colors in acidic, neutral and basic conditions.
Scheme 3.
The acidichromic switch of 5b and the corresponding colors in acidic, neutral and basic conditions.
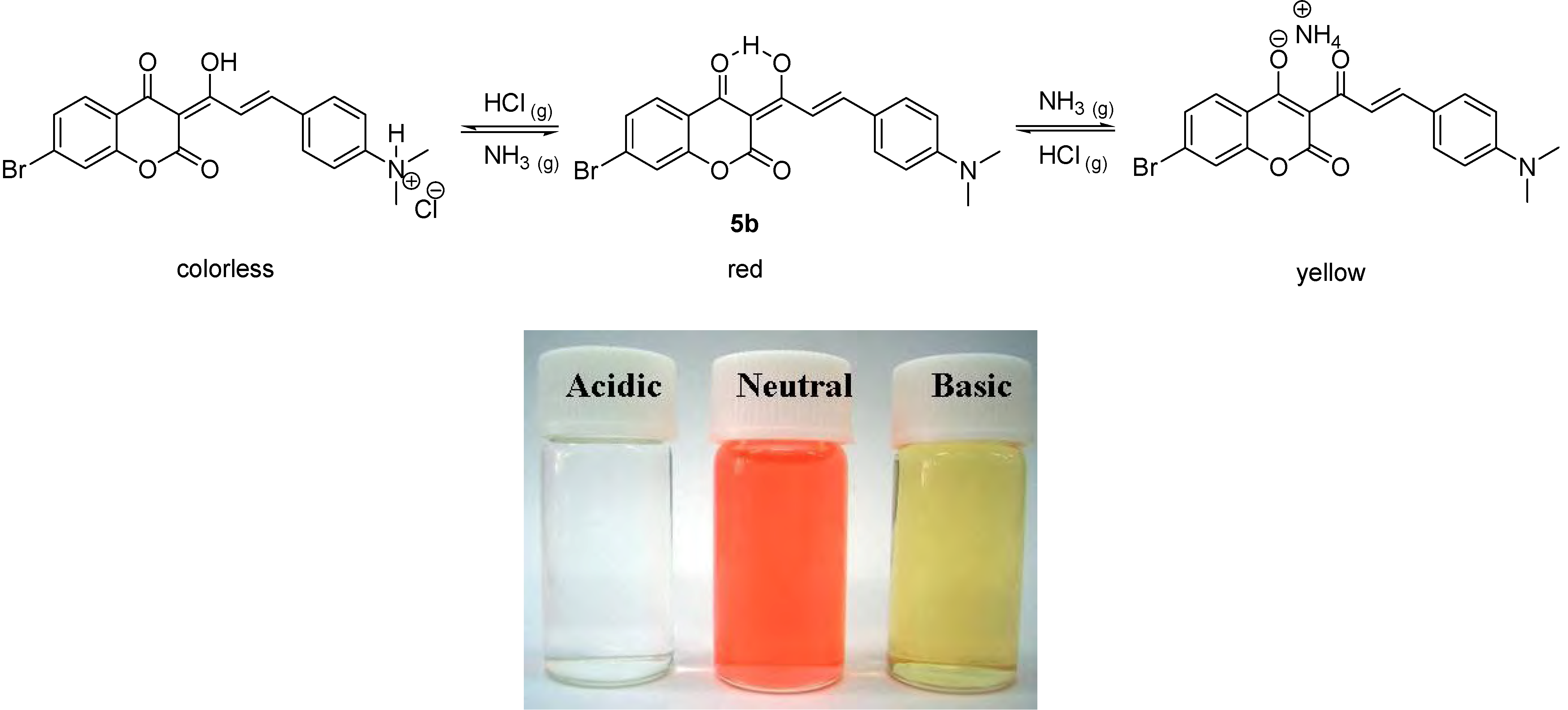
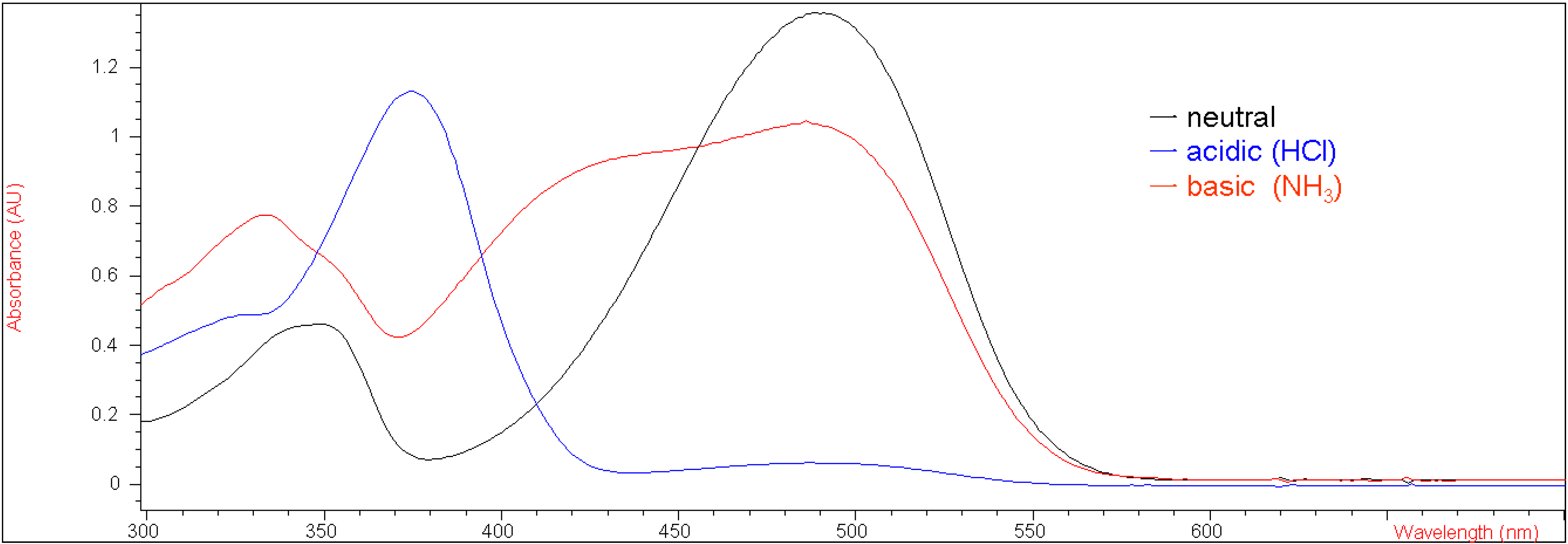
Figure 2 presents their corresponding UV-vis absorption spectra in methylene chloride under both acidic and basic conditions with the λmax values of 370 nm (ε = 38,970 M-1 cm-1) and 400 nm (ε = 34,610 M-1 cm-1), respectively. This result suggests that the electron-withdrawing group at the 7-position of the coumarin moiety rendered the enolic hydrogen on 5b more susceptible to deprotonation by a base than 1.
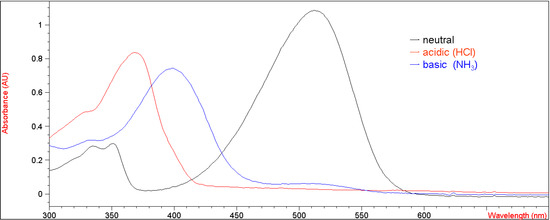
Figure 2.
UV-vis spectra of 5b (2.14 x 10-5 M in CH2Cl2) in neutral, acidic and basic conditions.
Figure 2.
UV-vis spectra of 5b (2.14 x 10-5 M in CH2Cl2) in neutral, acidic and basic conditions.
