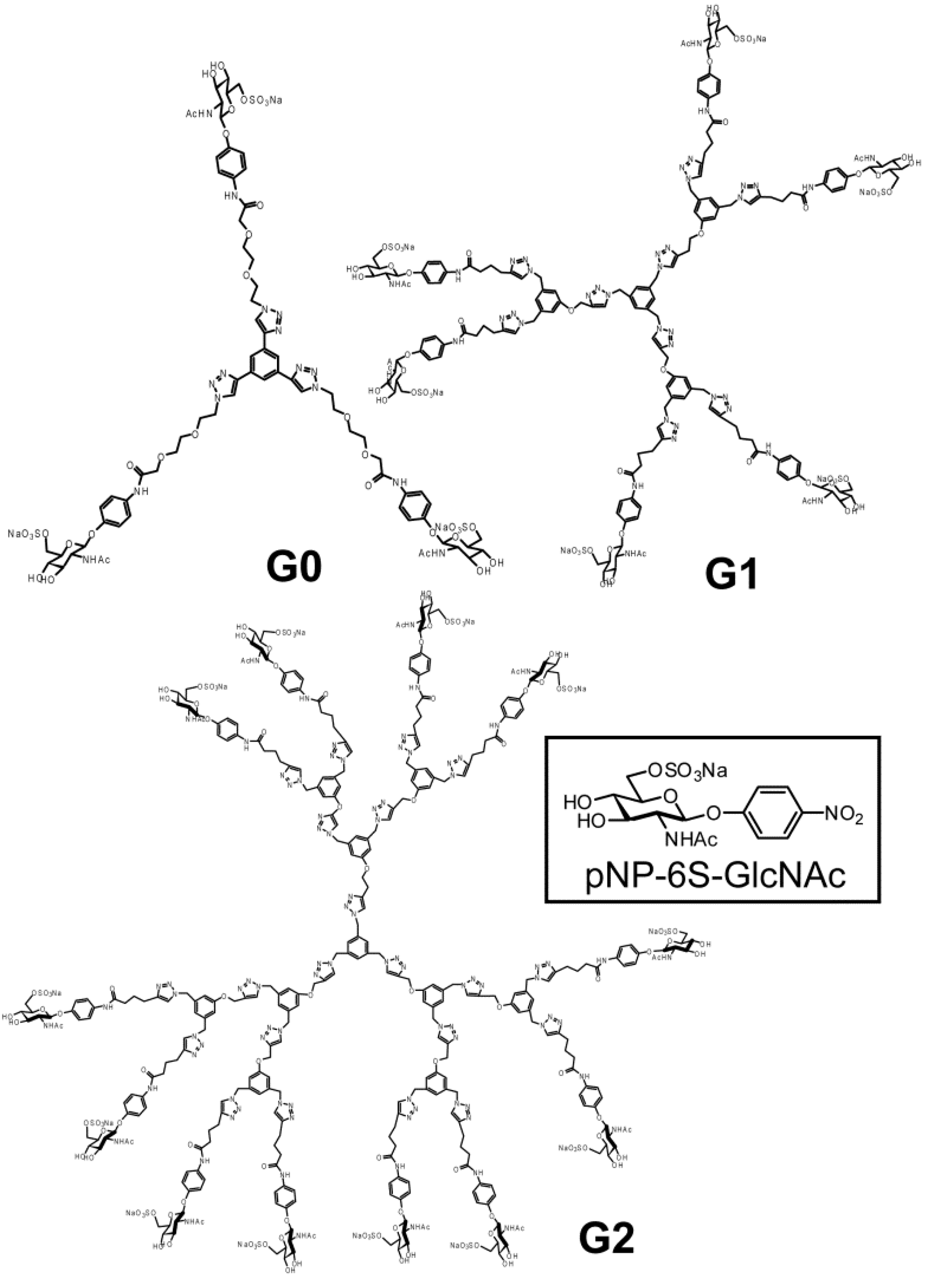
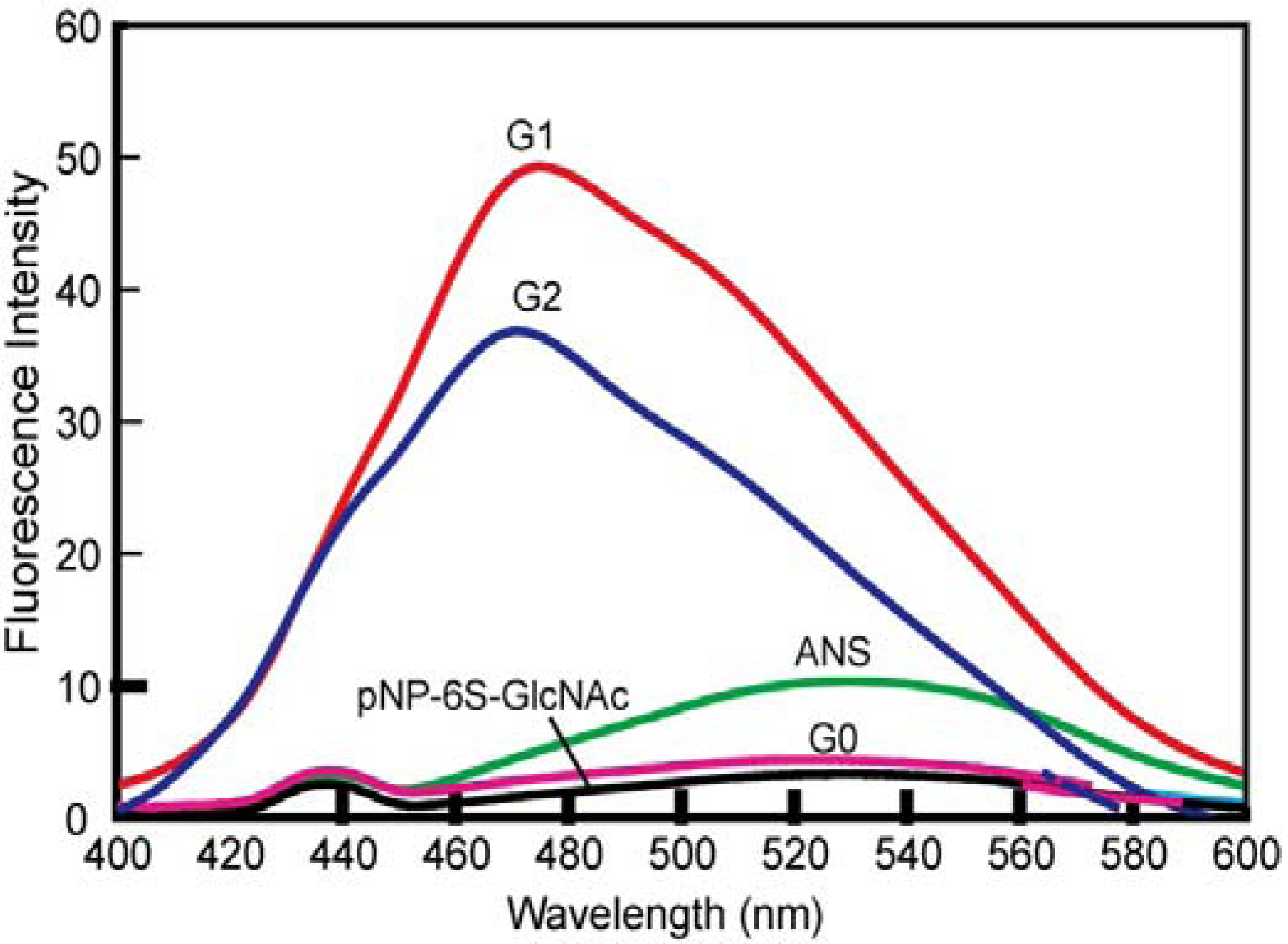
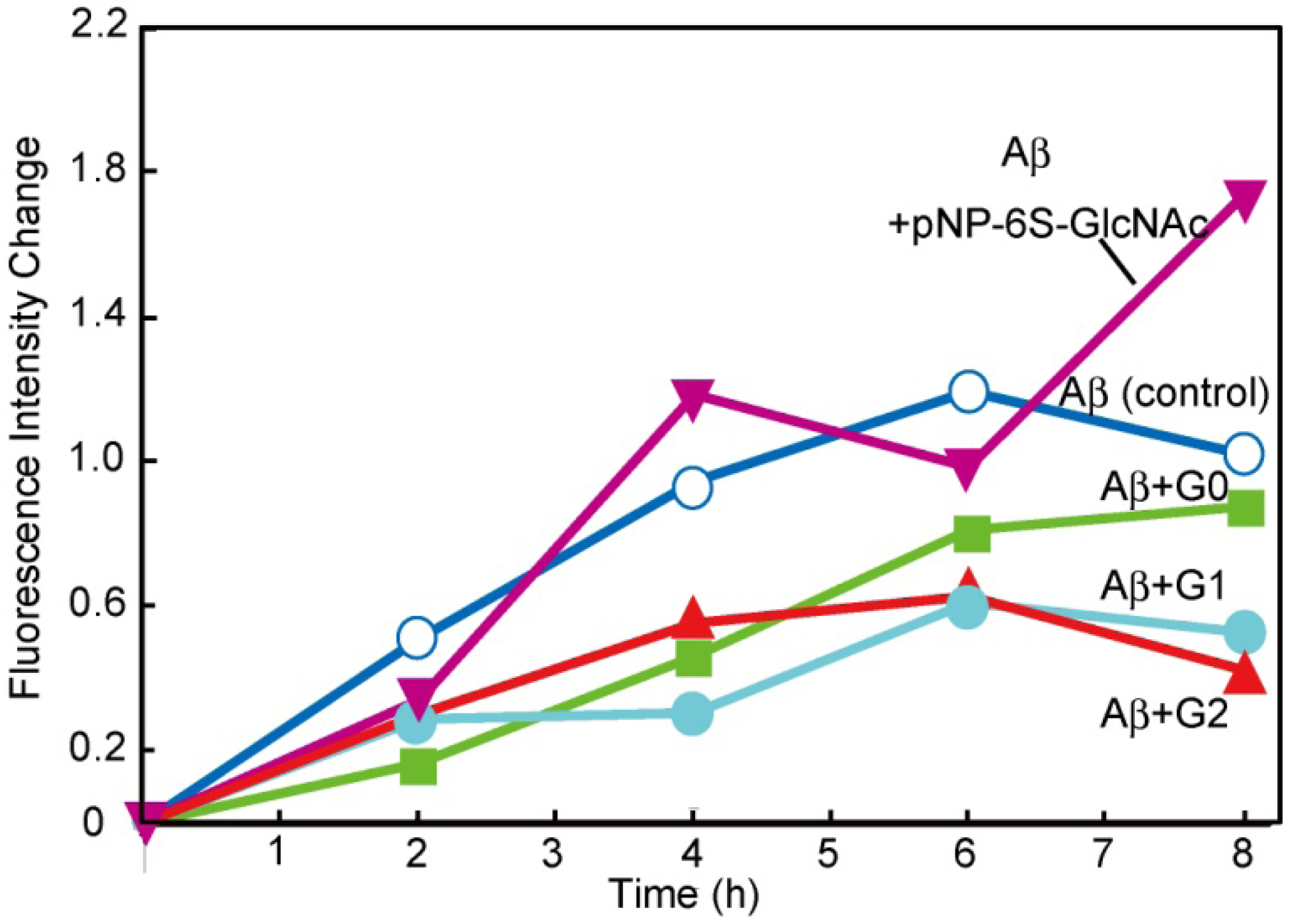
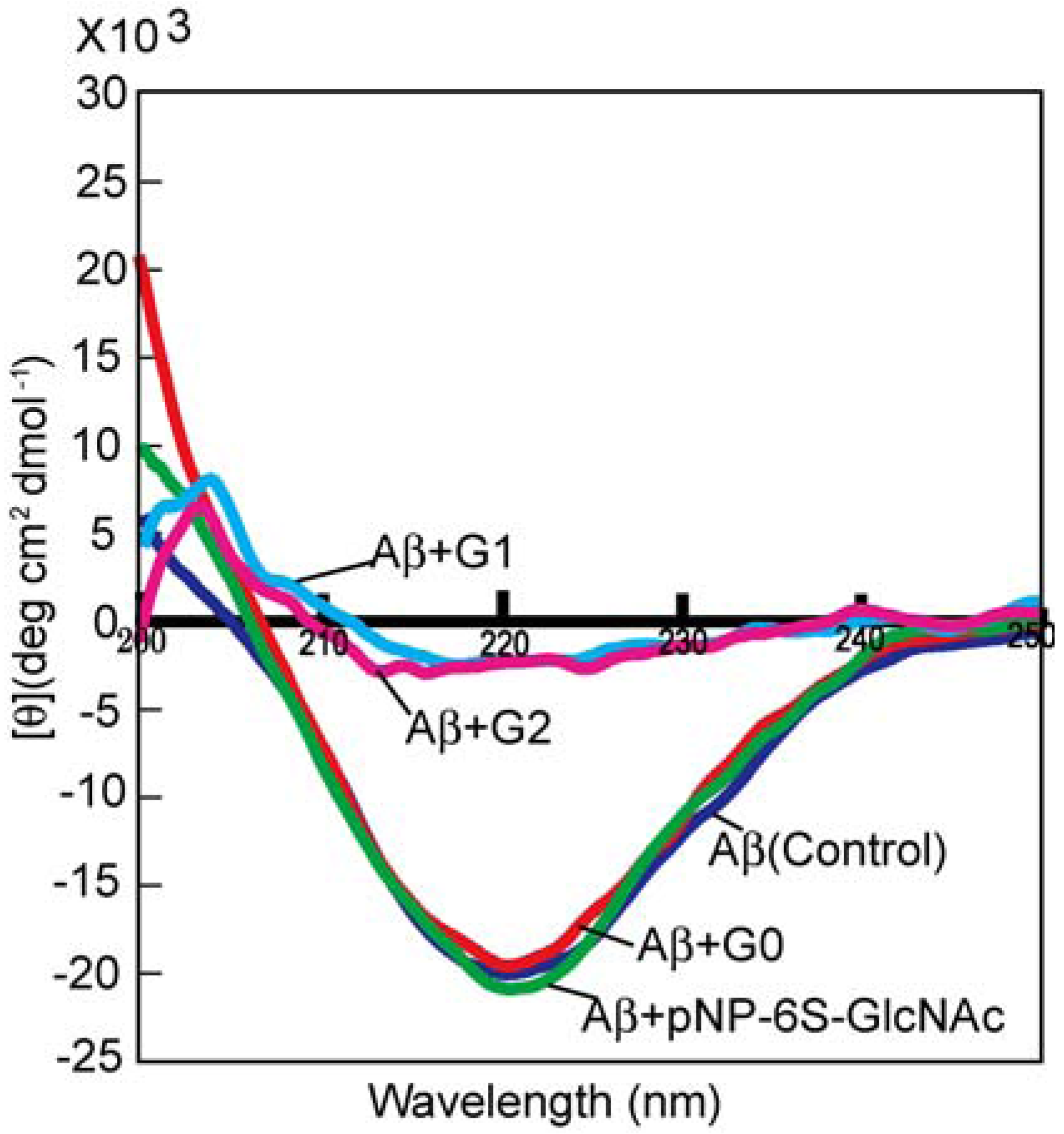
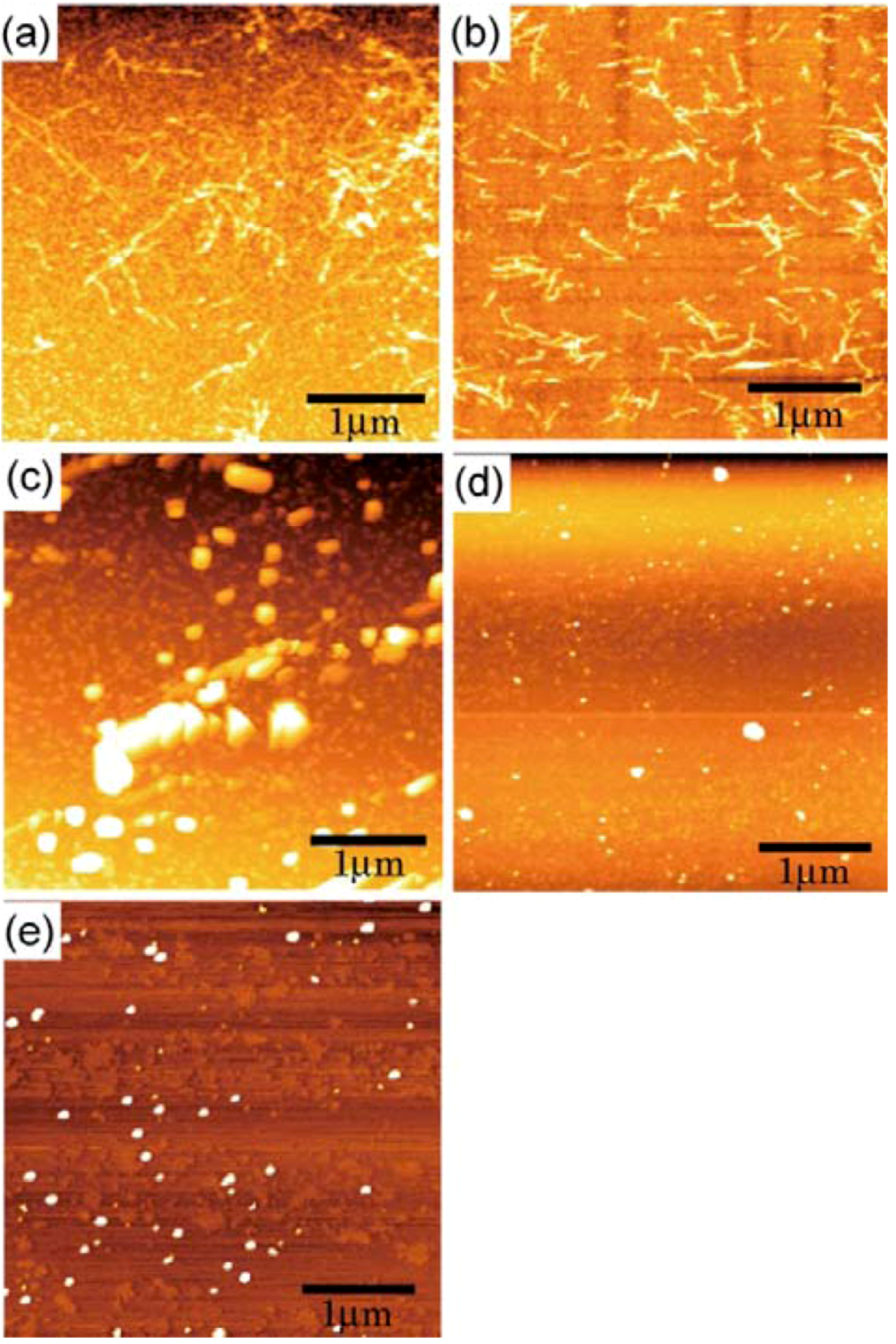
3.3. Syntheses
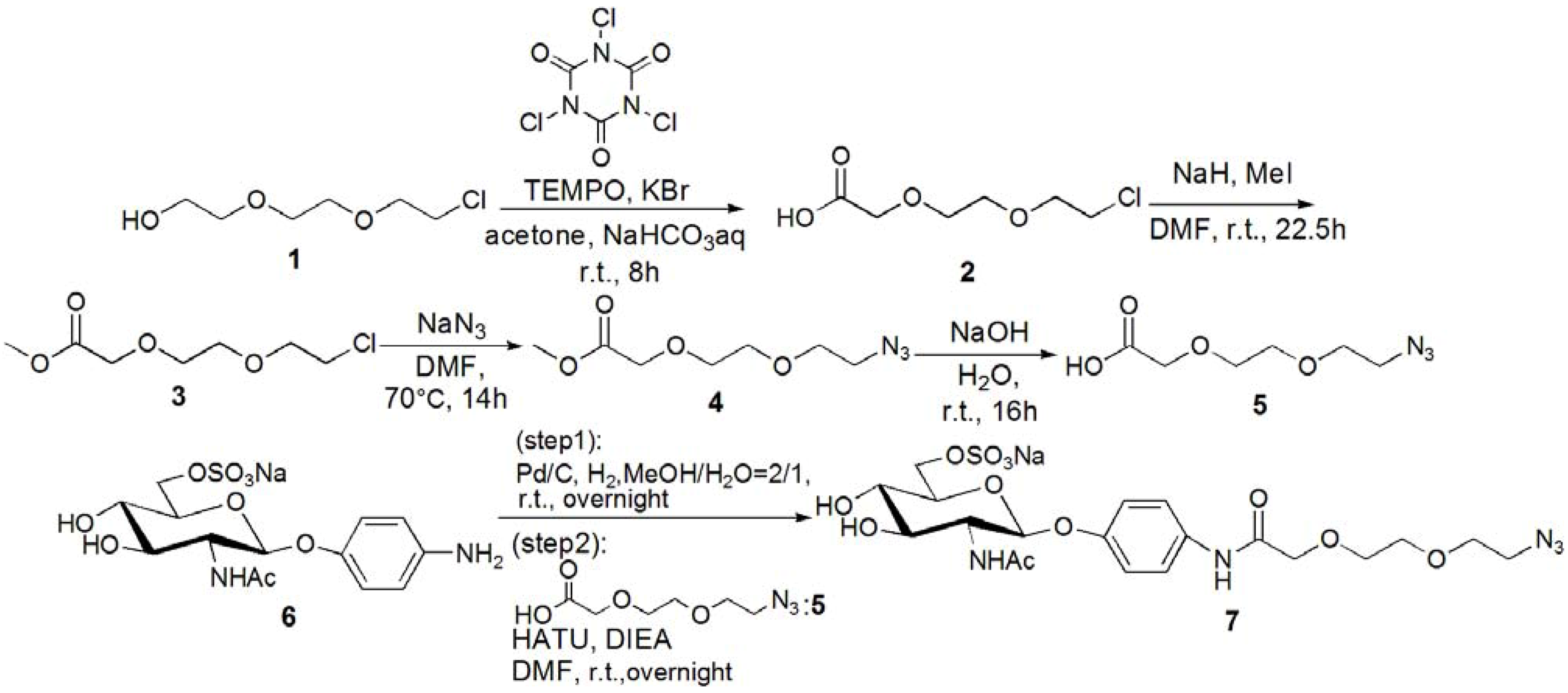
[2-(2-Chloroethoxy)ethoxy]acetic acid (2). [2-(2-Chloroethoxy)ethoxy]ethanol (1, 2.20 g, 13.0 mmol) was dissolved in acetone (30 mL), and 15% NaHCO3 aq. was added to the solution at 0 °C. KBr (0.312 g, 2.60 mmol) and TEMPO (0.040 g, 0.040 mmol) were added to the solution, and trichloroisocyanuric acid (6.10 g, 26.2 mmol) was added dropwise. The solution was allowed to warm up to room temperature, and stirred for 8 h. The reaction was confirmed by TLC (EtOAc–n-hexane = 3:1), and 2-propanol (10 mL) was added to quench the reaction. The reaction mixture was filtrated with Celite, and neutralized with sat. Na2CO3 aq. The solution was acidified with 1 N HCl, and extracted with CHCl3. The organic phase was dried over MgSO4, and MgSO4 was removed by filtration. The