Synthesis of New Liquid Crystalline Diglycidyl Ethers

Abstract
:1. Introduction
2. Results and Discussion
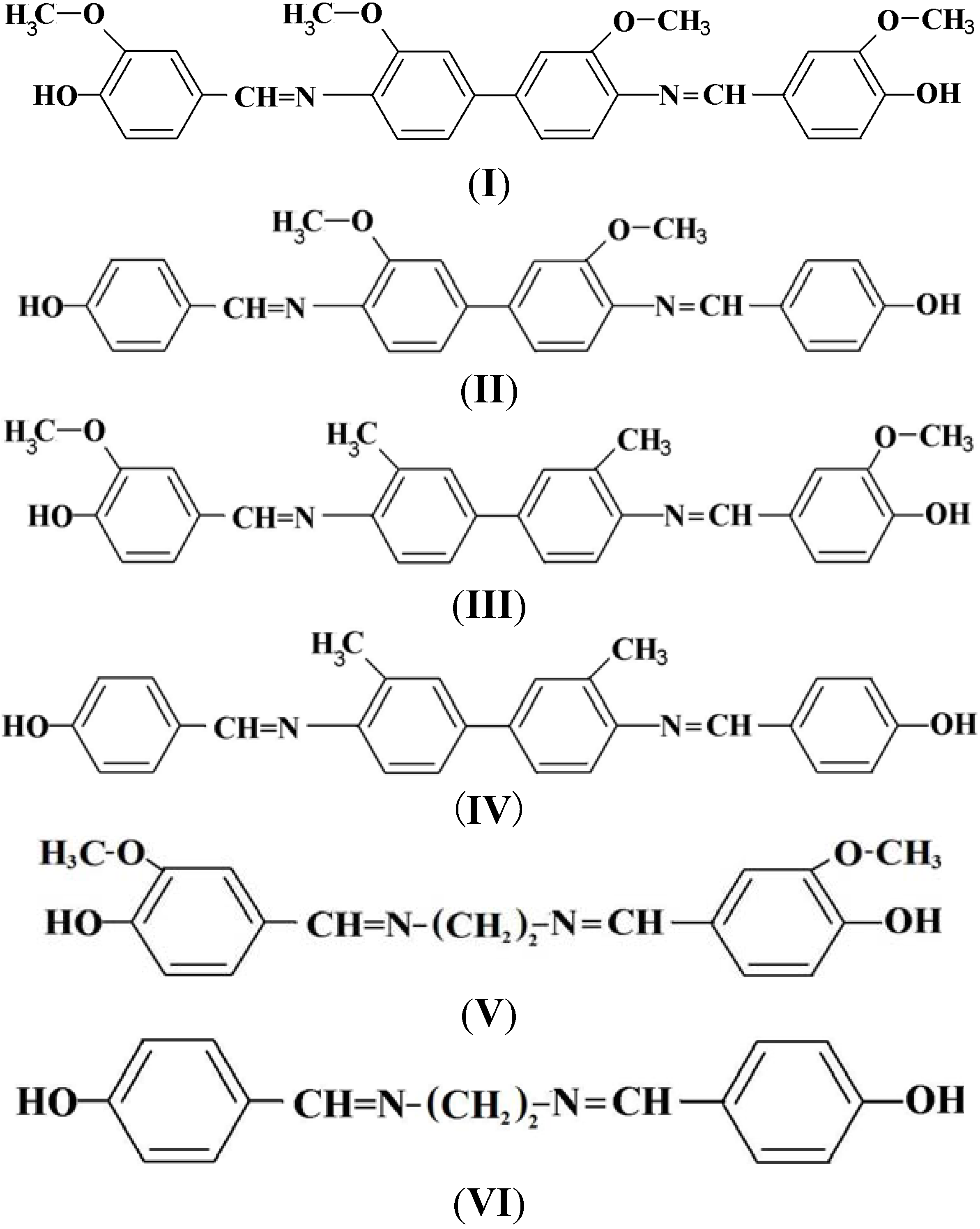
2.1. Characterization of the Phenolic Schiff Bases I–VI
