Photochemical Synthesis of Nucleoside Analogues from Cyclobutanones: Bicyclic and Isonucleosides

Abstract
: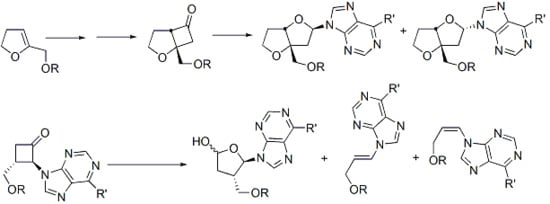
1. Introduction
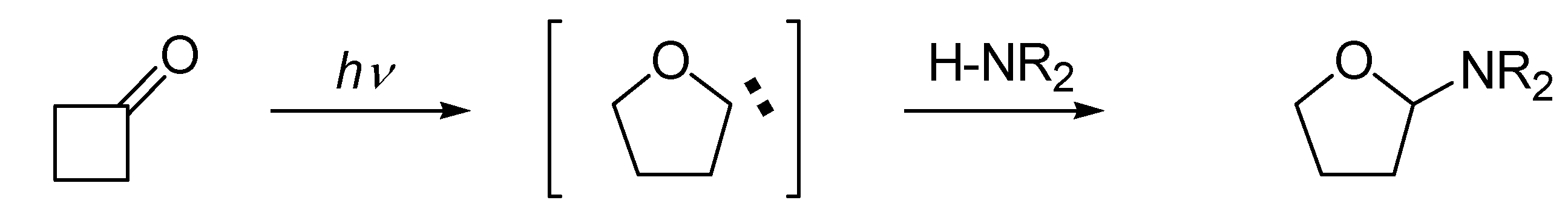
2. Results and Discussion
2.1. Photochemical Synthesis of a Bicyclic Nucleoside Analogue
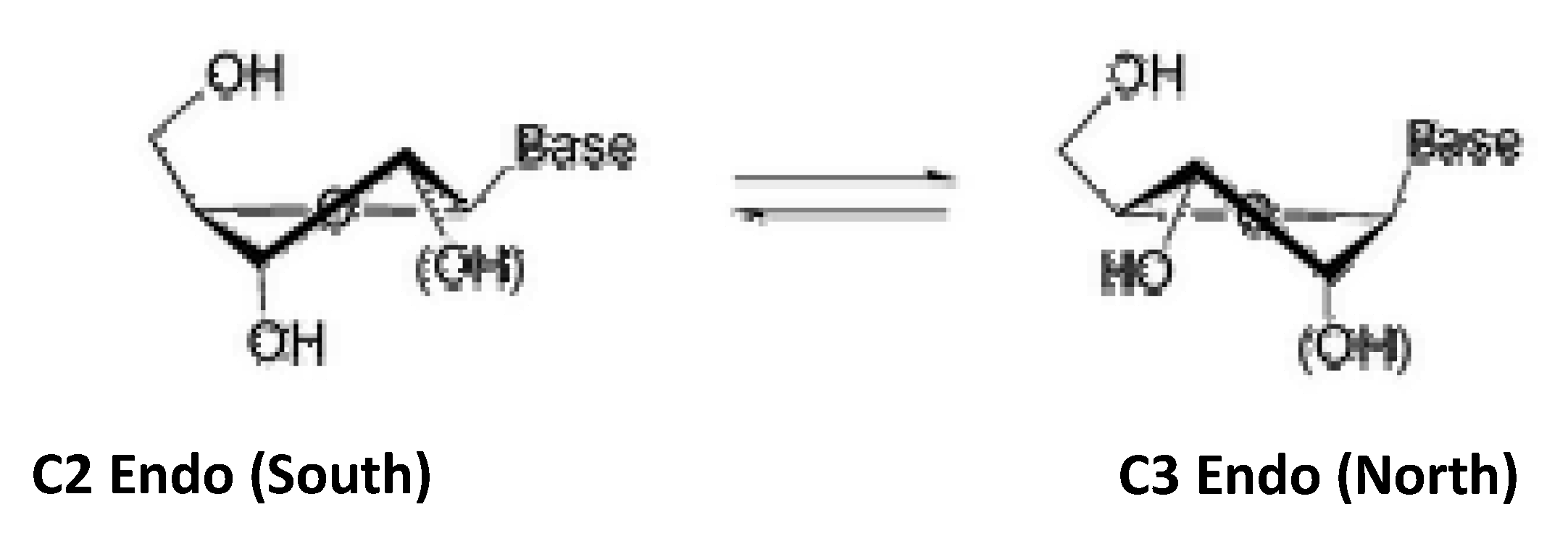


2.2. Synthesis of Acyclic and Isonucleoside Analogues from the same Cyclobutanone Precursor
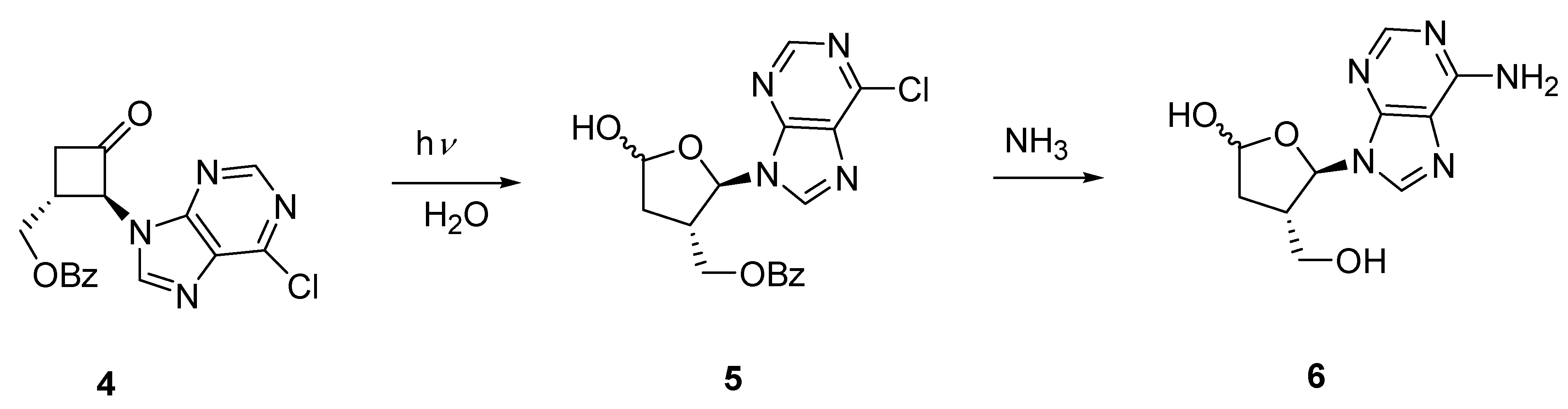


3. Experimental
3.1. General
3.2. 1-(Benzyloxymethyl)-7,7-dichloro-2-oxabicyclo[3.2.0]heptan-6-one
3.2.1. Method A
3.2.2. Method B
3.3. 1-(Benzyloxymethyl)- 2-oxabicyclo[3.2.0]heptan-6-one (3)
3.3.1. Method A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
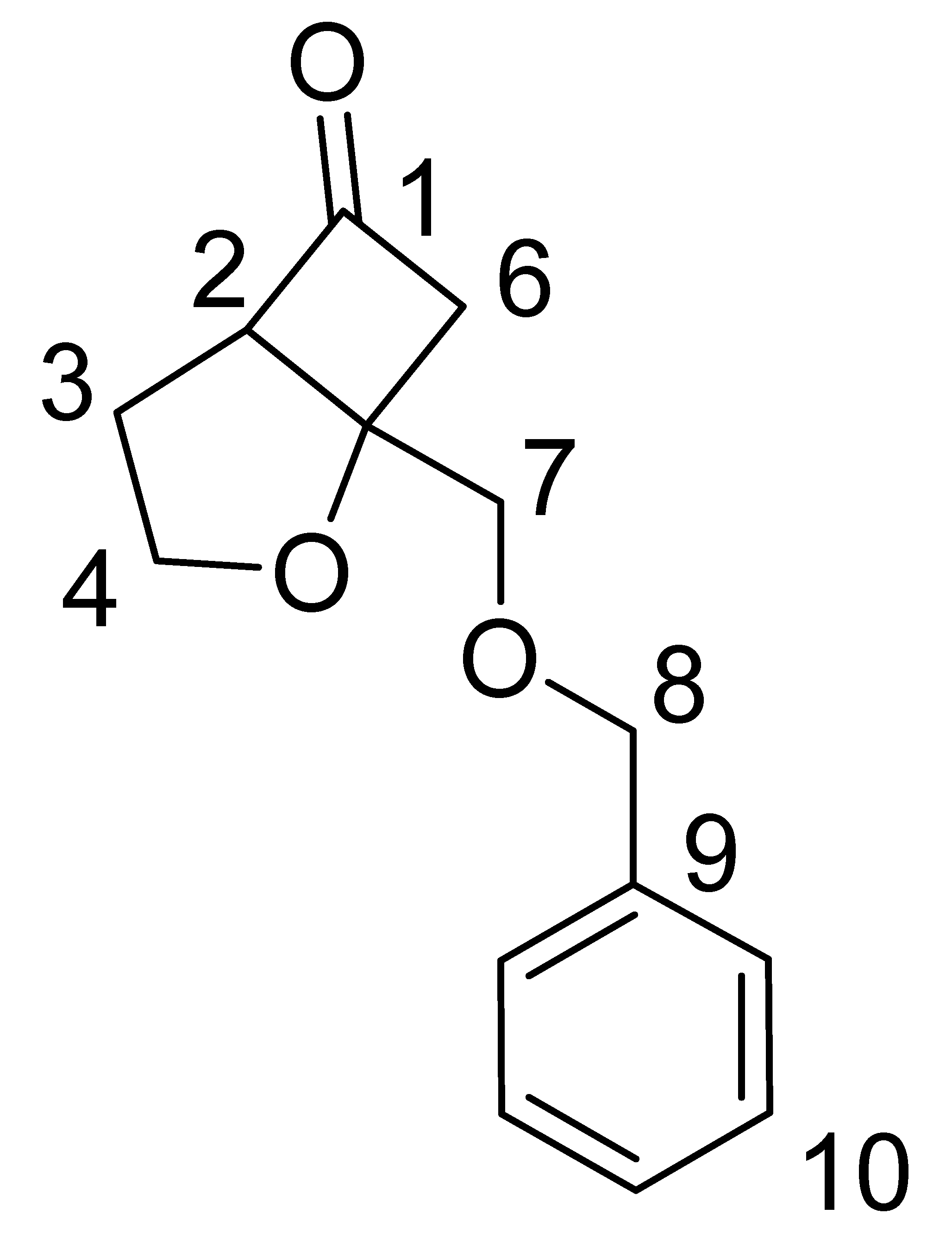 | ||
|---|---|---|
| Carbon No. | 13C δ (ppm) | 1H δ (ppm) |
| 1 | 214 | |
| 2 | 65.4 | 3.64–3.68 (m, 1H) |
| 3 | 29.37 | 1.90–2.22 (m, 2H) |
| 4 | 69.20 | 3.97–4.03 (m, 1H) 4.21-4.28 (m, 1H) |
| 5 | 29.9 | |
| 6 | 53.1 | 3.21–3.29 (dd, J = 5.4 Hz, 18Hz, 1H) 2.93–3.00 (dd, J = 2.1 Hz, 18 Hz, 1H) |
| 7 | 71.6 | 3.82–3.89 (m, 2H) |
| 8 | 73.5 | 4.66–4.70 (m, 2H) |
