Abstract
The reported structures of reaction products of enaminones with malononitrile in ethanolic piperidine are revised. A novel route to 2,3-dihydropyridazine-4-carboxylic acids 4a-c via reactions of 2-cyano-5-(dimethylamino)-5-arylpenta-2,4-dienamides 8a-c with nitrous acid or with benzenediazonium chloride is reported. Compounds 8a-c are converted to 1,2-dihydropyridine-3-carboxylic acid and 1,2-dihydropyridine-3-carbonitrile derivatives upon reflux in EtOH/ HCl and in AcOH.
Introduction
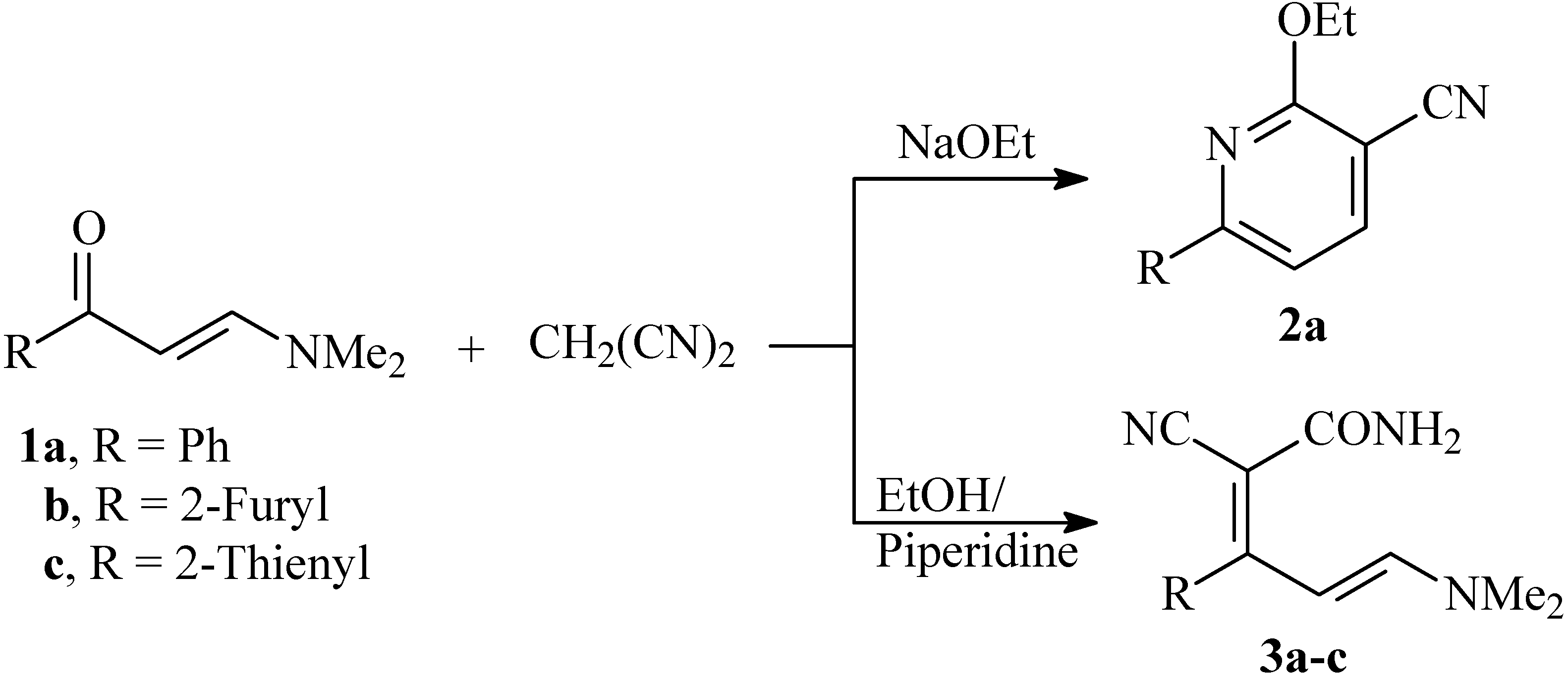
Enaminones are polydentate reagents that have been utilized extensively in this decade as building blocks in organic synthesis [1,2,3,4,5,6]. In previous work at our laboratories, we reported several efficient routes to polyfunctionally substituted heterocycles utilizing enaminones as starting materials [7,8,9,10,11]. We have also reported that the reaction of 1a with malononitrile in ethanolic sodium ethoxide afforded 2a in good yield [12], while reacting 1a-c with malononitrile in ethanolic piperidine was believed to afford 3a-c [13] (cf. Scheme 1). In continuation to this work, the chemical reactivity of the products believed to be 3a-c was reinvestigated. The work has led us to revise the initially proposed structures of these products.

Scheme 1.
Reported structures for the products of reaction of enaminones 1a-c and malononitrile.
Scheme 1.
Reported structures for the products of reaction of enaminones 1a-c and malononitrile.