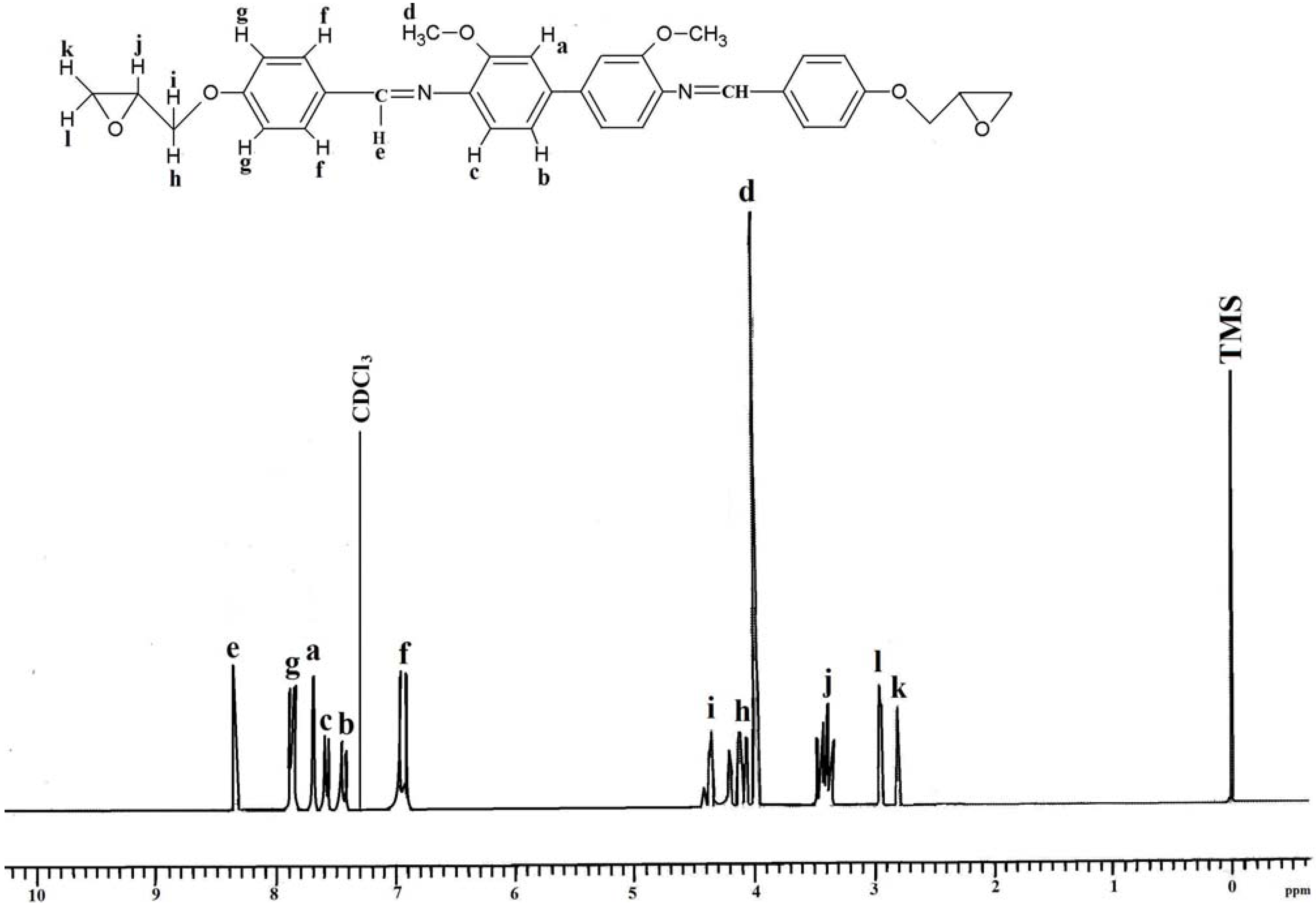
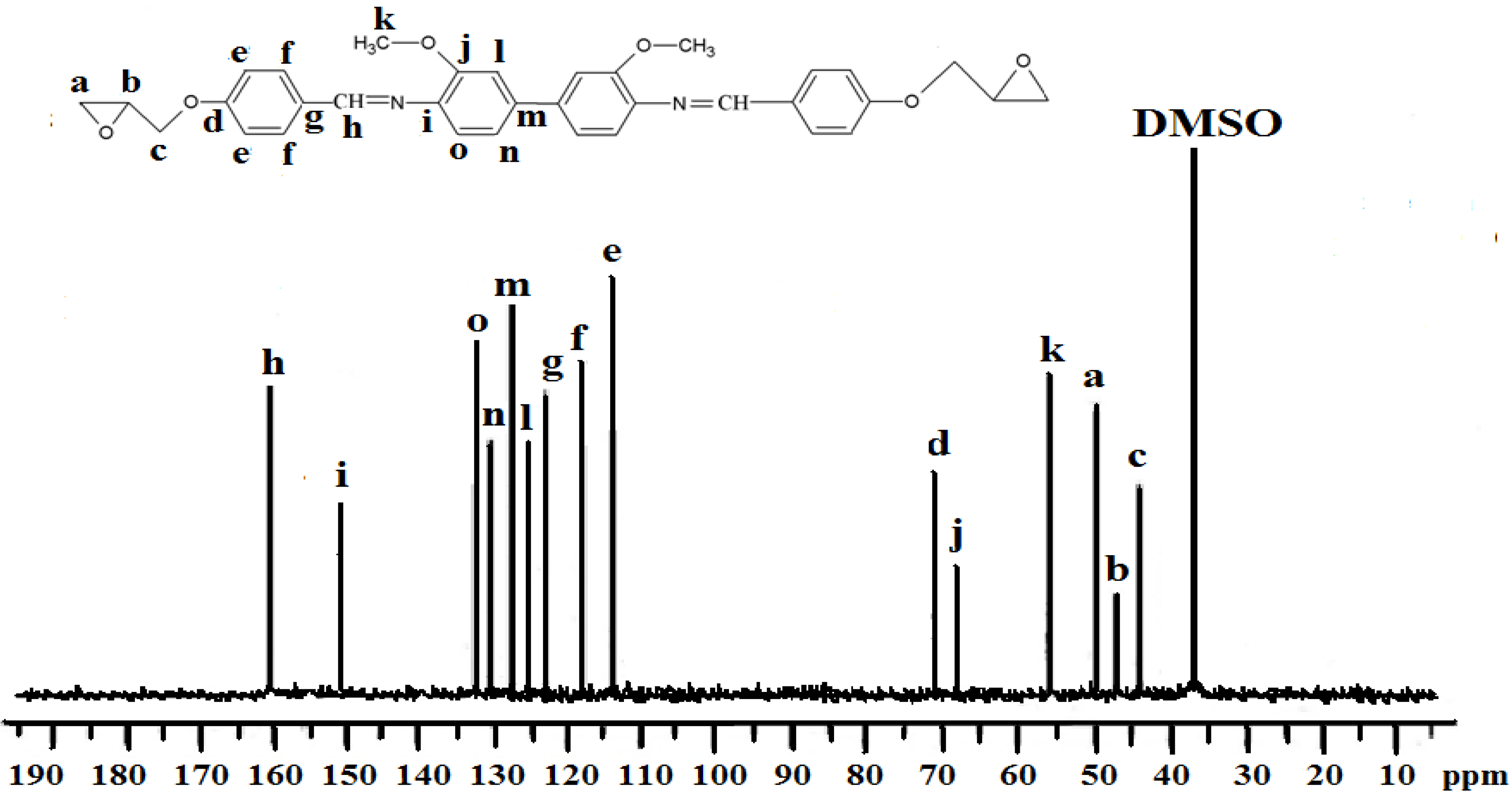
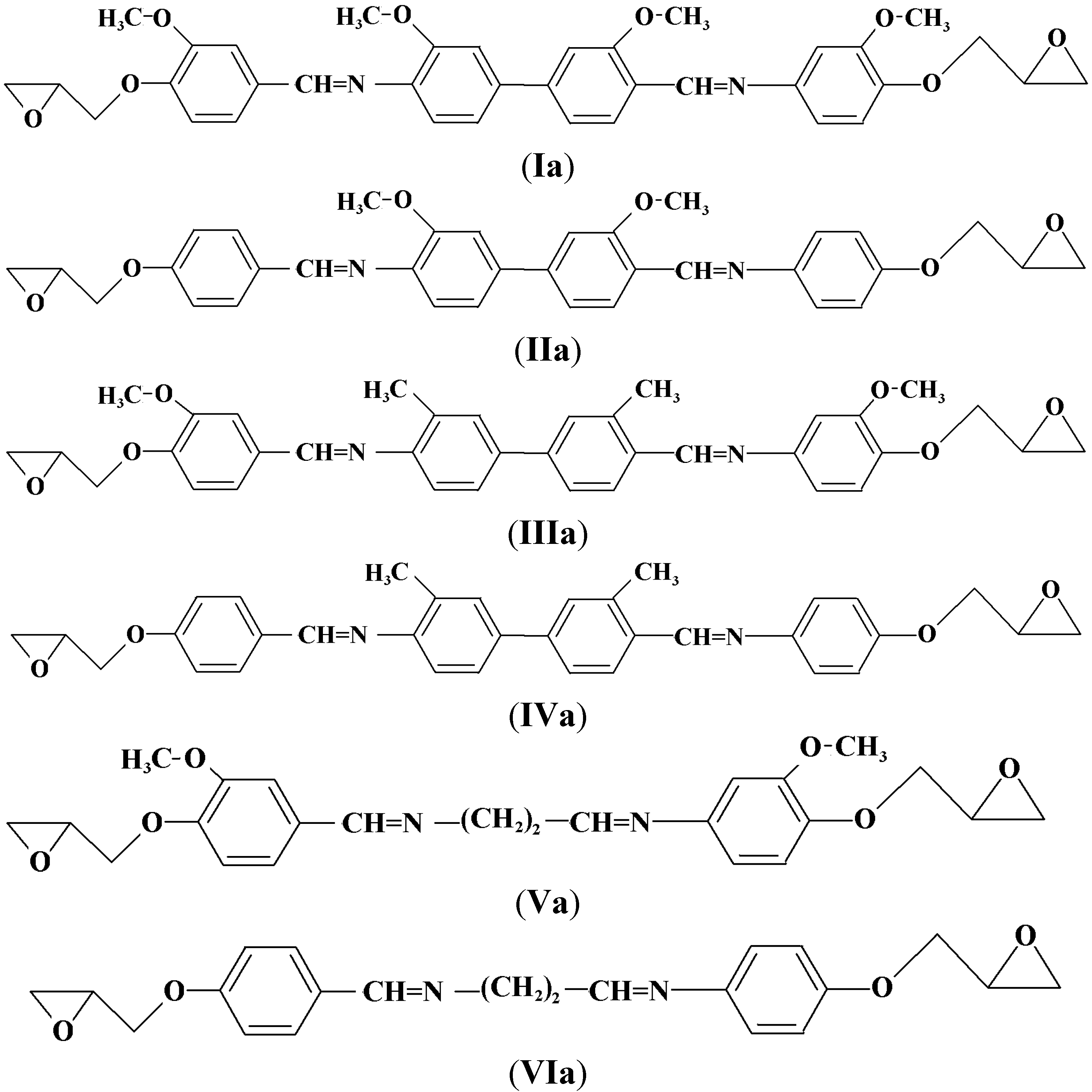
2.2. Characterization of the Diglycidyl Ethers Ia–VIa


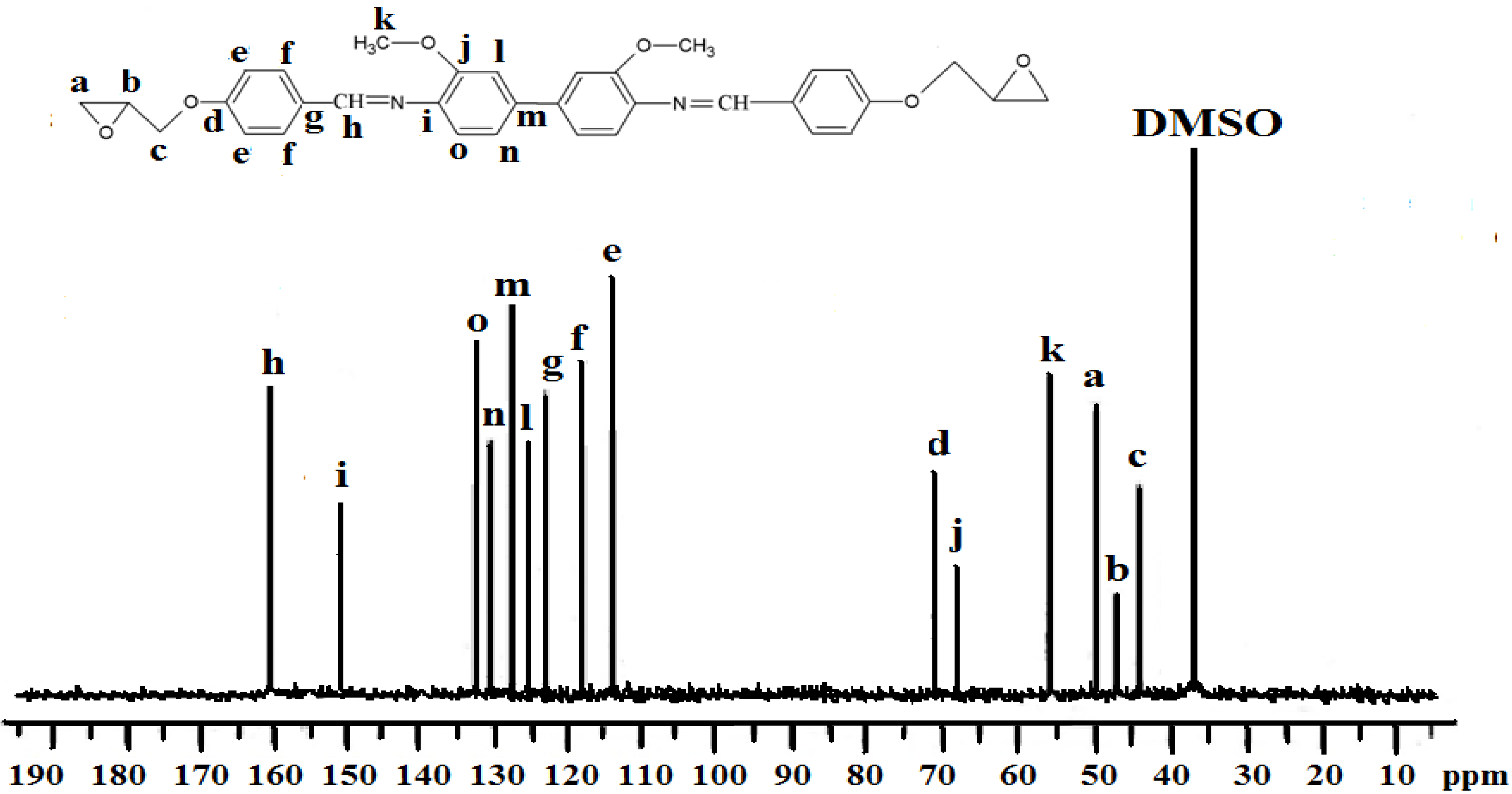