solution was evaporated to yield yellow oil; Yield 1.94 g, 88.6%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 3.58–3.60 (m, 2H, CH2), 3.64–3.68 (m, 2H, CH2), 3.69–3.70 (m, 2H, CH2), 3.71–3.76 (m, 2H, CH2), 4.18 (s, 2H, CH2). IR wavenumber [cm−1]: 3470 (OH) 1734 (C=O) 1112 (C-O). ESI-MS (negative): 181.2 [M−H]−.
Methyl 2-[2-(2-chloroethoxy)ethoxy]acetate (3). Compound 2 (0.312 g, 1.78 mmol) was dissolved in DMF (2 mL), and NaH (0.530 g, 2.22 mmol) was added to the solution at 0 °C. After stirring for 30 min, MeI (0.649 g, 3.33 mmol) was added to the solution. The reaction mixture was stirred for 22.5 h at room temperature. The reaction was confirmed by TLC (EtOAc–n-hexane = 1:3). The solution was evaporated, and the residue was dissolved in CHCl3. The solution was washed with 1 N HCl, sat. NaHCO3 aq. and brine. The solution was dried over MgSO4, and MgSO4 was removed by filtration. The residue was purified by column chromatography (EtOAc–n-hexane = 1:3) to yield colorless oil; Yield 0.242 g, 