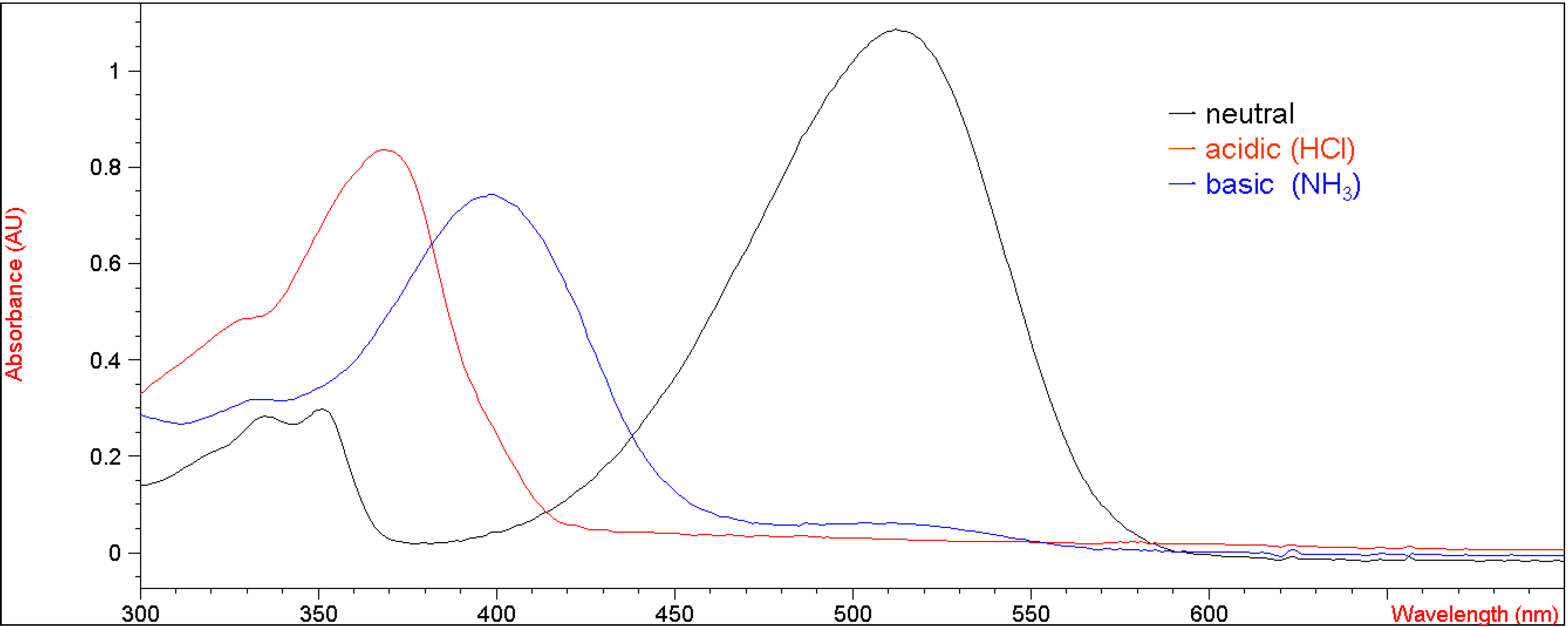
Figure 3 shows the UV-vis absorption changes of 5b every five seconds when bubbled with gaseous ammonia in methylene chloride. After bubbling with ammonia for 45 seconds, the UV absorbance at 515 nm decreased from 1.05 to the baseline, indicating that the enolic hydrogens on 5b were completely deprotonated by ammonia. We speculate that a shorter bubbling time would be required if an electron-withdrawing group stronger than a bromine atom were to be incorporated into the coumarin moiety of 5b. An unfocused isosbestic point was observed at 445-455 nm, suggesting interconversion of neutral and deprotonated forms of 5b. This unfocused isosbestic point was probably associated with the fluctuating concentration of 5b in methylene chloride during the bubbling of gaseous ammonia into the volatile solvent. When 5b was doped in a polycarbonate (PC) film [3], a similar acidichromic behavior was observed, as presented in Figure 4. It was red under neutral pH conditions, and turned colorless and yellow when exposed to gaseous hydrochloric acid and ammonia, respectively.
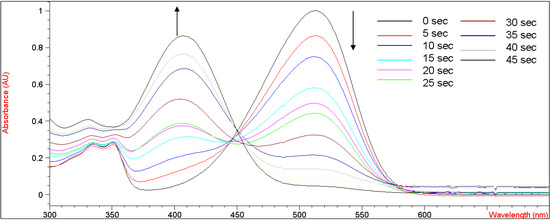
Figure 3.
UV-vis curves of titration of 5b (2.01 x 10-5 M in CH2Cl2) with increasing concentrations of NH3(g) in CH2Cl2.
Figure 3.
UV-vis curves of titration of 5b (2.01 x 10-5 M in CH2Cl2) with increasing concentrations of NH3(g) in CH2Cl2.