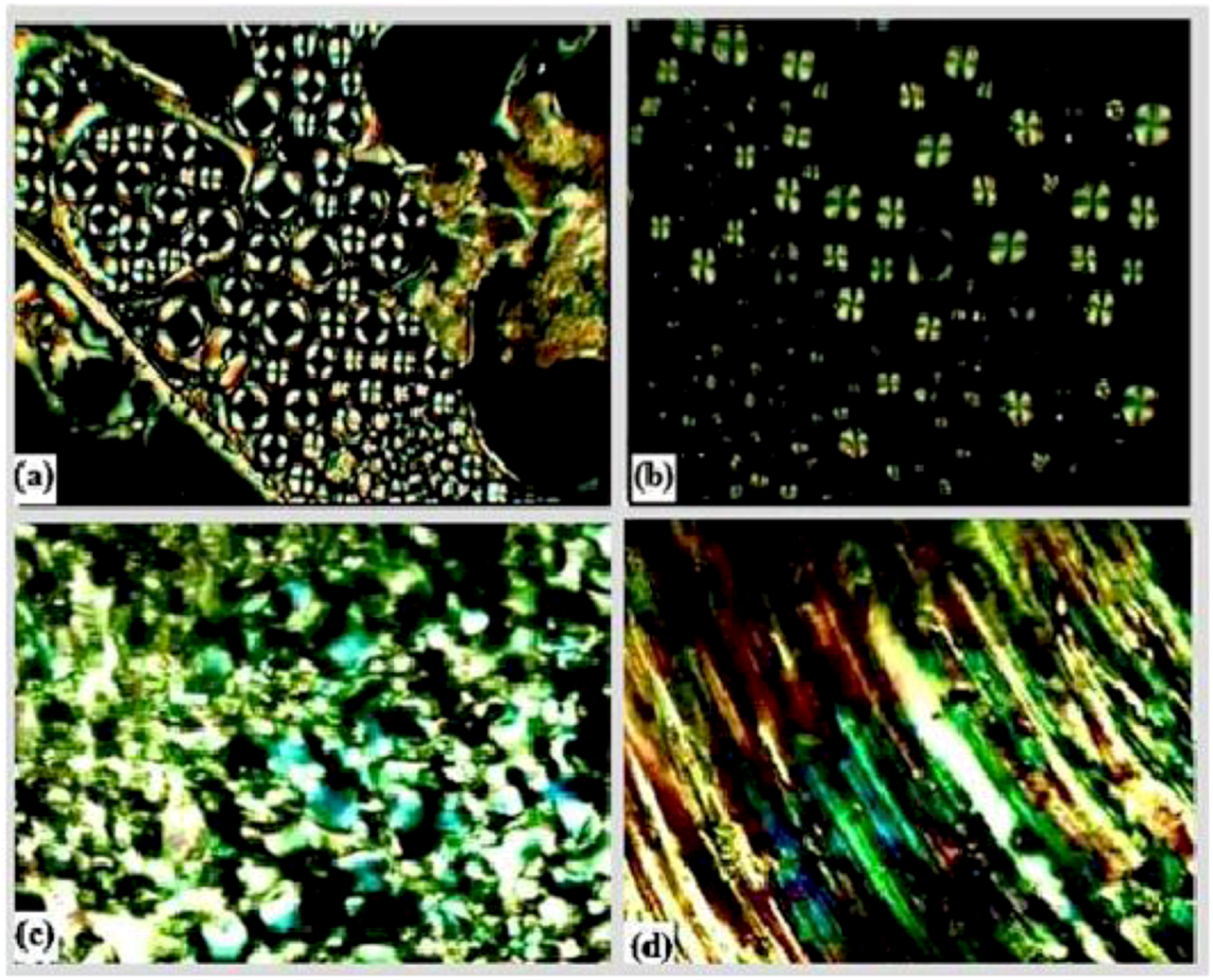
2.3. Properties of the Diglycidyl Ethers Ia–VIa

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diglycidyl ethers | Cr-N (°) * | N-I (°) * | ∆T/°C | Transition temperatures, DSC (°) | |
|---|---|---|---|---|---|
| Tm | Ti | ||||
| Ia | 158.0 | 251.0 | 148.0 | 149.8 | 257.0 |
| IIa | 160.1 | 275.2 | 115.1 | 157.0 | 285.1 |
| IIIa | 135.0 | 174.0 | 42.0 | 136.0 | 176.0 |
| IVa | 147.0 | 215.0 | 79.0 | 145.0 | 220.0 |
| Va | - | - | - | 138.0 | - |
| VIa | - | - | - | 140.0 | - |

3. Experimental
3.1. Materials
3.2. Instruments
3.3. General Procedure for the Preparation of the Phenolic Schiff Bases I–VI

3.4. General Procedure for the Preparation of the Diglycidyl Ethers Ia–VIa

4. Conclusions
Acknowledgments
- Sample Availability: Not available.