74.4%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 3.62 (t, 2H, J = 6 Hz, J = 6 Hz, CH2), 3.71–3.72 (m, 2H, CH2), 3.72–3.74 (m, 2H, CH2), 3.73 (s, 3H, CH3), 3.74–3.76 (m, 2H, CH2), 4.16 (s, 2H, CH2). ESI-MS (positive): 219.1 [M+Na]+.
Methyl 2-[2-(2-azidoethoxy)ethoxy]acetate (4). Compound 3 (0.242 g, 1.31 mmol) was dissolved in DMF (15 mL). NaN3 (0.511 g, 7.86 mmol) was added to the solution, and stirred for 14 h at 70 °C. The reaction was confirmed by TLC (EtOAc–n-hexane = 1:3). NaN3 was removed by filtration, and the solution was evaporated. The residue was dissolved in CHCl3, and the solution was washed with 1 N HCl, sat. NaHCO3 aq. and brine. The organic phase was dried over MgSO4, and MgSO4 was removed by filtration. The solution was evaporated to yield colorless oil; Yield 0.220 g, 87.8%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 3.38 (t, 2H, J = 5 Hz, J = 5 Hz, CH2), 3.65–3.67 (m, 2H, CH2), 3.67–3.67 (m, 2H, CH2), 3.70–3.72 (m, 2H, CH2), 3.73 (s, 3H, CH3), 4.16 (s, 2H, CH2). ESI-MS (positive) 226.1 [M+Na]+.