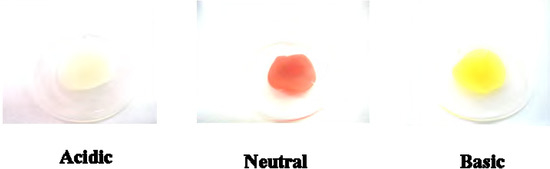
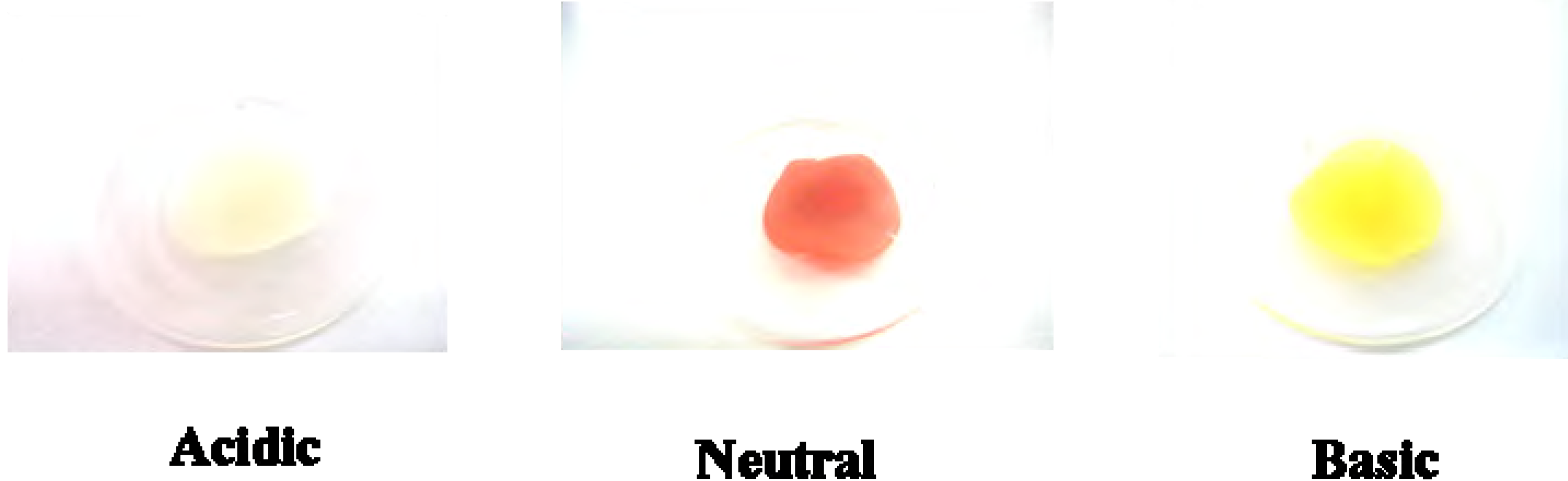
Figure 4.
Colors of 5b on PC film under acidic (exposed to HCl(g)), neutral and basic (exposed to NH3(g)) conditions.
Figure 4.
Colors of 5b on PC film under acidic (exposed to HCl(g)), neutral and basic (exposed to NH3(g)) conditions.
Conclusions
An acid and base-sensitive triketone functional group is introduced to design and synthesize an acidichromic colorant. An electron-withdrawing group at the 7-position of the coumarin moiety made the enolic hydrogen on 5b more susceptible to deprotonation by a base than in the unsubstituted compound 1. The 7-bromo-substituted 5b undergoes two distinct and reversible color changes under both gaseous hydrochloric acid and ammonia in solution, as well as on solid PC film. This readily available molecule may potentially function as an acid-base sensor or can be used in gas-controlled reversible color-change devices.
Experimental Section
General
Melting points were determined on a Mel-Temp melting point apparatus in open capillaries and are uncorrected. MS were performed on JOEL JMS-SX/SX 102A spectrometer. IR spectra were obtained using a 1725XFT-IR spectrophotometer. Absorption spectra were acquired using an HP8453 spectrophotometer and emission spectra were obtained on a Hitachi F-4500 fluorospectrometer. 1H- and 13C-NMR spectra were recorded at 300 and 75 MHz, respectively, on a Varian VXR300 spectrometer. Chemical shifts were reported in parts per million on the δ scale relative to an internal standard (tetramethylsilane, or appropriate solvent peaks) with coupling constants given in hertz. 1H NMR multiplicity data are denoted by s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60G-254 plates (25 mm) and developed with the solvents mentioned. Flash chromatography was performed in columns of various diameters with Merck silica gel (230-400 mesh ASTM 9385 Kieselgel 60H) by elution with the solvent systems given. Solvents, unless otherwise specified, were of reagent grade and distilled once prior to use. Preparation of 7-bromo-4-hydroxycoumarin has been previously reported [18,19].