References and Notes
- Theis, J.; Ritter, H. Formation of epoxide-amine oligo-adducts as OH-functionalized initiators for the ring-opening polymerization of ε-caprolactone. Beilstein J. Org. Chem. 2010, 6, 938–944. [Google Scholar] [CrossRef]
- Solomons, T.W.G.; Fryhle, C.B. Organic Chemistry, 9th ed; John Wiley and Sons Inc: New York, NY, USA, 2007. [Google Scholar]
- Bazhin, D.N.; Gorbunova, T.I.; Zapevalov, A.Y.; Kirichenko, V.E.; Saloutin, V.I. Features of reaction between fluorine-containing glycidyl ethers and alcohols in basic medium. Russ. J. Org. Chem. 2007, 43, 656–659. [Google Scholar] [CrossRef]
- Karadeniz, L.; Koz, G.; Aydin, K.; Astley, S.T. Co (III) catalysed asymmetric ring-opening of epichlorohydrin by salicylaldehyde derivatives: Reversal of enantioselectivity and rate acceleration on addition of AlCl3. Turk. J. Chem. 2010, 34, 711–718. [Google Scholar]
- Hanaoka, T.; Kawamura, N.; Hara, K.; Tsugane, S. Urinary bisphenol A and plasma hormone concentrations in male workers exposed to bisphenol A diglycidyl etherand mixed organic solvents. Occup. Environ. Med. 2002, 59, 625–628. [Google Scholar] [CrossRef]
- Yi, W.J.; Feng, Z.H.; Zhang, Q.F.; Zhang, J.; Li, L.D.; Zhu, W.; Yu, X.Q. Diol glycidyl ether-bridged cyclens: Preparation and their applications in gene delivery. Org. Biomol. Chem. 2011, 9, 2413–2421. [Google Scholar]
- Atta, A.M.; El-Kafrawy, A.F.; Aly, M.H.; Abdel-Azim, A.A. New epoxy resins based on recycled poly (ethylene terephthalate) as organic coatings. Prog. Org. Coat. 2007, 58, 13–22. [Google Scholar]
- Aroua, L.; Baklouti, A. Synthesis of α,ω-bis(oxazolidinone) polyoxyethylene via a lithium bromide-catalyzed reaction of oligoethylene glycol diglycidyl ethers with isocyanates. Synt. Commun. 2007, 37, 1935–1942. [Google Scholar] [CrossRef]
- Herweh, J.E.; Kauffman, W.J. 2-Oxazolidones via the lithium bromide catalyzed reaction of isocyanates with epoxides in hydrocarbon solvents. Tetrahedron Lett. 1971, 12, 809–812. [Google Scholar] [CrossRef]
- Huang, L.; Wah, S.; Chang, T.; Seo, W.; Rove, K. Microfabrication of Anisotropic Organic Materials via Self-Organization of an Ionic Perylenemonoimide. Adv. Mater. 2007, 19, 4149–4152. [Google Scholar] [CrossRef]
- Laschat, S. Progress in liquid crystal chemistry. Beilstein J. Org. Chem. 2009, 5. No. 48. [Google Scholar]
- Yang, Z.; Sun, P. Compare of three ways of synthesis of simple Schiff base. Molbank 2006, 2006, M514. [Google Scholar] [CrossRef]