Results and Discussion
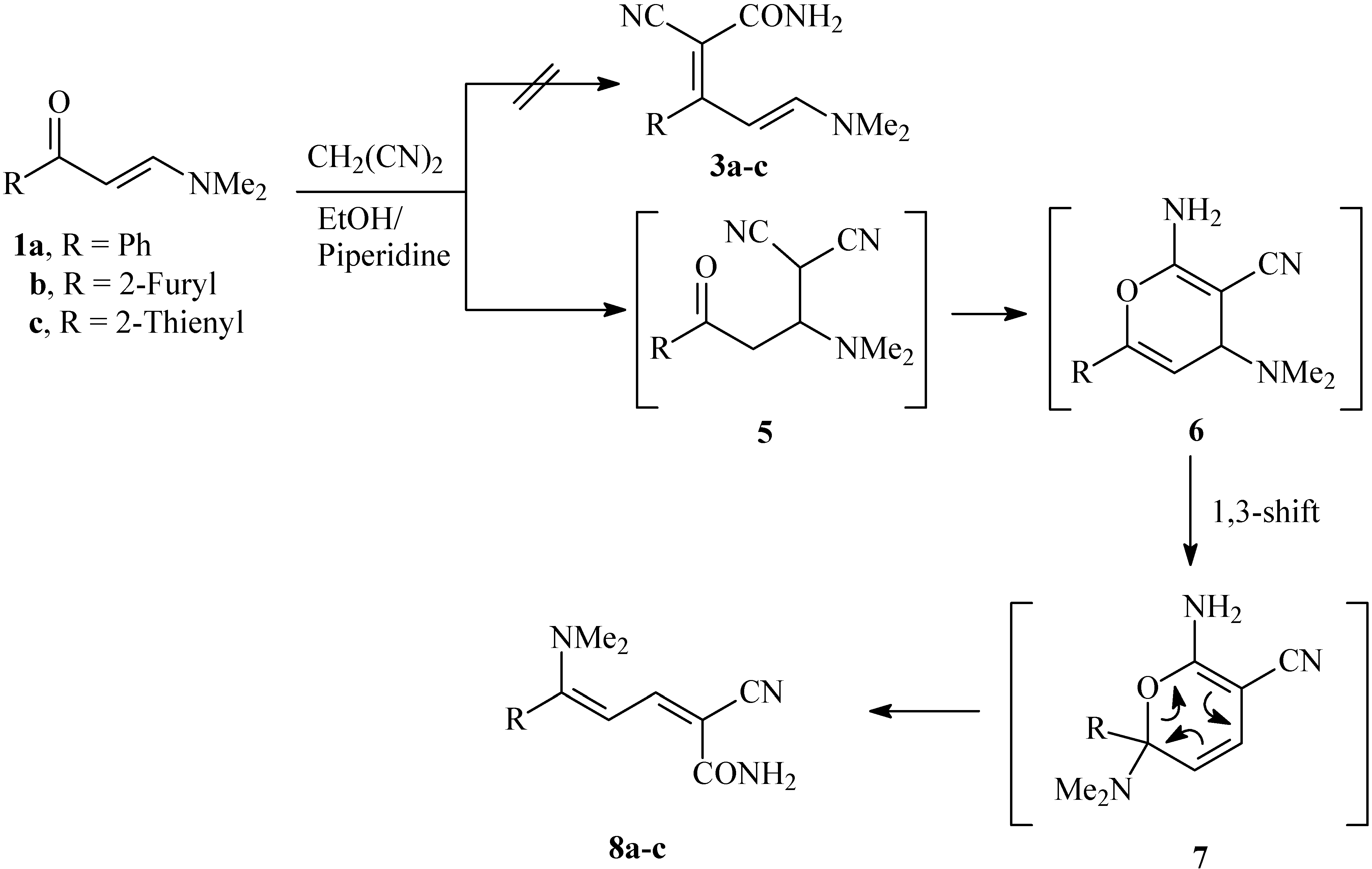
The reaction of 1a-c with malononitrile in ethanolic piperidine afforded products of molecular formulae corresponding to the formation of 1:1 adducts. As reported earlier [13], the reaction products showed in the 1H-NMR spectrum, in addition of the dimethylamino moiety, two olefinic proton doublets at δH = ca. 5.77 and 7.23 ppm with J = 13 Hz, which fits well with the previously assumed initial 1,2-addition of malononitrile at the carbonyl moiety. Subsequent water elimination and hydrolysis of one of the cyano groups into an amide yielded 3a-c. However, treating these reaction products with sodium nitrite in EtOH/HCl in presence of sodium acetate affords products for which structures 4a-c are assigned, based on X-ray crystal structure determination [14]. Although the described conditions may not normally lead to hydrolysis of nitriles, however a ready hydrolysis in this case may be prompted by the stabilization of products by potential hydrogen bonding and high reactivity of the nitrile group as part of a π-deficient system. Quite unexpectedly, coupling the products, obtained from the reaction of malononitrile with enaminones 1a-c, with benzenediazonium chloride in dioxane/AcONa resulted in the formation of the same products 4a-c, in good yields.
There is indication of extensive delocalization of N-1 lone-pair at carbonyl carbon. Thus N-3 bond angles are more like those of sp2 nitrogen, while those of N-7-C10-C8 are more like sp3 carbon (cf. Figure 1 and Table 1).
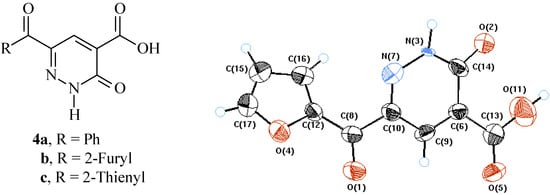
Figure 1.
X-Ray crystal structure of 4b.
Figure 1.
X-Ray crystal structure of 4b.

Table 1.
Selected bond lengths and angles for compound 4b.
| Bond | Bond length Ao | Bond | Bond angle |
|---|---|---|---|
| O2-C14 | 1.224 (4) | N7-N3-C14 | 122.5 (3) |
| N3-N7 | 1.390 (3) | N3-N7-C10 | 116.6 (3) |
| N3-C14 | 1.357 (4) | N7-C10-C8 | 114.6 (4) |
| C6-C14 | 1.447 (5) | ||
| N7-C10 | 1.306 (4) |
Disconnection of 4a-c and consideration of the reported data has led us to believe that the products initially thought to be 3a-c are in fact 8a-c, which are formed via initial 1,4-addition of malononitrile across the double bond to yield 5 that cyclized to 6 then rearranged to 7, that finally afforded 8 via anallowed 1,3-nitrogen shift (cf. Scheme 2). However, a possible conversion of 3 to 8 involving migration of R via a 1,3 shift should not be overlooked. We wish to state that both 8 and 3 have the same molecular formulae and same spectral data, which after further inspection, established the structures 8a-c. Thus, assuming that H-4 are shielded by nitrogen lone pair anisotropic effect, while H-3 are deshielded by electron attracting substituents; it is hence logic to assign the doublets at δH = ca. 5.68 ppm to H-4, while the doublets at δH = ca. 7.39 ppm would correspond to H-3. If the reaction products were 3a-c, then H-4 in these assumed structures are shielded and H-5 are deshielded. We note that the deshielded doublet for 8a at δH = ca. 7.39 ppm are correlated in the HMBC experiments with the amide carbonyl group at δC = ca. 164.90 ppm. If the reaction product was 3a, such a correlation should not exist. Moreover, the methyl protons at δH = ca. 2.99 ppm show a cross peak correlation with C-5 at δC = ca. 157.93 ppm. This carbon was proven by DEPT experiments to be quaternary, consistent with structure 8a. If, on the other hand, the reaction product were 3a, then the methyl protons should be correlated with a carbon bearing a proton.