[2-(2-Azidoethoxy)ethoxy]acetic acid (5). Compound 4 (0.220 g, 1.15 mmol) was dissolved in 1 N NaOHaq (15 mL), and the solution was stirred for 16 h. The reaction was confirmed by TLC (EtOAc–n-hexane = 1:3). The solution was acidified with 1 N HCl, and extracted with CHCl3. The solution was dried over MgSO4, and MgSO4 was removed by filtration. The solution was evaporated to yield colorless oil; Yield 0.172g, 84.4%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 3.41 (t, 2H, J = 4.8 Hz, J = 5.1 Hz, CH2), 3.67–3.69 (m, 2H, CH2), 3.70–3.71 (m, 2H, CH2), 3.75–3.78 (m, 2H, CH2), 4.16 (s, 2H, CH2). ESI-MS (negative): 188.6 [M−H]−.
p-N-[2-(2-Azidoethoxy)ethoxy]amidophenyl-2-acetamido-2-deoxy-6-sulfo-β-D-
glucopyranoside (
7)
. Compound
6 was synthesized by following the previous method [
5,
14]. Next
6 (0.142 g, 0.320 mmol) was dissolved in MeOH–H
2O = 2:1 (10 mL). Pd/C (20 mg) was added to the solution, and the solution was stirred overnight under H
2. Pd/C was removed by filtration, and the solution was evaporated. The residue was dissolved in DMF (5 mL), and
5 (86.2 mg, 0.456 mmol) was added to the solution. HATU (185 mg, 0.487 mmol) and DIEA (83 μL, 0.487 mmol) were added to the solution, and the solution was stirred overnight. The solution was evaporated, and the residue was purified by column chromatography (H
2O–MeOH = 5:1) to yield a brown solid; Yield 0.160 g, 83.2%.
1H-NMR (500 MHz, r.t., D
2O): δ/ppm 2.09 (s, 3H, OCOC
H3), 3.36–3.39 (m, 2H, CH
2), 3.48 (t, 1H,
JH3-H4 = 9.0 Hz,
JH4-H5 = 9.5 Hz, H4), 3.54 (t, 1H,
JH2-H3 = 10.0 Hz,
JH3-H4 = 9.5 Hz, H3), 3.57–3.62 (m, 2H, CH
2), 3.66–3.68 (m, 2H, CH
2), 3.71–3.73 (m, 2H, CH