| 9 | 138 | |
| 10 | 127.7–130.6 | 7.30–7.38 (m, 5H) |
3.3.2. Method B
3.4. 9-(3a-(Benzyloxymethyl)dihydrofuro[3,2-b]furan-2-yl)-6-chloro-9H-purine (1a and 1b)
| Major nucleoside 1a | ||
|---|---|---|
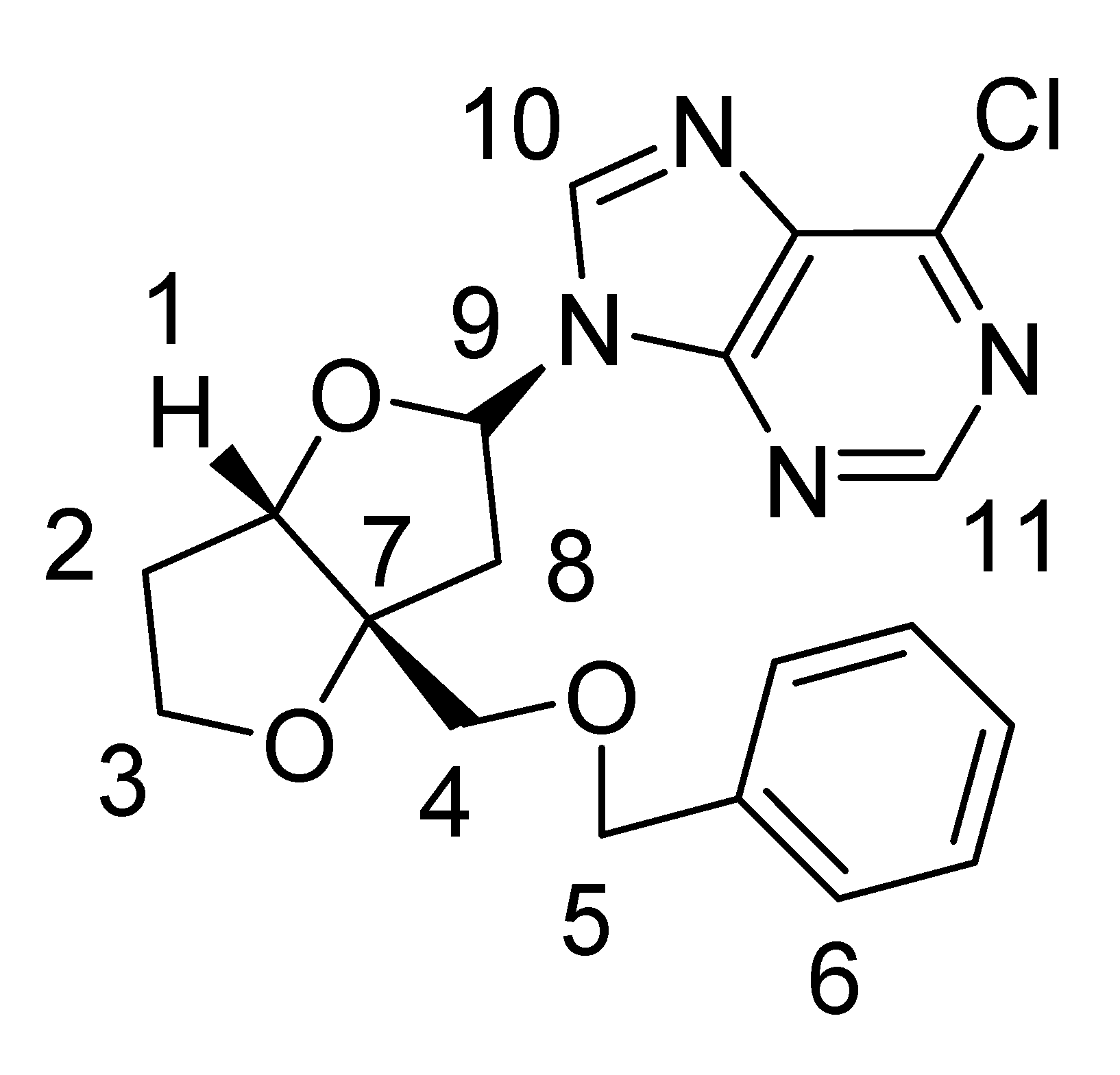 | ||
| Carbon No. | 13C δ (ppm) | 1H δ (ppm) |
| 1 | 92.2 | 4.90 (d, J = 4.4 Hz, 1H) |
| 2 | 33.2 | 2.12–2.23 (m, 2H) |
| 3 | 73.6 | 4.04–4.14 (m, 2H) |
| 4 | 67.9 | 3.66 (d, J = 9.6 Hz, 1H), 3.53 (d, J = 9.6 Hz, 1H) |
| 5 | 70.7 | 4.5 (AB q, J = 12 Hz, 2H) |
| 6 | 126.0-127.8 | 7.25-7.37 (m, 5H) |
| 7 | 76.6 | |
| 8 | 41.9 | 2.81 (dd, J = 7.2 Hz, 14.4 Hz, 1H)3.02 (dd, J = 5.2 Hz, 14.8 Hz, 1H) |
| 9 | 86.1 | 6.53-6.55 (dd, J = 5.2 Hz, 7.2 Hz, 1H) |
| 10 | 143.5 | 8.45 (s, 1H) |
| 11 | 151.9 | 8.70 (s, 1H) |
| Minor nucleoside 1b: | ||
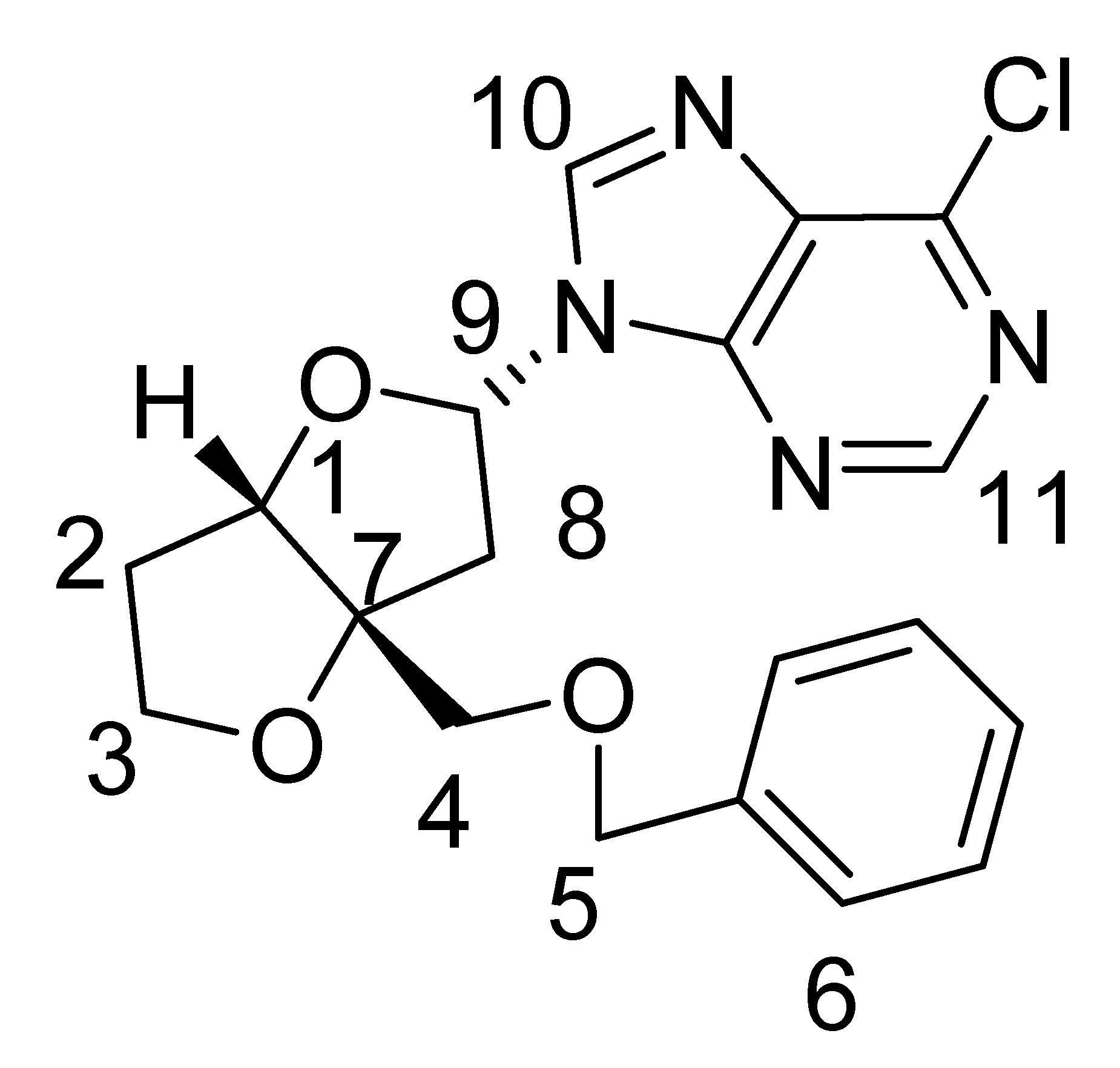 | ||
| Carbon No. | 13C δ (ppm) | 1H δ (ppm) |
| 1 | 87.3 | 4.73 (d, J =2.4 Hz, 1H) |
| 2 | 32.8 | 2.13–2.19 (m, 2H) |
| 3 | 73.7 | 4.09 (m, 2H) |
| 4 | 68.2 | 3.63 (d, J = 9.6 Hz, 1H), 3.54 (d, J = 9.6 Hz, 1H) |
| 5 | 70.9 | 4.63 (s, 2H) |
| 6 | 127.3–128.6 | 7.28–7.40 (m, 5H) |
| 7 | 76.0 | |