Scheme 2.
Proposed mechanism for the formation of 2-cyano-5-(dimethylamino)-5-aryl-penta-2,4-dienamides 8a-c.
Scheme 2.
Proposed mechanism for the formation of 2-cyano-5-(dimethylamino)-5-aryl-penta-2,4-dienamides 8a-c.
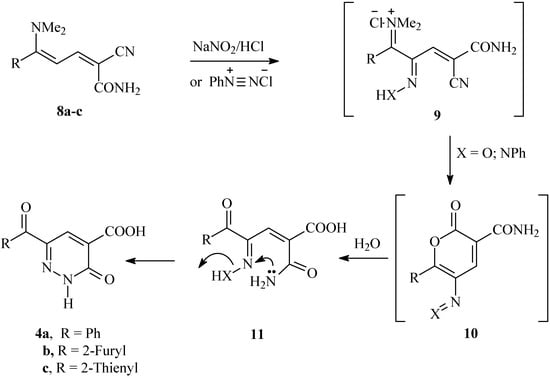
Scheme 3.
Proposed mechanism for the nitrozation and coupling reactions of 2-cyano-5-(dimethylamino)-5-phenylpenta-2,4-dienamides 8a-c.
Scheme 3.
Proposed mechanism for the nitrozation and coupling reactions of 2-cyano-5-(dimethylamino)-5-phenylpenta-2,4-dienamides 8a-c.
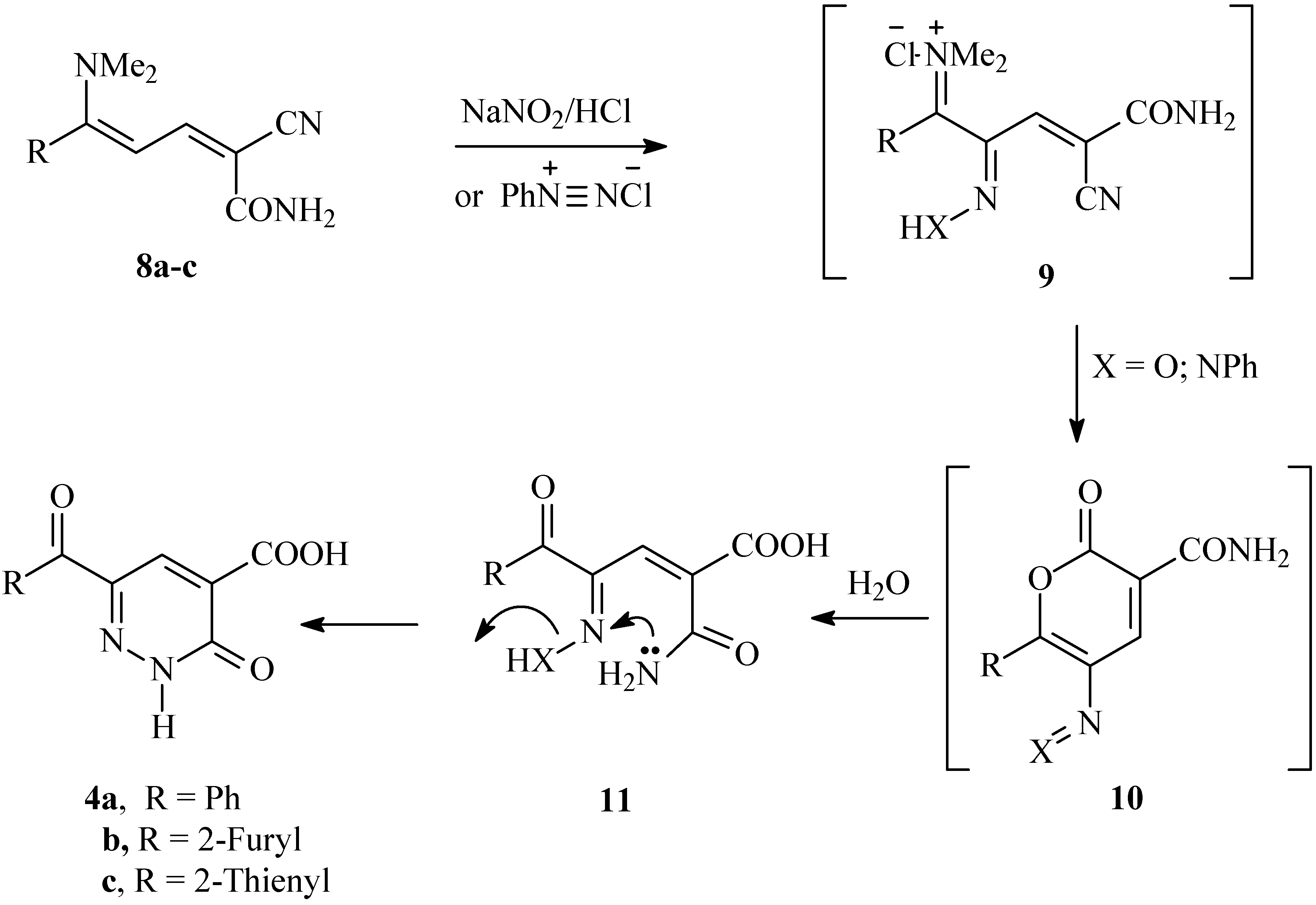
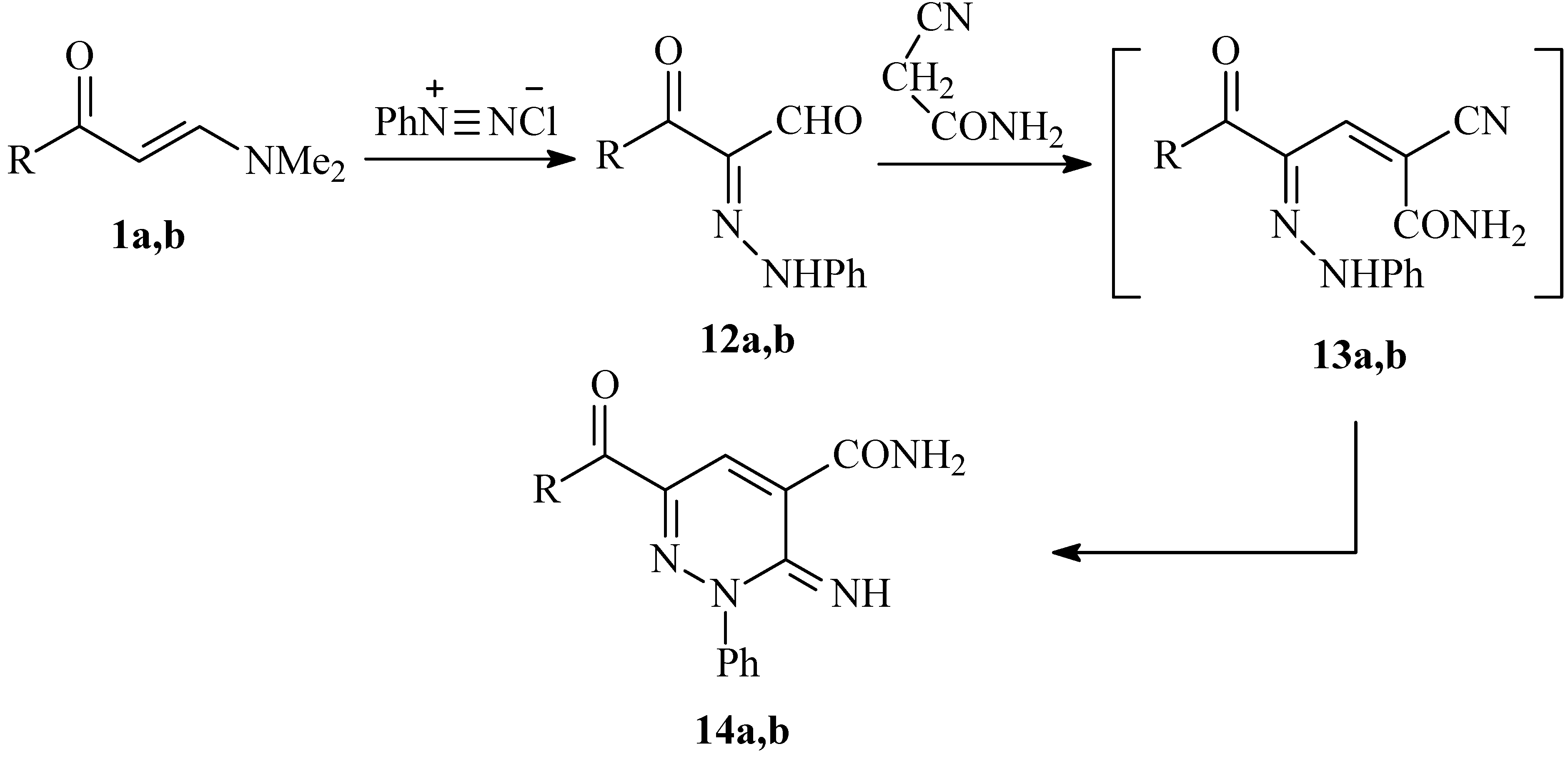
Consequently, a plausible mechanism for the formation of compounds 4a-c is illustrated in Scheme 3. It is assumed that the initially formed 9 is subject to an intramolecular cyclization to 10, which is further hydrolysed into 11 under the reaction conditions. Finally, the lone pairs on the amide nitrogen then react with the oxime