General procedure for preparation of compounds 3a-b
To a mixture of 4-hydroxy-7-methoxycoumarin (50mg, 0.3mmol) or 7-bromo-4-hydroxycoumarin (78 mg, 0.3 mmol) and triethylamine (40 mg, 0.4 mmol) in methylene chloride (5 mL) was added acetyl chloride (31 mg, 0.4 mmol) at 0 oC. The resulting mixture was stirred for 30 min. After completion of the reaction, this mixture was poured into water. The product was extracted with methylene chloride twice. The combined organic extracts were then dried over MgSO4, filtered, and concentrated. The crude compound was purified by column chromatography (EtOAc-hexanes = 1:7) to give the desired product.
4-Acetoxy-7-methoxycoumarin (3a). White solid; Yield 95%; Rf = 0.40 (30% EtOAc-hexanes); Mp 127–128 oC; 1H-NMR (CDCl3) δ 7.51 (d, J = 9.0 Hz, 1H), 6.88−6.84 (m, 2H), 6.34 (s, 1H), 3.89 (s, 3H), 2.43 (s, 3H); 13C-NMR (CDCl3) δ 166.6, 163.5, 162.0, 158.7, 155.5, 123.7, 112.7, 108.6, 102.0, 100.8, 55.8, 21.2; HRMS (EI) m/z calcd. for C12H10O5 234.0528, found 234.0520 (M+); IR ν (KBr) 3143, 2979, 2942, 1716, 1560, 1300, 1100, 895, 751 cm-1.
4-Acetoxy-7-bromocoumarin (3b). White solid; Yield 93%; Rf = 0.50 (30% EtOAc-hexanes); Mp 129–130 oC; 1H-NMR (CDCl3) δ 7.53 (d, J = 1.5 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.43 (dd, J = 8.4, 1.5 Hz, 2H), 6.55 (s, 1H), 2.45 (s, 3H); 13C-NMR (CDCl3) δ 166.3, 160.7, 157.7, 153.7, 127.7, 126.8, 123.8, 120.3, 114.3, 105.1, 21.3; HRMS (EI) m/z calcd. for C11H7BrO4 281.9558, found 281.9528 (M+); IR ν (KBr) 3084, 1744, 1599, 1196, 1157, 1085, 561 cm-1.
General procedure for preparation of compounds 4a-b
To a solution of compound 3 (0.2 mmol) in methylene chloride (5 mL) were added KCN (13.9 mg, 0.2 mmol), Et3N (21.6 mg, 0.2 mmol) and a catalytic amount of 18-crown-6 at room temperature. The resulting mixture was stirred at room temperature for 72 h. After completion of the reaction (monitored by TLC), the solvent was concentrated in vacuo. This mixture was poured into water and the product was then extracted twice with methylene chloride. The combined organic extracts were dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (EtOAc -hexanes = 1:5) to give the desired product.
3-Acetyl-4-hydroxy-7-methoxycoumarin (4a). White solid; Yield 88%; Rf = 0.60 (30% EtOAc- hexanes); Mp 183–184 oC; 1H-NMR (CDCl3) δ 17.77 (s, 1H), 7.95 (d, J = 9.0 Hz, 1H), 6.89 (dd, J = 9.0, 2.1 Hz, 1H), 6.74 (d, J = 2.1 Hz, 1H), 3.91(s, 3H), 2.75 (s, 3H); 13C-NMR (CDCl3) δ 205.7, 178.7, 166.6, 160.6, 157.2, 127.2, 113.6, 108.5, 100.4, 100.1, 56.2, 30.2; HRMS (EI) m/z calcd. for C12H10O5 234.0528, found 234.0526 (M+); IR ν (KBr) 3445, 3080, 2991, 2946, 1731, 1618, 1543, 1421, 1109, 829, 548 cm-1.