| 8 | 40.1 | 2.99 (dd, J =7.8 Hz, 14.7 Hz, 1H)2.76 (dd, J = 3.6 Hz, 14.7 Hz, 1H) |
| 9 | 84.2 | 6.47–6.49 (dxd, J = 3.9 Hz, 7.8 Hz, 1H) |
| 10 | 144.0 | 8.59 (s, 1H) |
| 11 | 152.2 | 8.79 (s, 1H) |
3.5. Cyclobutanone 4
3.6. Photolysis of Ketone 4 in Acetonitrile spiked with Water
3.7. Photolysis of Ketone 4 in Methanol
3.8. Photolysis of Ketone 4 in Acetone
3.9. Photolysis of Ketone 4 in Benzene
4. Conclusions
Acknowledgements
References and Notes
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537. [Google Scholar] [CrossRef]
- Bellezza, I.; Tucci, A.; Minelli, W. 2-Chloroadenosine and human prostate cancer cells. 2008; 8, 783–789. [Google Scholar]
- De Clerq, E.; Neyts, J. DNA RNA Chain Terminators. In Handbook of Experimental Pharmacology; Springer-Verlag: Heidelberg, Germany, 2009; Volume 189, pp. 53–84. [Google Scholar]
- Ohno, H.; Terui, T.; Kitawaki, T.; Chida, N. Total synthesis of dapiramicin B. Tetrahedron Lett. 2006, 47, 5747–5750. [Google Scholar]
- Angelini, M.P.; Lee-Ruff, E. A novel photochemical synthesis of dideoxyfuranosyl disaccharides. Tetrahedron Lett. 1998, 39, 8783–8786. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Wan, W-Q.; Jiang, J.-L. A Novel Approach toward the Synthesis of Chiral 2,3-Dideoxy Nucleosides and Their Carbocyclic Analogs. J. Org. Chem. 1994, 59, 2114–2118. [Google Scholar]