2), 3.74–3.75 (m, 1H, H5), 3.88 (t, 1H,
JH1-H2 = 9.5 Hz,
JH2-H3 = 9.5 Hz, H2), 4.11 (s, 2H, CH
2), 4.13 (dd, 1H,
JH5-H6proR = 4.0 Hz,
JH6proR-H6proS = 6.0 Hz, H6proR), 4.28 (d, 1H,
J = 10 Hz H6proS), 5.01 (d,
J = 8.5 Hz, H1), 6.98(d, 2H,
Jo-m=7.0 Hz, Ph-o), 7.27 (d, 2H,
Jo-m = 9.0 Hz, Ph-m). ESI-MS (negative): 421.1 [M−Na]
−.
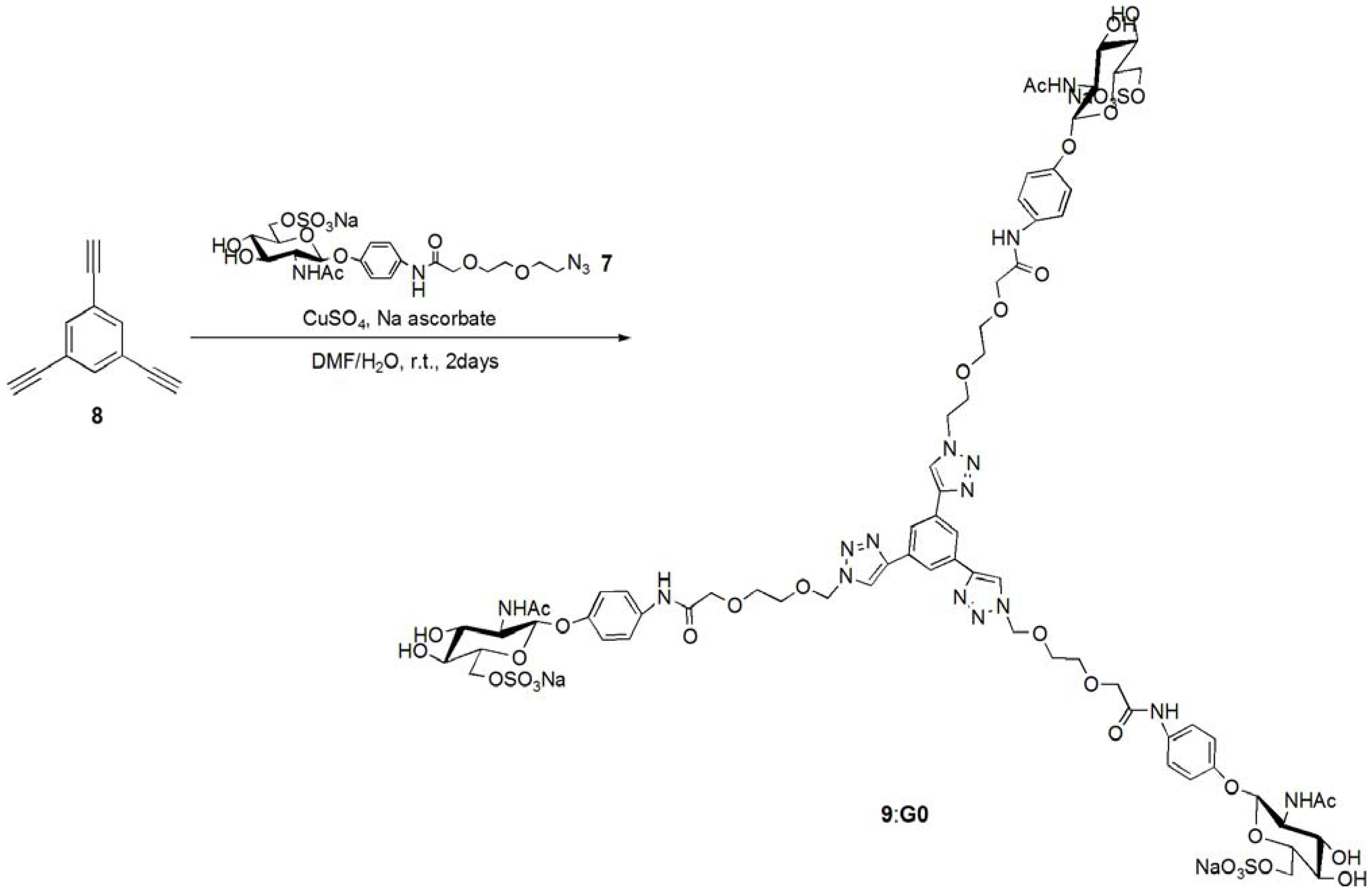
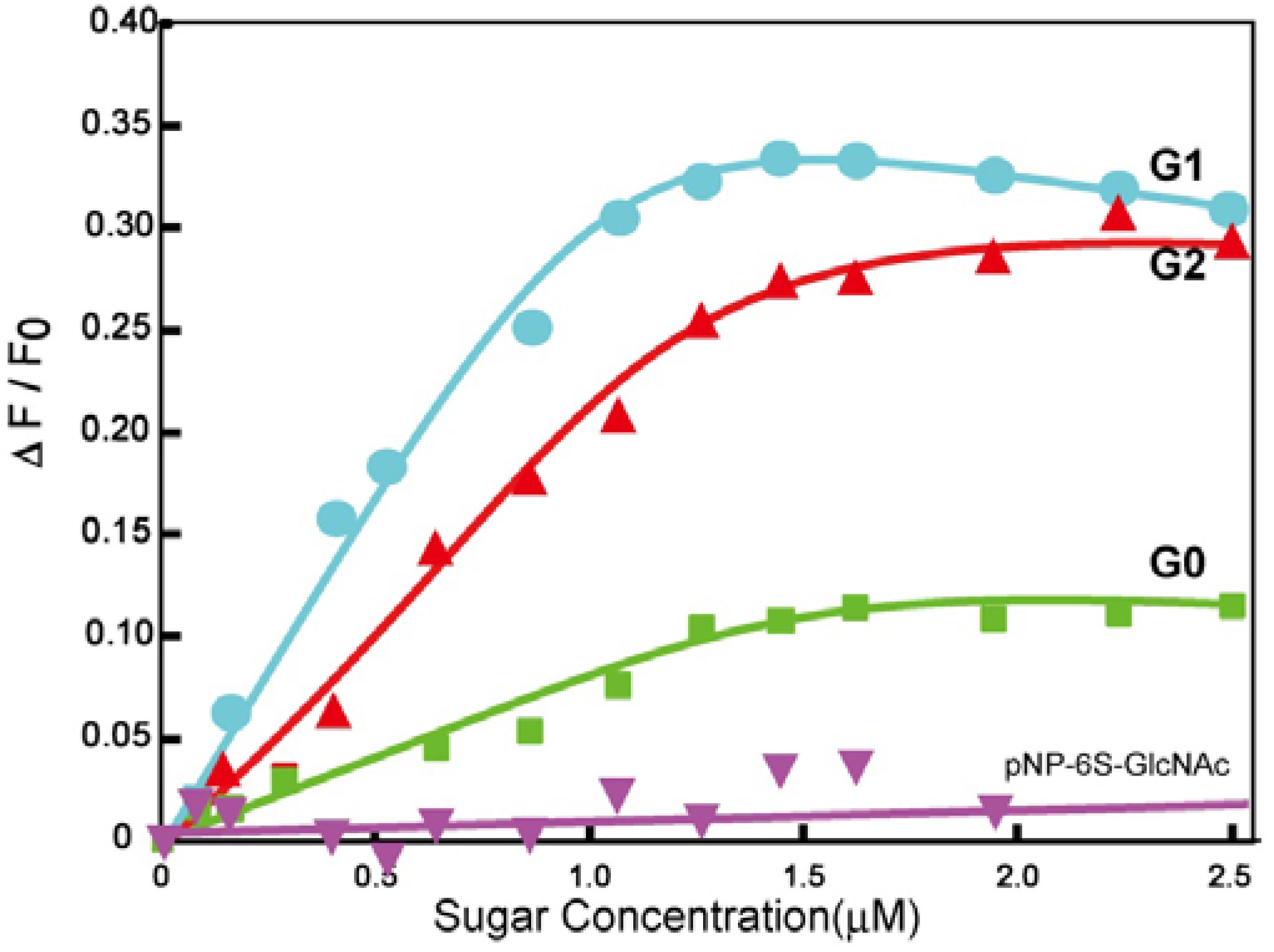
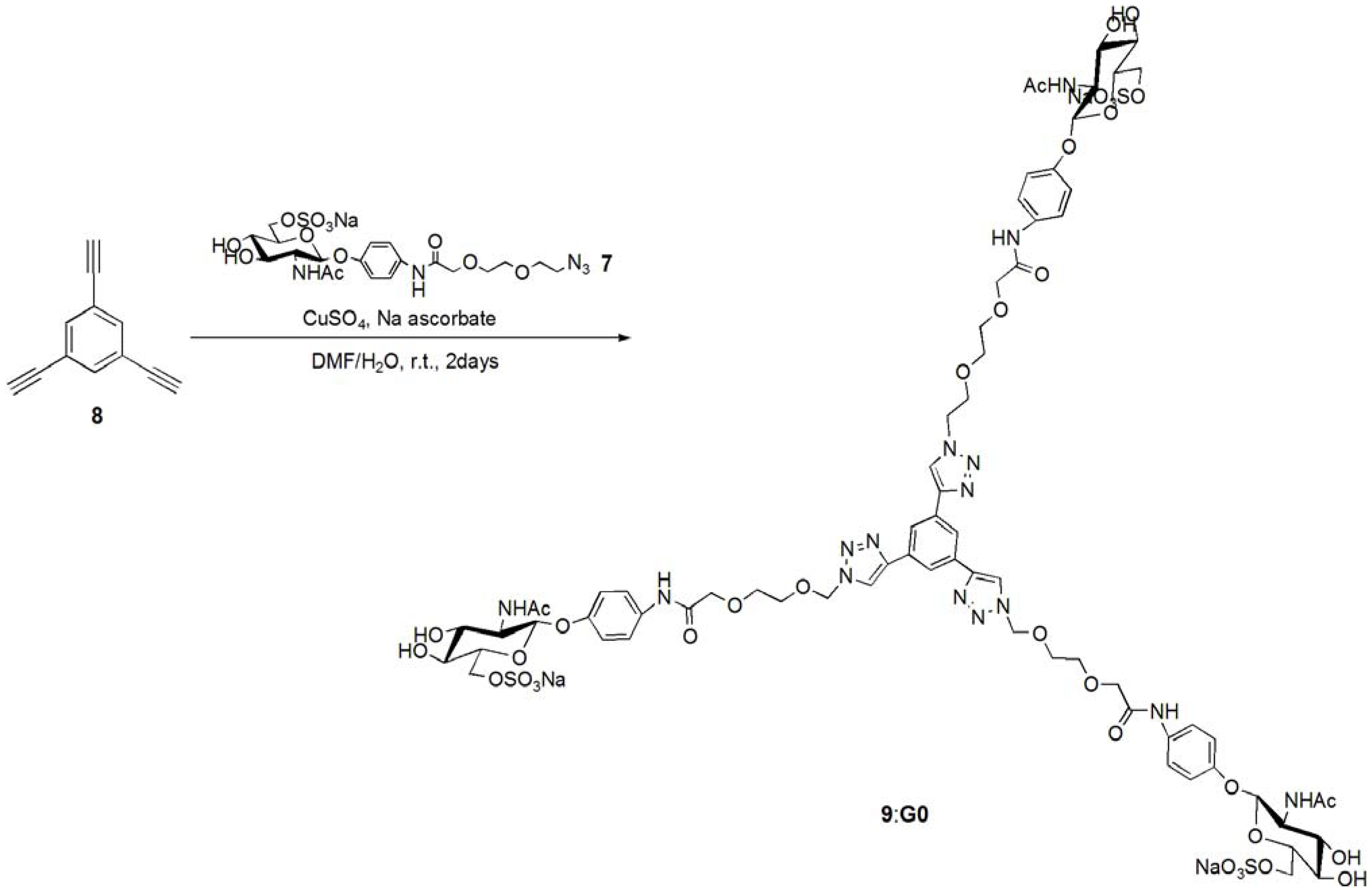
pNP 6-Sulfo-GlcNAc trimer (G0:9). Compound 7 (91.8 mg, 0.157 mmol) and 1,3,5-triethylbenzene (8) (5.12 mg, 0.0341 mmol) were dissolved in DMF (20 mL). To the solution, sodium ascorbate (12.2 mg, 0.6 eq.) and CuSO4 (4.91 mg, 0.3 eq.) was added. The solution was stirred for 2 days, and the reaction was confirmed by reversed phase TLC (H2O–MeOH = 5:1). Copper was removed by centrifugation, and the residue was purified by column chromatography (H2O–MeOH = 5:1) to yield colorless solid; Yield 0.0320g, 49.2%. 1H-NMR (300 MHz, r.t., D2O):δ/ppm 1.86(s, 9H, CH3), 3.41–3.47 (overlap, 6H, CH2), 3.41–3.47 (overlap, 3H, H4), 3.52–3.57 (overlap, 3H, H3), 3.52–3.57 (overlap, 12H, CH2 × 2), 3.66 (broad, 3H, 5H), 3.66 (broad, 6H, CH2), 3.75–3.77 (overlap, 3H, H2), 3.77 (s, 6H, CH2), 3.98 (s, 6H, NH), 4.05–4.07 (m, 3H, H6proR), 4.16 (d, 3H, J = 11.5 Hz H6proS), 4.61–4.62 (overlap, 3H, H1), 6.47 (d, 6H, Jo-m = 9.0 Hz, Ph-o), 6.77 (d, 6H, Jo-m = 8.5 Hz, Ph-m), 7.53 (s, 3H, Ar), 8.18 (s, 3H, C=CH). 13C-NMR (75 MHz, r.t., D2O): δ/ppm 21.6, 49.5, 53.7, 54.8, 66.2, 67.8, 68.7, 68.8, 69.2, 72.9, 73.3, 99.3, 116.1, 121.2, 121.7, 122.6, 130.0, 130.6, 153.2, 169.2, 174.0. ESI-MS: m/z 976.1 [M+2Na]2+, (LCMS, negative) m/z 822.9 [M−3Na]3−.