nitrogen or the hydrazone nitrogen kicking out either a water molecule or aniline, thus producing 4a-c. To our knowledge, this is the first reported cyclization via aniline elimination in such a system. It is worth mentioning that condensing 3-aroyl-2-(2-phenylhydrazono)propanals 12a,b with cyanoacetamide has been reported to yield the 2,3-dihydropyridazine-4-carboxamides 14a,b [15] (cf. Scheme 4).
Scheme 4.
Reported synthesis of 2,3-dihydropyridazine-4-carboxamides 14a,b from the condensation of arylhydrazonals 12a,b with cyanoacetamide.
Scheme 4.
Reported synthesis of 2,3-dihydropyridazine-4-carboxamides 14a,b from the condensation of arylhydrazonals 12a,b with cyanoacetamide.
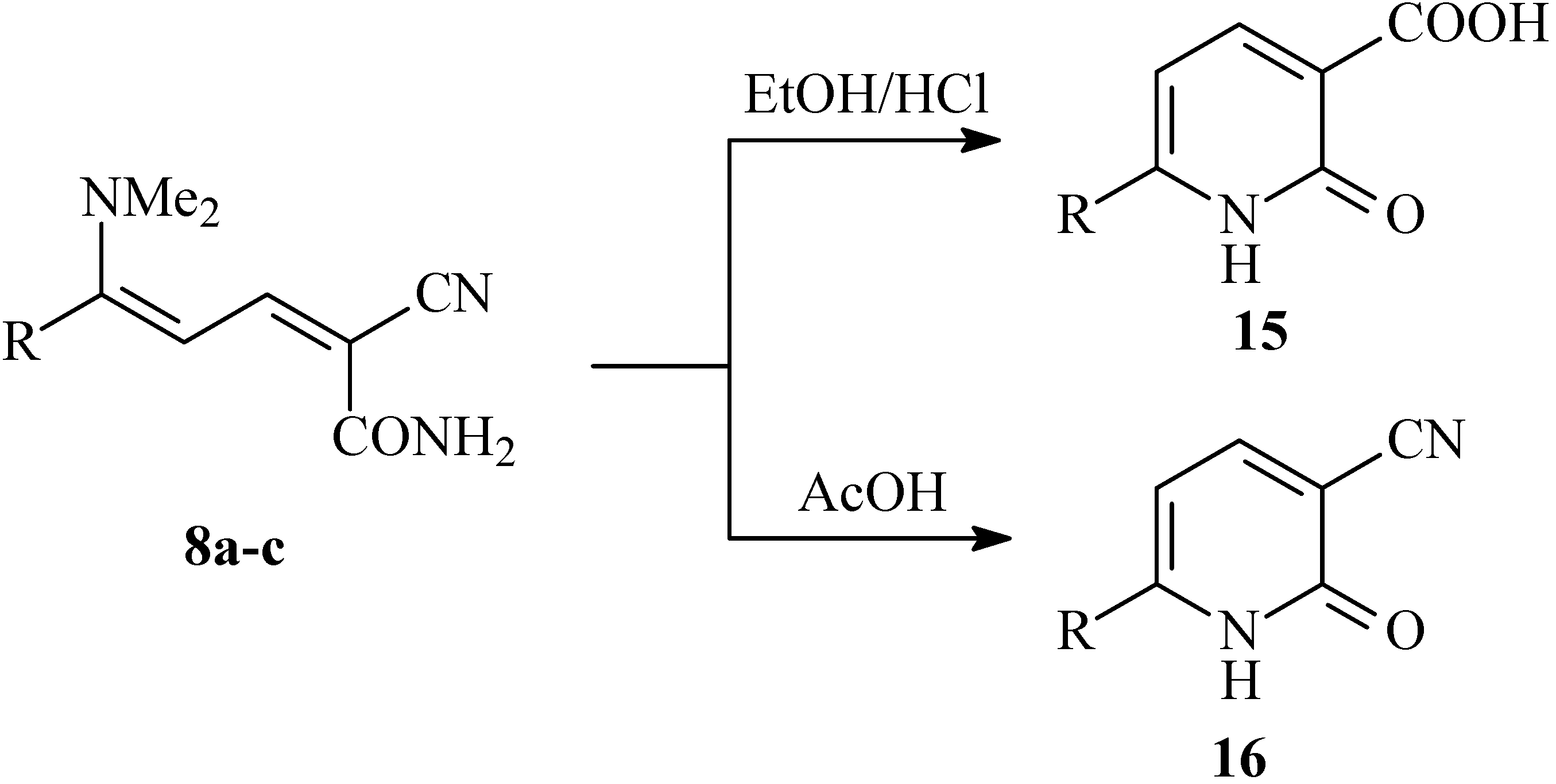
Conversions of 8a-c into nicotinic acid derivatives 15a-c were achieved by boiling in EtOH/HCl. When however compounds 8a-c are heated under reflux in AcOH, nicotinic nitrile derivatives 16a-c are obtained (cf. Scheme 5).