- Lee-Ruff, E.; Xi, F.; Qie, J.H. Enantioselective Preparation of 2',3'-Dideoxynucleosides and Their Analogs from Ring-Expansion of Cyclobutanones. 2. Synthesis of 2',3'-Dideoxyribosides and (1S,3R)-1-Amino-3-(hydroxymethyl)cyclopentane. J. Org. Chem. 1996, 61, 1547–1550. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Ostrowski, M.; Ladha, A.; Stynes, D. V.; Vernik, I.; Jiang, J.-L.; Wan, W.-Q.; Ding, S.-F.; Joshi, S. Synthesis and HIV Inhibition Activity of 2',3'-Dideoxy-3'-C-hydroxymethyl Nucleosides. J. Med. Chem. 1996, 39, 5276–5280. [Google Scholar]
- Lee-Ruff, E.; Margau, R. Photochemical synthesis of novel dideoxynucleosides. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 185–196. [Google Scholar] [CrossRef]
- Zhong, J.-H.; Fishman, A.; Lee-Ruff, E. Photochemistry on Soluble Polymer Supports: Synthesis of Nucleosides. Org. Lett. 2002, 4, 4415–4417. [Google Scholar] [CrossRef]
- Ghazi, H.; Lee-Ruff, E. Synthesis of fluorocyclobutanones and their use in the synthesis of fluoronucleosides. J. Fl. Chem. 2005, 126, 1565–1569. [Google Scholar] [CrossRef]
- Mladenova, G.; Lee-Ruff, E. Cyclobutanone photoadducts of HCN and malononitrile: useful intermediates for the synthesis of C-nucleosides. Tetrahedron Lett. 2007, 48, 2787–2789. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Wells, D. Bicyclic Nucleoside Synthesis-A Photochemical Approach. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 484–494. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Mladenova, G. Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev. 2003, 103, 1449–1483. [Google Scholar]
- Lee-Ruff, E. Synthesis of cyclobutanes. Chem. Cyclobutanes 2005, 1, 281–355. [Google Scholar]
- Lee-Ruff, E.; Hayes, I.E.E.; Kazarians-Moghaddam, H. Photocycloeliminations of bicyclic cyclobutanones. I. Solvent effects on distribution of products. Struct. Chem. 1991, 2, 175–83. [Google Scholar] [CrossRef]
- Mathe, C.; Perigaud, C. Recent approaches in the synthesis of conformationally restricted nucleoside analogues. Eur. J. Org. Chem. 2008, 1489–1505. [Google Scholar]