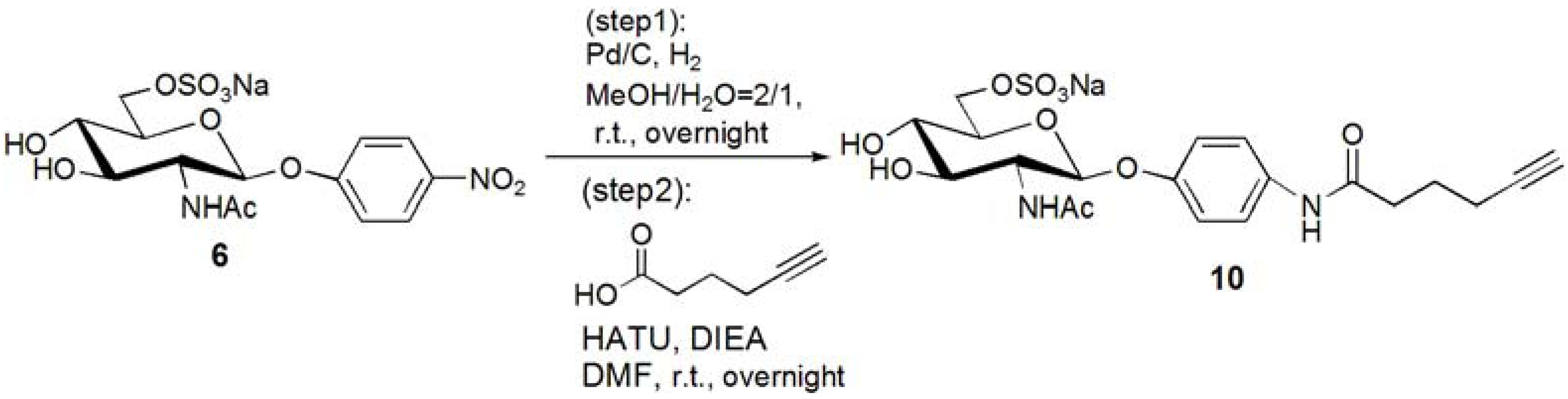
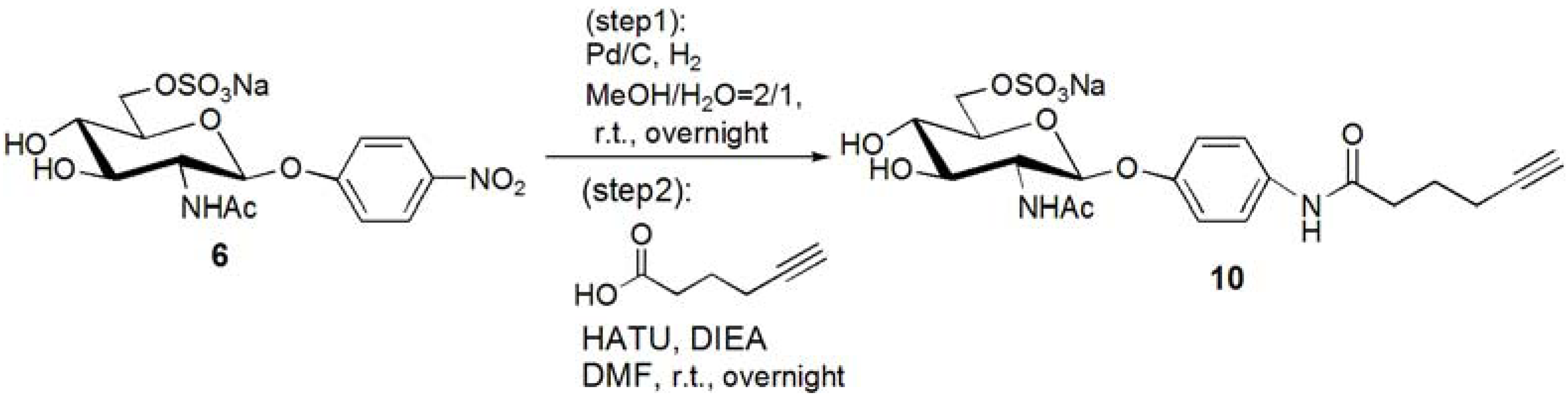
p-N-(5-Hexynoic)amidophenyl-2-acetoamido-2-deoxy-6-sulfo-β-D-glucopyranoside (10). Compound 6 was dissolved in MeOH–H2O = 2:1. Pd/C was added to the solution, and the solution was stirred overnight under H2. Pd/C was removed by filtration, and the solution was evaporated. The residue, p-aminophenyl-2-acetoamido-2-deoxy-6-sulfo-β-D-glucopyranoside (0.353 g, 0.853 mmol) was dissolved in DMF (20 mL), and 5-hexynoic acid (0.143 g, 1.28 mmol) was added to the solution at 0 °C. HATU (0.486 g, 1.28 mmol) and DIEA (217 μL, 1.28 mmol) was added to the solution, and the solution was stirred overnight. DMF was evaporated, and the residue was purified by reversed phase column chromatography (H2O–MeOH = from 5:1 to 2:1) to yield white solid; Yield 0.366g, 81.6%. 1H-NMR (500 MHz, r.t., D2O): δ/ppm 1.74 (t, J = 7.0Hz, 2H, CH2), 1.90(s, 3H, OCOCH3), 2.15–2.18 (m, 2H, CH2), 2.25 (t, J = 2.5Hz, 1H, C≡CH), 2.36–2.39 (m, 2H, CH2), 3.47 (t, 1H, JH3-H4 = 9.5 Hz, JH4-H5 = 9.5 Hz, H4), 3.52–3.55 (m, 1H, H3), 3.70–3.73 (m, 1H, H5), 3.87 (t, 1H, JH1-H2 = 8.5Hz, JH2-H3 = 10 Hz, H2), 4.11 (dd, 1H, JH5-H6proR = 5.5Hz, JH6proR-H6proS = 5.0 Hz, H6proR), 4.25 (d, 1H, J = 11.5 Hz, H6proS), 4.99 (d, J = 8.5 Hz, H1), 6.93 (d, 2H, Jo-m = 9.5 Hz, Ph-o), 7.27 (d, 2H, Jo-m = 9.5 Hz, Ph-m).
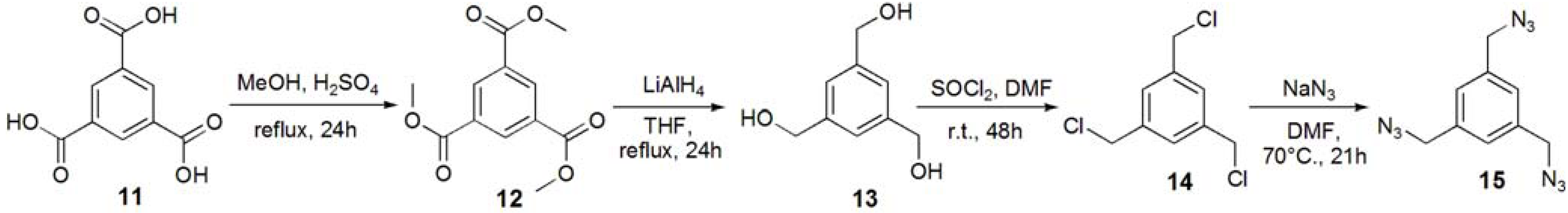
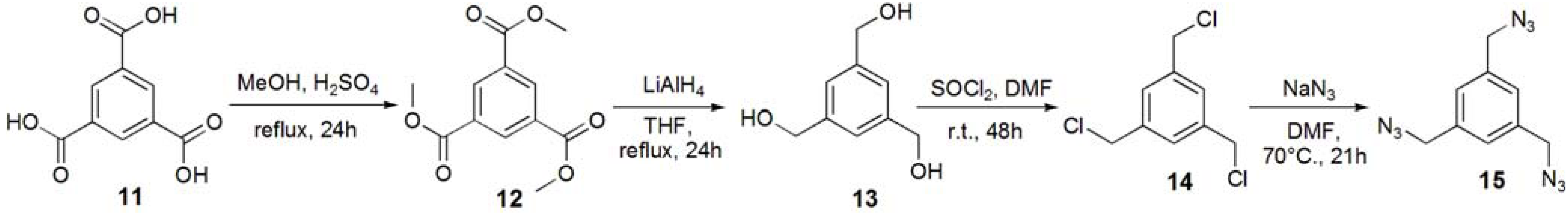
Trimethyl 1,3,5-benzenetricarboxylate (12). Trimesic acid (11, 8.00 g, 38.1 mmol) was dissolved in MeOH (140 mL), and conc. H2SO4 (2 mL) was added to the reaction mixture. The solution was refluxed for 24 h, and the solution was evaporated. The residue was dissolved in CHCl3, and washed with sat. Na2CO3aq. The solution was dried with MgSO4, and removed by filtration. The solution was evaporated to obtain white solid; Yield 9.09 g, 94.7%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 3.96 (s, 9H, OMe), 8.83 (s, 3H, Ar).
1,3,5-Trihydroxymethylbenzene (13). Compound 12 (1.00 g, 3.97 mmol) was dissolved in THF (70 mL). LiAlH4 (35.7 mmol) was added to the solution, and the solution was refluxed for 24 h. The reaction was confirmed by TLC (EtOAc–n-hexane=1:1), and water was added to the reaction in order to quench the reaction. The solution was dried over MgSO4 and evaporated to yield white solid; Yield 0.571 g, 85.6%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.60 (s, 6H, CH2), 7.25 (s, 3H, Ar).
1,3,5-Tribenzylchloride (14). Compound 13 (0.200 g, 1.19 mmol) was dissolved with SOCl2 (5 mL) at 0 °C and to the solution the catalytic amount of DMF was added. The solution was stirred for 48 h at room temperature. The reaction was confirmed by TLC (EtOAc–n-hexane = 1:1). The reaction mixture was poured into ice-water, and extracted with CHCl3. The organic phase was washed with water, and dried over MgSO4. The solution was evaporated to yield white solid; Yield 0.241 g, 93.9%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.57 (s, 6H, CH2), 7.36 (s, 3H, Ar).
1,3,5-Tribenzylazide (15). Compound 14 (0.214 g, 0.964 mmol) was dissolved in DMF (10 mL). NaN3 (1.13 g, 17.4 mmol, 18 eq.) was added to the solution, and the solution was stirred at 70 °C for 21 h. The solution was evaporated, and the residue was dissolved with chloroform. The solution was washed with 1 N HCl aq., sat. NaHCO3, and brine. The solution was dried over MgSO4. The solution was evaporated to yield colorless oil; Yield 0.218 g, 93.0%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.38 (s, 6H, CH2), 7.23 (s, 3H, Ar).