3-Acetyl-4-hydroxy-7-bromocoumarin (4b). White solid; Yield 85%; Rf = 0.35 (25% EtOAc-hexanes); Mp 188–190 oC; 1H-NMR (CDCl3) δ 17.83 (s, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.49−7.46 (m, 2H), 2.78 (s, 3H); 13C-NMR (CDCl3) δ 205.9, 178.2, 159.4, 154.7, 130.8, 128.1, 126.7, 120.3, 114.2, 101.3, 30.0; HRMS(EI) m/z calcd. for C11H7BrO4 281.9528, found 281.9532 (M+); IR ν (KBr) 3445, 3082, 1729, 1600, 1031, 779 cm-1.
General procedure for preparation of compounds 5a-b
To a solution of compound 4 (0.2 mmol) and 4-N,N-dimethylaminobenzaldehyde (31.6 mg, 0.2 mmol) in benzene (25 mL) was added piperidine (18.0 mg, 0.2 mmol). The mixture was then refluxed in a Dean-Stark trap. After completion of the reaction within 3 h, it was cooled down to room temperature and the solvent was concentrated in vacuo. The resulting residue was poured into water, and the product was then extracted twice with methylene chloride. The combined organic extracts were dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (EtOAc-hexanes = 1:6) to give the desired product.
3-((E)-3-(4-N,N-Dimethylaminophenyl)acryloyl)-4-hydroxy-7-methoxy-2H-chromen-2-one (5a). Dark solid; Yield 73%; Rf = 0.50 (30% EtOAc-hexanes); Mp 267−268 oC; 1H-NMR (CDCl3) δ 19.25 (s, 1H), 8.23 (d, J = 14.7 Hz, 1H), 8.08 (d, J = 14.7 Hz, 1H), 7.98 (d, J = 9.0 Hz, 2H), 7.64 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 10.5 Hz, 1H), 6.71 (m, 2H), 3.90 (s, 3H), 3.09 (s, 6H); 13C-NMR (CDCl3) δ 190.4, 181.8, 165.8, 161.0, 156.6, 152.8, 148.8, 132.0, 127.2, 125.2, 122.6, 118.5, 116.0, 112.8, 111.7, 100.1, 55.9, 40.1; HRMS (EI) calcd. for C21H19NO5 365.1263, found 365.1270 (M+); IR (KBr) v 1712, 814 cm-1.
(3E)-7-Bromo-3-((E)-3-(4-N,N-dimethylaminophenyl)-1-hydroxyallylidene)-3H-chromen-2,4-dione (5b). Dark solid; Yield 72%; Rf = 0.40 (25% EtOAc-hexanes); Mp 251−252 oC; 1H-NMR (CDCl3) δ 19.15 (s, 1H), 8.20 (d, J = 15.3 Hz, 1H), 8.13 (d, J = 15.3 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.44 (m, 2H), 6.69 (d, J = 9.0 Hz, 2H), 3.10 (s, 6H); 13C-NMR (CDCl3) δ 190.0, 181.9, 160.1, 154.6, 153.1, 150.1, 132.4, 129.7, 127.6, 126.9, 122.5, 120.1, 116.5, 115.1, 111.8, 99.7, 40.1; HRMS (EI) calcd. for C20H16BrNO4 413.0263, found 413.0262 (M+); IR (KBr) v 3447, 1713, 1477, 1165, 814 cm-1.
Preparation of PC film of compound 5b
The PC film (thickness ca. 1 x 10-5 m) was fabricated with a CH2Cl2 solution of 5b (2 x 10-5 M) in PC matrix on quartz substrate by the vertical dipping method.
Acknowledgments
The authors would like to thank the National Science Council of Republic of China, Taiwan for financially supporting this research under Contract No. NSC 94-2113-M-029-001.