- Dyguda, E.; Szefczyk, B.; Sokalski, W.J. The Mechanism of Phosphoryl Transfer Reaction and the Role of Active Site Residues on the Basis of Ribokinase-like Kinases. Int. J. Mol. Sci. 2004, 5, 141–153. [Google Scholar]
- Miyazaki, H.; Ohkawa, N.; Nakamura, N.; Ito, T.; Sada, T.; Oshima, T.; Koike, H. Lactone and cyclic ether analogs of platelet-activating factor. Synthesis and biological activities. Chem. Pharm. Bull. 1989, 37, 2379–2390. [Google Scholar] [CrossRef]
- Machiguchi, T.; Okamoto, J.; Takachi, J.; Hasegawa, T.; Yamabe, S.; Minato, T. Exclusive Formation of α-Methyleneoxetanes in Ketene-Alkene Cycloadditions. Evidence for Intervention of Both an α-Methyleneoxetane and the Subsequent 1,4-Zwitterion. J. Am. Chem. Soc. 2003, 125, 14446–14448. [Google Scholar]
- Nair, V. Antiviral isonucleosides: discovery, chemistry and chemical biology. Recent Adv. Nucleoside: Chem. Chemther. 2002, 149–166. [Google Scholar] [CrossRef]
- Zhang, S.; Switzer, C.; Chaput, J.C. The Resurgence of Acyclic Nucleic Acids. Chem. Biodivers. 2010, 7, 245–258. [Google Scholar] [CrossRef]
- Ebead, A.; Lee-Ruff, E. York University, Toronto, Canada. Unpublished work.