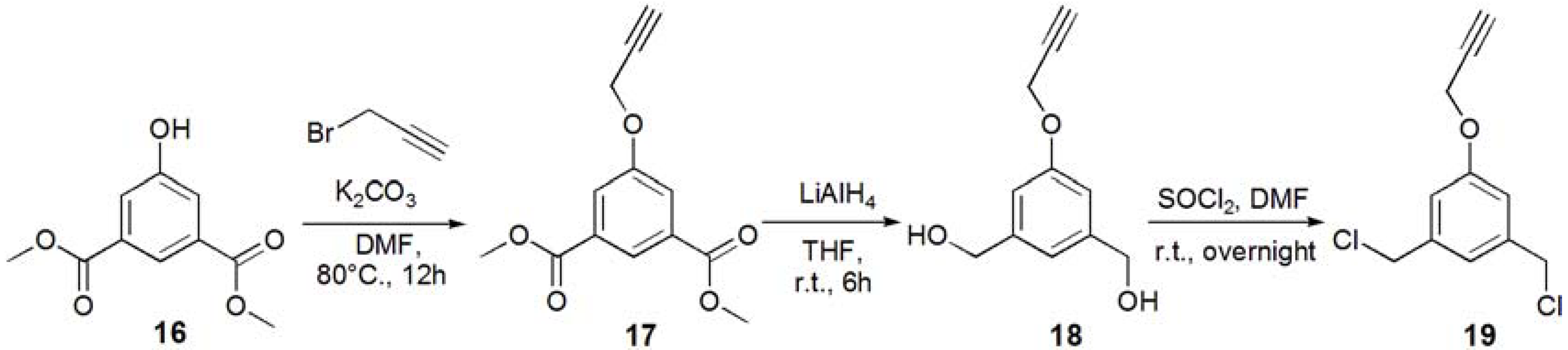
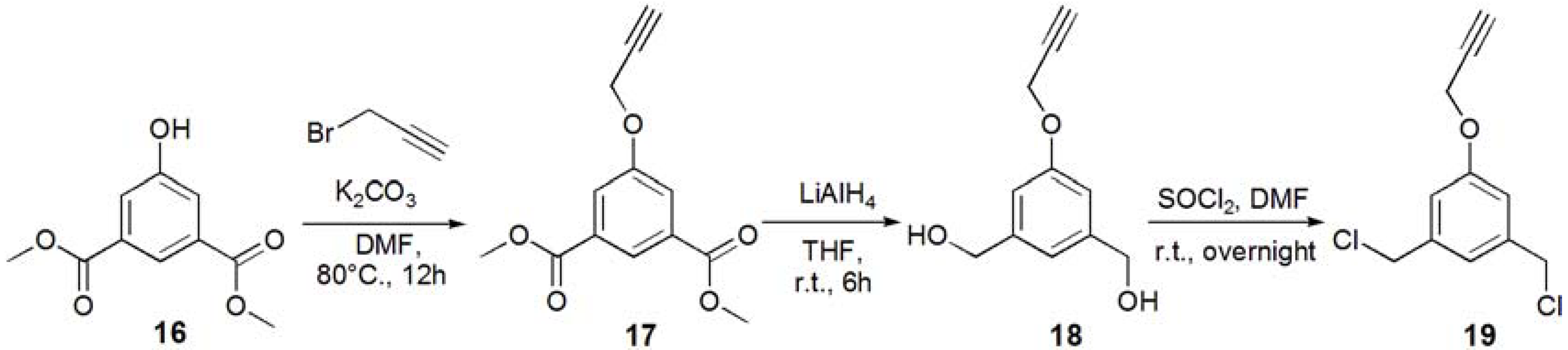
1-(3-Acetyl-5-prop-2-ynyloxy-phenyl)ethanone (17). Dimethyl 5-hydroxyisophtalate (16, 5.01 g, 23.8 mmol) and K2CO3 (93 g, 35.7 mmol, 1.5 eq.) were dissolved in DMF (100 mL), and the solution was degassed with bubbling of N2. The solution was heated up to 80 °C, and propargyl bromide (2.80 mL, 35.7 mmol, 1.5 eq.) was added under N2. The solution was stirred for overnight. The solution was allowed to be cooled to room temperature. K2CO3 was removed by filtration, and the solution was evaporated. The residue was recrystallized in EtOH to yield white solid; Yield: 4.52 g, 76.5%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 2.53 (t, J = 2.1 Hz, J = 2.4 Hz, 1H, C≡CH), 3.92 (s, 6H, CH3), 4.76 (d, J = 2.4 Hz, 2H, CH2), 7.80 (d, J = 1.5 Hz, 2H, ArH), 8.30 (t, J = 1.5 Hz, J = 1.2 Hz, 1H, ArH).
(3-Hydroxymethyl-5-prop-2-ynyloxy-phenyl)methanol (18). Compound 17 (0.500 g, 2.01 mmol) was dissolved in THF (35.0 mL), and degassed. LiAlH4 (12.1 mmol) was added to the solution. The reaction mixture was stirred for 6 h, and the reaction was confirmed by TLC (EtOAc–n-hexane = 1:1). Then, water was added to the solution in order to quench the reaction. The solution was dried over MgSO4, and MgSO4 was removed by filtration. The solution was evaporated to yield white solid; Yield 0.425 g, 99%. 1H-NMR (300 MHz, r.t., CD3OD): δ/ppm 2.91 (t, J = 2.7 Hz, J = 2.4 Hz, 1H, C≡CH), 4.57 (s, 4H, CH2OH), 4.72 (d, J = 2.4 Hz, 2H, CH2), 6.89 (s, 2H, ArH), 8.30 (d, J = 0.6 Hz, 1H, ArH).