Scheme 5.
Synthesis of 1,2-dihydropyridine-3-carboxylic acids 15a-c and 1,2-dihydropyridine-3-carbonitriles 16a-c.
Scheme 5.
Synthesis of 1,2-dihydropyridine-3-carboxylic acids 15a-c and 1,2-dihydropyridine-3-carbonitriles 16a-c.
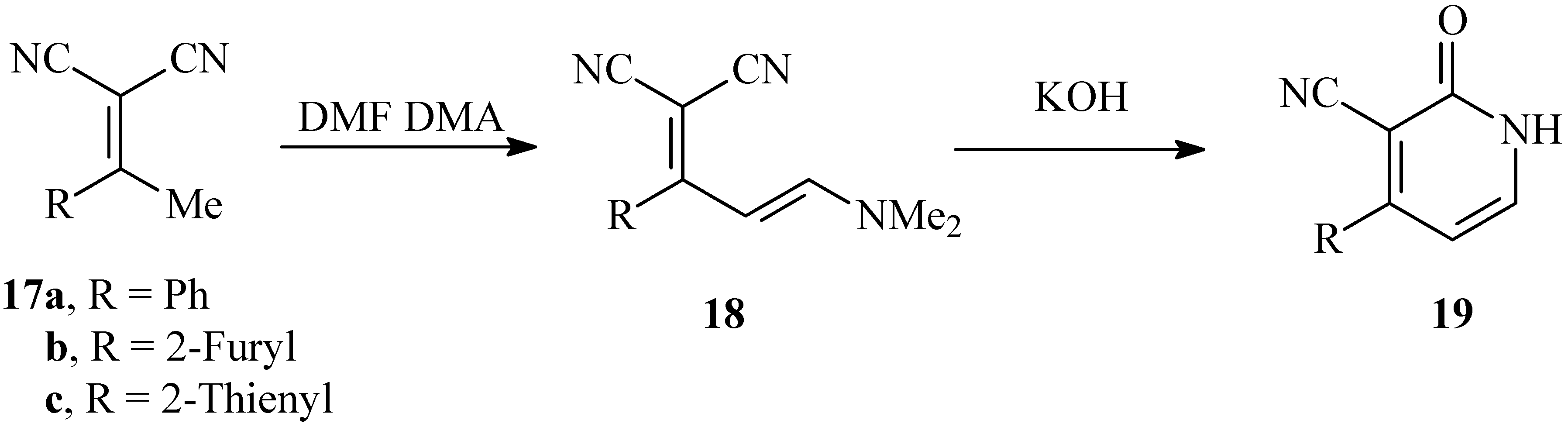
It has been reported earlier [16] that hydrolysis of 18 obtained via condensation of 17 with dimethylformamide dimethylacetal afforded the pyridone derivatives 19. We have repeated this experiment and came to the conclusion that hydrolysis of 18 in KOH affords in fact isomeric pyridone 19, which in turn gives spectra very similar to those of 16. Indeed, the mixed m.p. of the two products proves that they are different (cf. Scheme 6).
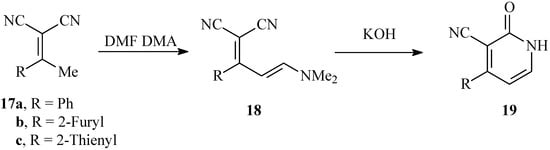
Scheme 6.
Reported synthesis of isomeric pyridones 13a-c.
Scheme 6.
Reported synthesis of isomeric pyridones 13a-c.
Conclusions
We are now able to correct a previously reported initial 1,2-addition of malononitrile at the carbonyl moiety of enaminones 1a-c and suggest instead the novel compounds 8a-c as precursors for syntheses of pyridazinones and pyridones derivatives.
Experimental
General
Melting points were determined on a Shimadzu-Gallenkamp apparatus and are uncorrected. Elemental analyses were obtained by means of a LECO CHNS-932 Elemental Analyzer. NMR spectra were measured in DMSO-d6 using a Bruker DPX 400 MHz superconducting spectrometer; HMQC, DEPT and NOE spectra were measured using Bruker Avance II 600 MHz superconducting spectrometer, and FT-IR measurements were from a Perkin Elmer 2000 FT-IR system. Mass spectrometric analysis was carried out on a VG-Autospec-Q high performance tri-sector GC/MS/MS.
General procedure for the preparation of compounds 8a-c
A mixture of equimolecular amounts of each of enaminones 1a-c (10 mmol) and malononitrile (10 mmol, 066 g) in EtOH (10 mL) was refluxed for 1 hr in the presence of few drops of piperidine. Upon cooling to r.t. a solid product precipitated, which was collected by filtration and crystallized from dioxane.