- Haas, Y. Photochemical α-cleavage of ketones: revisiting acetone. Photochem. Photobiol. Sci. 2004, 3, 6–16. [Google Scholar]
- Smith, R.D.; Simmons, H.E. Norcarane. Org. Synth. 1973, Coll. Vol. 5, 855, 1961, 41, 72.. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors;
Share and Cite
Jaffer, M.; Ebead, A.; Lee-Ruff, E. Photochemical Synthesis of Nucleoside Analogues from Cyclobutanones: Bicyclic and Isonucleosides. Molecules 2010, 15, 3816-3828. https://doi.org/10.3390/molecules15063816
Jaffer M, Ebead A, Lee-Ruff E. Photochemical Synthesis of Nucleoside Analogues from Cyclobutanones: Bicyclic and Isonucleosides. Molecules. 2010; 15(6):3816-3828. https://doi.org/10.3390/molecules15063816
Chicago/Turabian StyleJaffer, Mileina, Abdelaziz Ebead, and Edward Lee-Ruff. 2010. "Photochemical Synthesis of Nucleoside Analogues from Cyclobutanones: Bicyclic and Isonucleosides" Molecules 15, no. 6: 3816-3828. https://doi.org/10.3390/molecules15063816
APA StyleJaffer, M., Ebead, A., & Lee-Ruff, E. (2010). Photochemical Synthesis of Nucleoside Analogues from Cyclobutanones: Bicyclic and Isonucleosides. Molecules, 15(6), 3816-3828. https://doi.org/10.3390/molecules15063816