- Jarrahpour, A.A.; Jalbout, A.F.; Rezaei, S.; Trzaskowski, B. Synthesis and Physical Character of 2-((E)-1-(3-((E)-1-(2-hydroxyphenyl) ethylideneamino)-2-methylphenylimino ethyl) phenol. Molbank 2006, 2006, M455. [Google Scholar] [CrossRef]
- Issam, A.M.; Ismail, J. Improvement of thermal stability of new heteroaromatic poly(azomethine urethane)s. J. Appl. Polym. Sci. 2006, 100, 1198–1204. [Google Scholar] [CrossRef]
- Lee, H.L.; Issam, A.M.; Belhami, M.; Assouar, M.B.; Rinnert, H.; Alnot, M. Thermal and optical properties of CdS nanoparticles in thermotropic liquid crystal monomers. Materials 2010, 3, 2069–2086. [Google Scholar] [CrossRef]
- Issam, A.M.; Sankar, G.; Khairuddean, M.; Bakar, M.A. Synthesis and liquid crystalline properties of new diols containing azomethine groups. Molecules 2010, 15, 3260–3269. [Google Scholar] [CrossRef]
- Singh, S.; Dunmur, D.A. Liquid Crystals: Fundamentals; World Scientific Publishing CO. Pte. Ltd.: London, UK, 2002. [Google Scholar]
- Jieh, S.S.; Chou, C.T. Studies on thermotropic liquid crystalline polyurethanes.III. Synthesis and properties polyurethane elastomers by using various mesogenic units as chain extender. J. Polym. Sci. A Polym. Chem. 1996, 34, 771–779. [Google Scholar] [CrossRef]
- Ha, S.T.; Ng, M.Y.; Subramaniam, T.R.; Ito, M.M.; Saito, A.; Watanabe, M.; Lee, S.L.; Bonde, N.L. Mesogenic azomethine esters with different end groups: Synthesis and thermotropic properties. Int. J. Phys. Sci. 2010, 5, 1256–1262. [Google Scholar]
- Jin, J.I.L.; Kang, C.S. Effect of methoxy substituent on mesophase forming capacity of main chain aromatic polyesters having polymethylene spacers. Polymer 1993, 34, 2407–2412. [Google Scholar] [CrossRef]
- Li, C.H.; Chang, T.C. Thermotropic liquid crystalline polymer.III. Synthesis and properties of poly(amide-azomethine-ester). J. Polym. Sci. A Polym. Chem. 1991, 29, 361–367. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mohammed, I.A.; Hamidi, R.M. Synthesis of New Liquid Crystalline Diglycidyl Ethers. Molecules 2012, 17, 645-656. https://doi.org/10.3390/molecules17010645
Mohammed IA, Hamidi RM. Synthesis of New Liquid Crystalline Diglycidyl Ethers. Molecules. 2012; 17(1):645-656. https://doi.org/10.3390/molecules17010645
Chicago/Turabian StyleMohammed, Issam Ahmed, and Rashidah Mohamed Hamidi. 2012. "Synthesis of New Liquid Crystalline Diglycidyl Ethers" Molecules 17, no. 1: 645-656. https://doi.org/10.3390/molecules17010645
APA StyleMohammed, I. A., & Hamidi, R. M. (2012). Synthesis of New Liquid Crystalline Diglycidyl Ethers. Molecules, 17(1), 645-656. https://doi.org/10.3390/molecules17010645