2-Cyano-5-(dimethylamino)-5-phenylpenta-2,4-dienamide (8a): Yellow crystals, yield (72 %, 1.77 g); mp 257-258 oC; IR (cm-1): 3435 and 3334 (NH2), 2195 (CN) and 1666 cm-1 (CO); MS m/z (M)+ = 241; 1H-NMR: d = 2.78 (s, 3H, NCH3), 3.14 (s, 3H, NCH3), 5.64 (d, 1H, J = 12.8 Hz, H-4), 6.82 (s, 2H, NH2), 7.13 (d, 1H, J = 12.8 Hz, H-3), 7.25-7.28 (m, 2H, phenyl-H), 7.34-7.65 (m, 3H, phenyl-H); 13C-NMR: d = 165.08 (CONH2), 164.59 (C-5), 153.27 (C-3), 133.99, 129.56, 128.76, 128.72, 118.90, 96.91, 87.57, 41.92 (N(CH3)2); Anal. calcd. for C14H15N3O: (241.12): C, 69.69; H, 6.27; N, 17.41. Found: C, 69.55; H, 6.07; N, 17.29.
2-Cyano-5-(dimethylamino)-5-(furan-2-yl)penta-2,4-dienamide (8b): Brownish red crystals, yield (75 %, 1.73 g); mp 245-246 oC; IR (cm-1): 3331 and 3292 (NH2), 2190 (CN) and 1671 cm-1 (CO); MS m/z (M)+ = 231; 1H-NMR: d = 2.97 (s, 6H, N(CH3)2), 5.59 (d, 1H, J = 12.5 Hz, H-4), 6.72 (d, 1H, J = 5.0 Hz, furyl H-3), 6.78 (d, 1H, J = 5.0 Hz, furyl H-5), 7.02 (s, 2H, NH2), 7.56 (d, 1H, J = 12.5 Hz, H-3), 7.99 (t, 1H, J = 5.0 Hz, furyl H-4); 13C-NMR: d = 164.49 (CONH2), 153.40 (C-5), 151.73, 145.37, 144.80, 118.38, 115.75, 111.53, 98.31, 90.40, 40.77; Anal. calcd. for C12H13N3O2: (231.10): C, 62.33; H, 5.67; N, 18.17. Found: C, 61.97; H, 5.61; N, 18.21.
2-Cyano-5-(dimethylamino)-5-(thiophen-2-yl)penta-2,4-dienamide (8c): Yellow crystals, yield (72 %, 1.77 g); mp 258-259 oC; IR (cm-1): 3403 and 3328 (NH2), 2196 (CN) and 1669 cm-1 (CO); MS m/z (M)+ = 247; 1H-NMR: d = 2.99 (s, 6H, N(CH3)2), 5.68 (d, 1H, J = 12.5 Hz, H-4), 6.94 (s, 2H, NH2), 7.18 (d, 1H, J = 5.0 Hz, thienyl H-3), 7.23 (t, 1H, J = 5.0 Hz, thienyl H-4), 7.39 (d, 1H, J = 12.5 Hz, H-3), 7.88 (d, 1H, J = 5.0 Hz, thienyl H-5); 13C-NMR: d = 164.90 (CONH2), 157.93 (C-5), 153.06 (C-3), 133.66, 131.21, 130.16, 128.14, 119.04 (CN), 99.29 (C-4), 89.99 (C-2), 41.46 (N(CH3)2); Anal. calcd. for C12H13N3OS: (247.08): C, 58.28; H, 5.30; N, 16.99; S, 12.97. Found: C, 58.23; H, 5.29; N, 16.85; S, 12.74.
General procedure for the preparation of compounds 4a-c
Procedure A: To a solution of each of compound 8a-c (10 mmol) in dioxane (15 mL) and HCl (2 mL), was added dropwise a prepared solution of NaNO2 (0.85 g, 10 mmol) and sodium acetate (15 mmol) in water (10 mL). The mixture was stirred for 1h. and allowed to warm up to r.t. During this time a precipitate is formed. The reaction mixture is then filtered off and recrystallized from dioxane.
Procedure B: Coupling reaction was carried out following procedure described earlier [17], which involves coupling each of compounds 8a-c with phenyldiazonium chloride in dioxane /AcONa.
6-Benzoyl-3-oxo-2,3-dihydropyridazine-4-carboxylic acid (4a): Greenish crystals, yield (78 %, 1.90 g); mp 186-187 oC; IR (cm-1): 3420 (OH), 3240 (NH), 1737, 1678 and 1661 cm-1 (3 CO); MS m/z (M)+ = 244; 1H-NMR: d = 7.59-7.62 (t, 2H, J = 7.2 Hz, phenyl-H), 7.75 (t, 2H, J = 7.2 Hz, phenyl-H), 7.96 (br. s, 1H, OH, D2O exchangeable), 8.03 (s, 1H, pyridazinyl H-5), 8.05-8.06 (m, 2H, phenyl-H), 8.25 (br. s, 1H, NH, D2O exchangeable); 13C-NMR: d = 187.43 (ketone CO), 162.42 (carboxylic acid CO), 161.22 (ring CO), 154.14, 134.93, 134.46, 132.70, 129.10, 129.01, 128.72; Anal. calcd. for C12H8N2O4: (244.05): C, 59.02; H, 3.30; N, 11.47. Found: C, 58.95; H, 3.29; N, 11.52
6-(Furan-2-carbonyl)-3-oxo-2,3-dihydropyridazine-4-carboxylic acid (4b): Beige crystals, yield (75 %,1.75 g); mp 213-214 oC; IR (cm-1): 3419 (OH), 3202 (NH), 1744, 1688 and 1657 cm-1 (3 CO); MS m/z (M)+ = 234; 1H-NMR: d = 6.86 (t, 1H, J = 4.5 Hz, furyl H-4), 7.77 (d, 1H, J = 4.3 Hz, furyl H-3), 7.96 (br. s, 1H, OH, D2O exchangeable), 8.03 (s, 1H, pyridazinyl H-5), 8.26 (br. s, 1H, NH, D2O exchangeable), 8.27 (d, 1H, J = 4.5 Hz, furyl H-5); Anal. calcd. for C10H6N2O5: (234.03): C, 51.29; H, 2.58; N, 11.96. Found: C, 51.34; H, 2.61; N, 12.03.