References and Notes
- Yin, M.; Gong, H.; Zhang, B.; Liu, M. Photochemical reaction, acidichromism, and supramolecular nanoarchitectures in the Langmuir-Blodgett films of an amphiphilic styrylquinoxaline derivative. Langmuir 2004, 20, 8042–8048. [Google Scholar] [CrossRef]
- Siri, O.; Braunstein, P.; Rohmer, M.M.; Benard, M.; Welter, R. Novel "potentially antiaromatic", acidichromic quinonediimines with tunable delocalization of their 6π -electron subunits. J. Am. Chem. Soc. 2003, 125, 13793–13803. [Google Scholar]
- Luo, Q.; Li, X.; Jing, S.; Zhu, W.; Tian, H. A novel bisthienylethene as acidichromic and photochromic yellow dye. Chem. Lett. 2003, 32, 1116–1117. [Google Scholar] [CrossRef]
- Gong, H.; Wang, C.; Liu, M.; Fan, M. Acidichromism in the Langmuir-Blodgett films of novel photochromic spiropyran and spirooxazine derivatives. J. Mater. Chem. 2001, 11, 3049–3052. [Google Scholar]
- Miliani, C.; Romani, A.; Favaro, G. Acidichromic effects in 1,2-di- and 1,2,4-tri-hydroxyanthraquinones. A spectrophotometric and fluorimetric study. J. Phys. Org. Chem. 2000, 13, 141–150. [Google Scholar]
- Pina, F.; Melo, M.J.; Maestri, M.; Ballardini, R.; Balzani, V. Photochromism of 4'-Methoxyflavylium Perchlorate. A "Write-Lock-Read-Unlock-Erase" Molecular Switching System. J. Am. Chem. Soc. 1997, 119, 5556–5561. [Google Scholar] [CrossRef]
- Sun, X.D.; Fan, M.G.; Meng, X.J.; Knobbe, E.T. Acidichromic effects in spiro(1,3,3-trimethylindolo-2,3'-naphth[1,2-b]-1,4-oxazine), a photochromic compound I. Absorption characteristics. J. Photochem. Photobiol. A 1997, 102, 213–216. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M. Monolayers Langmuir-Schaefer films and acidichromism of a nonamphiphilic acetone derivative containing carbazole. New J. Chem. 2002, 26, 180–183. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M. Langmuir-Blodgett film and acidichromism of a long chain carbazole-containing Schiff base. Thin Solid Films 2002, 415, 248–252. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Wen, T.C.; Gopalan, A. Tuning the optical sensing of pH by poly(diphenylamine). Sens. Actuators. B Chem. 2003, 96, 646–657. [Google Scholar] [CrossRef]
- Hilborn, E.H.; Grafstein, D. Fluidic-thermochromic display device. U.S. Patent 03,516,185, 1970. [Google Scholar]
- Lin, C.T.; Shih, J.H.; Chen, C.L.; Yang, D.Y. Synthesis of novel triketone-based acidichromic colorants. Tetrahedron Lett. 2005, 46, 5033–5037. [Google Scholar] [CrossRef]
- Chen, Y. S.; Kuo, P. Y.; Shie, T. L.; Yang, D. Y. Structure, reactivity, and application of some triketone derivatives. Tetrahedron 2006, 62, 9410–9416. [Google Scholar] [CrossRef]
- Kincal, D.; Kumar, A.; Child, A.D.; Reynolds, J.R. Conductivity switching in polypyrrole-coated textile fabrics as gas sensors. Synth. Met. 1998, 92, 53–55. [Google Scholar]
- Yuan, J.; El-Sherif, M.A.; MacDiarmid, A.G.; Jones, W.E., Jr. Fiber optic chemical sensors using a modified conducting polymer cladding. Proc. SPIE - Int. Soc. Opt. Eng. 2001, 4205, 170–179. [Google Scholar]
- Sengupta, P.; Barik, S.; Adhikari, B. Polyaniline as a gas-sensor material. Mater. Manuf. Process 2006, 21, 263–270. [Google Scholar] [CrossRef]
- Montes, I.F.; Burger, U. The cyanide catalyzed isomerization of enol esters derived from cyclic 1,3-diketones. Tetrahedron Lett. 1996, 37, 1007–1010. [Google Scholar] [CrossRef]
- Li, H.Y.; Boswell, G.A. A facile synthesis of fluorinated 4-hydroxycoumarins. Tetrahedron Lett. 1996, 37, 1551–1554. [Google Scholar]
- Buckle, D.R.; Cantello, B.C.C.; Smith, H.; Spicer, B.A. Antiallergic Activity of 4-Hydroxy-3-nitrocoumarins. J. Med. Chem. 1975, 18, 391–394. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.