3-Oxo-6-(thiophene-2-carbonyl)- 2,3-dihydropyridazine-4-carboxylic acid (4c): Yellow crystals, yield (72 %, 1.80 g); mp 201-202 oC; IR (cm-1): 3434 (OH), 3220 (NH), 1776, 1746 and 1693 cm-1 (3 CO); MS m/z (M)+ = 250; 1H-NMR: d = 7.37 (t, 1H, J = 4.3 Hz, thienyl H-4), 7.97 (br. s, 1H, OH, D2O exchangeable), 8.04 (s, 1H, pyridazinyl H-5), 8.21 (d, 1H, J = 4.3 Hz, thienyl H-3), 8.27 (m, 2H, thienyl H-5 and NH); Anal. calcd. for C10H6N2O4S: (250.00): C, 48.00; H, 2.42; N, 11.20; S, 12.81. Found: C, 48.05; H, 2.40; N, 11.01; S, 12.72.
General procedure for the preparation of compounds 15a-c
Each of compounds 8a-c (10 mmol) was refluxed in an EtOH/HCl mixture (3:1, 10 mL) for 30 min. Upon cooling to r.t. a solid product precipitated that was collected by filtration and recrystallized from EtOH.
2-Oxo-6-phenyl-1,2-dihydropyridine-3-carboxylic acid (15a): Beige crystals, yield (89 %, 1.91 g); mp 260-262 oC; IR (cm-1): 3380 (NH), and 1725 cm-1 (CO); MS m/z (M)+ = 215; 1H-NMR: d = 6.84 (d, 1H, J = 8.8 Hz, pyridyl H-5), 7.50-7.627 (m, 3H, phenyl-H), 7.79-7.80 (m, 2H, phenyl-H), 8.20 (d, 1H, J = 8.8 Hz, pyridyl H- 4), 12.79 (s, 1H, NH); Anal. calcd. for C12H9NO3: (215.20): C, 66.97; H, 4.22; N, 6.51. Found: C, 67.03; H, 4.20; N, 6.56.
6-(Furan-2-yl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid (15b): Brownish crystals, yield (90 %, 1.85 g); mp 268-269 oC; IR cm-1: 3387 (NH), and 1730 cm-1 (CO); MS m/z (M)+ = 205; 1H-NMR: d = 6.81 (t, 1H, J = 4.3 Hz, furyl H-4), 6.98 (d, 1H, J = 7.6 Hz, pyridyl H-5), 7.34 (d, 1H, J = 4.3 Hz, furyl H-3), 7.84 (br. s, 1H, OH, D2O exchangeable), 7.94 (br. s, 1H, NH, D2O exchangeable), 8.07 (d, 1H, J = 4.5 Hz, furyl H-5), 8.41 (d, 1H, J = 7.6 Hz, pyridyl H-4); Anal. calcd. for C10H7NO4: (205.04): C, 58.54; H, 3.44; N, 6.83. Found: C, 58.31; H, 3.44; N, 7.05.
2-Oxo-6-(thiophen-2-yl)-1,2-dihydropyridine-3-carboxylic acid (15c): Brownish crystals, yield (93 %, 2.0 g); mp 266-268 oC; IR (cm-1): 3398 (NH) and 1721 cm-1 (CO); MS m/z (M)+ = 221; 1H-NMR: d = 7.18 (d, 1H, J = 7.4 Hz, pyridyl H-5), 7.30 (t, 1H, J = 4.5 Hz, thienyl H-4), 7.82 (br. s, 1H, OH, D2O exchangeable), 7.94 (br. s, 1H, NH, D2O exchangeable), 7.95 (d, 1H, J = 4.5 Hz, thienyl H-3), 7.99 (d, 1H, J = 4.5 Hz, thienyl H-5), 8.39 (d, 1H, J = 7.4 Hz, pyridyl H-4). Anal. calcd. for C10H7NO3S: (221.01): C, 54.29; H, 3.19; N, 6.33; S, 14.49. Found: C, 54.14; H, 3.39; N, 6.63; S, 14.56.
General procedure for the preparation of compounds 16a-c
Each of compounds 8a-c (10 mmol) was refluxed in AcOH (10 mL) for 30 min. Upon cooling to r.t. a solid product precipitated that was collected by filtration and crystallized from AcOH.
2-Oxo-6-phenyl-1,2-dihydropyridine-3-carbonitrile (16a): White crystals, yield (92 %, 1.80 g); mp 292-294 oC; IR (cm-1): 3151 (NH), 2226 (CN) and 1660 cm-1 (CO); MS m/z (M)+ = 196; 1H-NMR: d = 6.74 (d, 1H, J = 8.8 Hz, pyridyl H-5), 7.51-7.57 (m, 3H, phenyl-H), 7.80-7.82 (m, 2H, phenyl-H), 8.22 (d, 1H, J = 8.8 Hz, pyridyl H- 4), 12.81 (s, 1H, NH); Anal. calcd. for C12H8N2O: (196.06): C, 73.46; H, 4.11; N, 14.28. Found: C, 73.43; H, 4.00; N, 14.20.
6-(Furan-2-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (16b): Beige crystals, yield (94 %, 1.75 g); mp 298-300 oC; IR (cm-1): 3163 (NH), 2230 (CN) and 1661 cm-1 (CO); MS m/z (M)+ = 186; 1H-NMR: d = 6.72 (d, 1H, J = 8.8 Hz, pyridyl H-5), 6.78 (t, 1H, J = 5.0 Hz, furyl H-4), 7.62 (d, 1H, J = 5.0 Hz, furyl H-3), 8.03 (d, 1H, J = 5.0 Hz, furyl H-5), 8.16 (d, 1H, J = 8.8 Hz, pyridyl H-4), 12.82 (s, 1H, NH). Anal. calcd. for C10H6N2O2: (186.04): C, 64.52; H, 3.25; N, 15.05. Found: C, 64.48; H, 3.14; N, 15.10.
2-Oxo-6-(thiophen-2-yl)-1,2-dihydropyridine-3-carbonitrile (16c): Yellowish crystals, yield (95 %, 1.92 g); mp 300-302 oC; IR (cm-1): 3093 (NH), 2226 (CN) and 1651 cm-1 (CO); MS m/z (M)+ = 202; 1H-NMR: d = 6.70 (d, 1H, J = 8.6 Hz, pyridyl H-5), 7.25 (t, 1H, J = 4.5 Hz, thienyl H-4), 7.88 (d, 1H, J = 4.5 Hz, thienyl H-3), 8.01 (d, 1H, J = 4.5 Hz, thienyl H-5), 8.13 (d, 1H, J = 8.6 Hz, pyridyl H-4), 12.82 (s, 1H, NH); Anal. calcd. for C10H6N2OS: (202.02): C, 59.39; H, 2.99; N, 13.85; S, 15.86. Found: C, 59.35; H, 3.09; N, 13.90; S, 15.79.
Acknowledgements
This research was done by the financial support of the Public Authority for Applied Education and Training (Transform grant TS-07-11) of Kuwait.
References and Notes
- Ferraz, H. M. C.; Goncalo, E. R. S. Recent preparations and synthetic applications of enaminones. Quim. Nova 2007, 30, 957–964. [Google Scholar] [CrossRef]
- Lue, P.; Greenhill, J.V. Enamines in heterocyclic synthesis. Adv. Heterocycl. Chem. 1997, 67, 207–215. [Google Scholar]
- Elassar, A.Z.A.; El-Khair, A.A. Recent Developments in the chemistry of enaminones. Tetrahedron 2003, 59, 8463–8480. [Google Scholar] [CrossRef]
- Stanovnik, B.; Svete, J. Synthesis of heterocycles from alkyl 3-(dimethylamino) propenoates and related enaminones. Chem. Rev. 2004, 104, 2433–2480. [Google Scholar] [CrossRef]
- Yermolayev, S. A.; Gorobets, N. Y.; Lukinova, E. V.; Shishkin, O. V.; Shishkina, S. V.; Desenko, S. M. An efficient synthesis of N1-substituted 2,5-dioxo-1,2,5,6,7,8-hexahydro-3-quinolinecarboxamide via enolate salts. Tetrahedron 2008, 64, 4649–4655. [Google Scholar] [CrossRef]
- Gorobets, N.Y.; Yousefi, B.H.; Belaj, F.; Kappe, C.O. Rapid microwave-assisted solution phase synthesis of substituted 2-pyridone libraries. Tetrahedron 2004, 60, 8633–8644. [Google Scholar] [CrossRef]
- Al-Saleh, B.; Abdel-Khalik, M. M.; Al-Enzy, A.; Elnagdi, M. H. Synthesis of new azoloazine derivatives: new routes to 1,2,4-triazolo[4,3-a]pyrimidines, pyrazolo[1,5-a]pyridines and pyrazolo[3,4-b]pyridinones. J. Chem. Res. 1999. [Google Scholar]
- Abdel-Khalik, M. M.; Elnagdi, M. H.; Agamy, S. M. Studies with functionally substituted heteroaromatics: the chemistry of n-phenylhydrazonylalkylpyridinium salts and of phenylhydrazonylalkylbenzoazoles. Synthesis 2000, 1166–1169. [Google Scholar] [CrossRef]
- Abdel-Khalik, M. M.; Agamy, S. M.; Elnagdi, M. H. Studies with 2-Arylhydrazono-3-Oxopropanals: A novel route to 4-aroyl-2-aryl-1,2,3-triazoles, 3-substituted 4-arylazopyrazoles, 2-substituted glyoxalonitrile and 3-oxoalkanonitriles. Z. Naturforsch 2000, 55b, 1211–1215. [Google Scholar]
- Agamy, S. M.; Abdel-Khalik, M. M.; Mohamed, M. H.; Elnagdi, M. H. Enaminones as building blocks in heterocyclic synthesis: a new one pot synthesis of polyfunctional substituted pyridines. Z. Naturforch. 2001, 56b, 1074–1078. [Google Scholar]
- Al-Qalaf, F.; Abdelkhalik, M. M.; Al-Enezi, A.; Al-Ajmi, J. R. Studies with functionally substituted enamines: synthesis of 2-aroyl-3-dimethylamino-2-propenenitrile and their reactivity toward nitrogen nucleophiles. Heterocycles 2008, 75, 145–156. [Google Scholar] [CrossRef]
- Al-Omran, F.; Al-Awadi, N.; El-Khair, A. A.; Elnagdi, M. H. Synthesis of new aryl and heteroaromatic substituted pyridines, pyrazoles, pyrimidines and pyrazolo[3,4-d]pyridazine. Org. Prep. Proced. Int. 1997, 29, 285–292. [Google Scholar] [CrossRef]
- Al-Omran, F.; Al-Awadi, N.; Abdel-Khalik, M. M.; Kaul, K.; El-Khair, A. A.; Elnagdi, M. H. Substituted 3-dimethylaminoprop-2-en-1-ones as building blocks in heterocyclic synthesis : routes to 6-aryl and 6-heteroaryl-2H-pyran-2-ones and 6- and 4-aryl-1,2-dihydropyridine-2(1H)-ones. J. Chem. Res. 1997. [Google Scholar]
- CCDC 705064 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
- Agamy, S. M. 1-Substituted 3-dimethylaminoprop-2-en-1-ones as building blocks in heterocyclic synthesis: new routes to 6-aroylpyridazin-3-ones, 4,6-diaroylpyridazin-3-imines and 3-aroylpyrazolo[5,1-c][1,2,4]triazines. J. Chem. Res. 2001. [Google Scholar] [CrossRef]
- Al-Omran, F.; Abdel-Khalik, M. M.; Elnagdi, M. H. Studies with polyfunctionally substituted heteroaromatics: new routes for the synthesis of polyfunctionally substituted pyridines and 1,2,4-triazolo[1,5-a]pyridines. J. Heteroatom Chem. 1995, 6, 545–551. [Google Scholar] [CrossRef]
- Al-Omran, F.; Abdel-Khalik, M. M.; El-Khair, A. A.; Elnagdi, M. H. Studies with functionally substituted heteroaromatics: a novel route for the synthesis of 1-aryl-6-oxopyridazinones, 1-arylpyridazine-6-imines and 1-aryl-6-imino-4-pyridazinals. Synthesis 1997, 91–94. [Google Scholar]
- Sample Availability: Samples of the compounds 4a-c, 8a-c, 15a-c and 16a-c are available from authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).