1,3-Bischloromethyl-5-prop-2-ynyloxy-benzene (19). Compound 18 (0.425 g, 2.21 mmol) was dissolved in CH2Cl2, and SOCl2 was added to the solution with catalytic amount of DMF at 0 °C. The reaction mixture was stirred for overnight, and the reaction was confirmed by TLC (EtOAc–n-hexane = 1:1). The reaction mixture was poured into ice-water, and extracted with CHCl3. The organic phase was washed with water, and dried over MgSO4. MgSO4 was removed by filtration, and the solution was evaporated to yield white solid; Yield. 0.462 g, 91.2%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 2.52 (t, J = 2.4 Hz, J = 2.4 Hz, 1H, C≡CH), 4.53 (s, 4H, CH2), 4.70 (d, J = 2.4 Hz, 2H, CH2), 6.95 (d, J = 1.5 Hz, 2H, ArH), 7.02 (t, J = 0.6 Hz, J = 0.6 Hz, 1H, ArH).
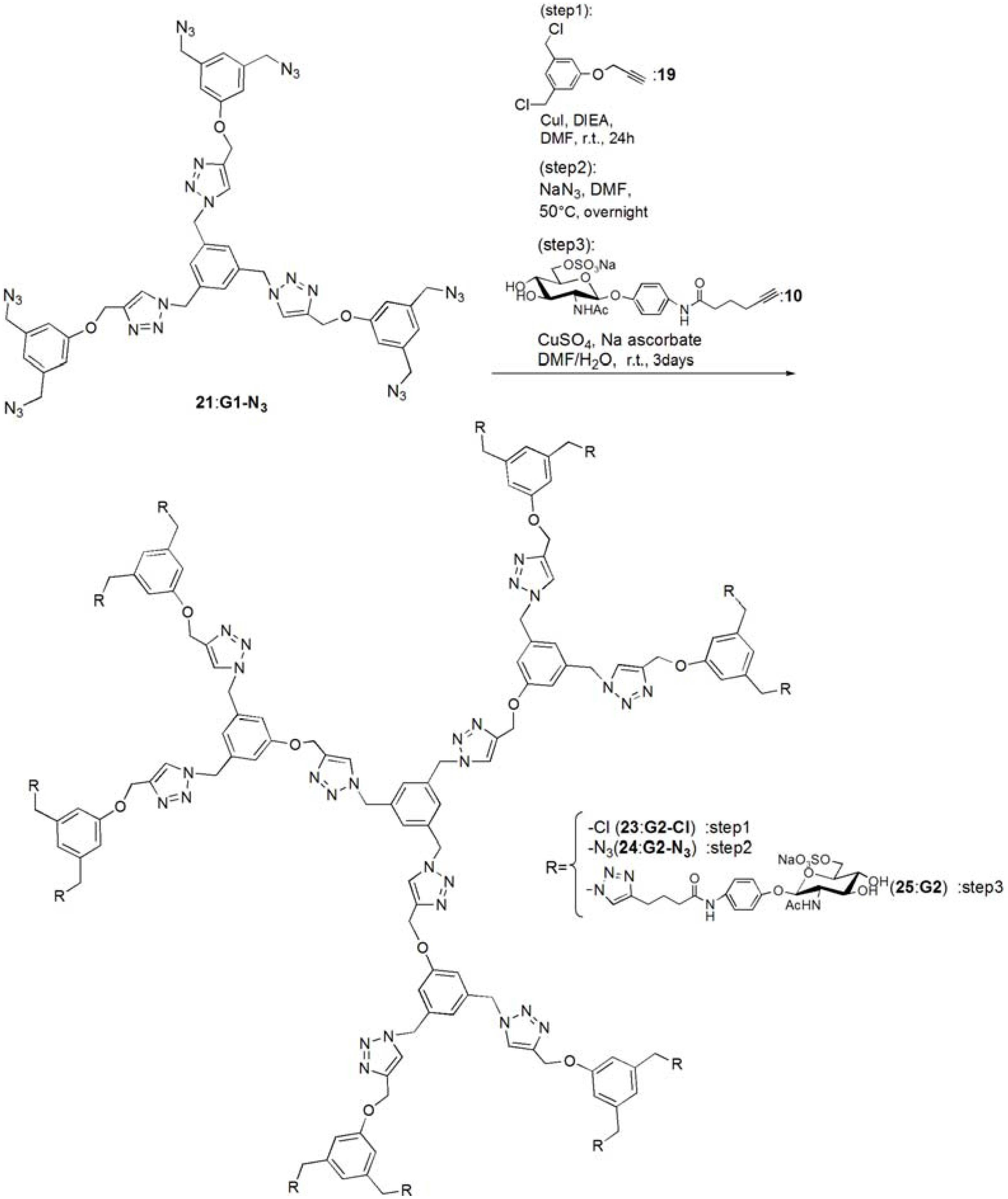
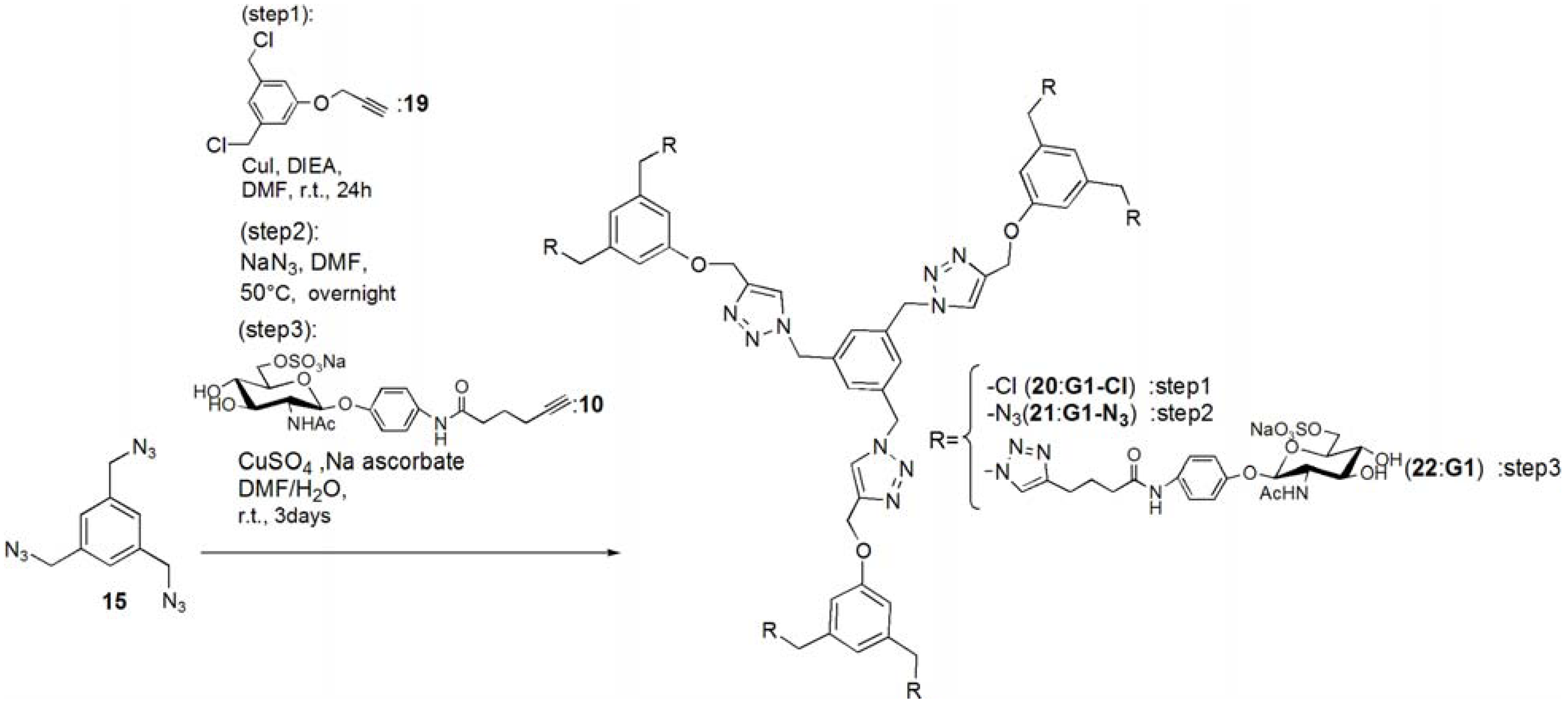
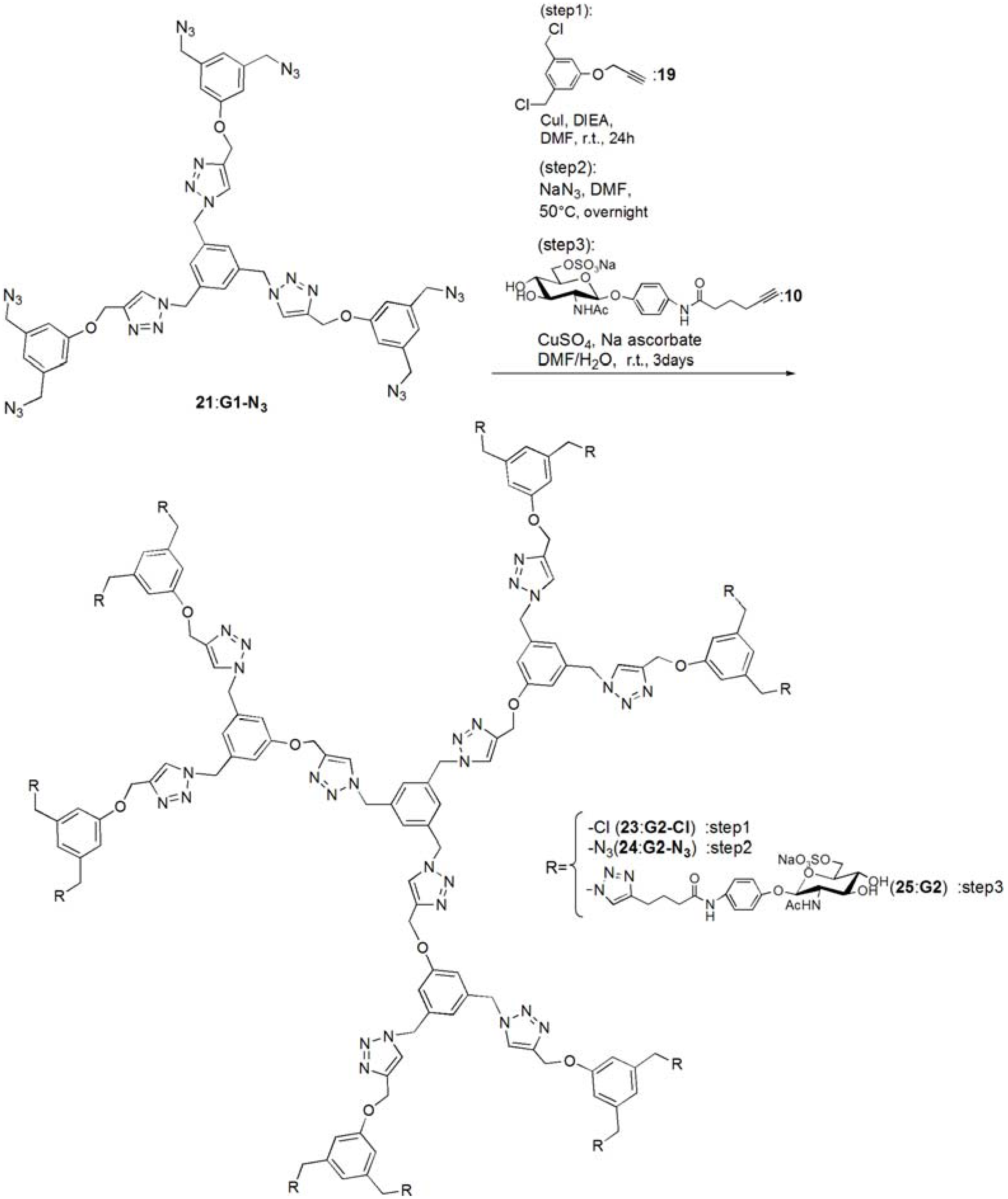
G1-Cl (20). Compounds 15 (95.0 mg, 0.391 mmol) and 19 (0.420 g, 1.83 mmol) was dissolved in DMF (3 mL). To the reaction mixture, CuI (111 mg, 0.586 mmol) was added and suspended. DIEA (100 mL, 0.588 μmol) was added to the solution. The solution was stirred for 24 h. The reaction mixture was evaporated, and the residue was purified by column chromatography (CHCl3–MeOH = 60:1) to yield white solid; Yield 0.268 g, 73.7%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.50 (s, 12H, CH2), 5.20 (s, 6H, CH2), 5.46 (s, 6H, CH2), 6.94 (s, 6H, ArH), 6.99 (s, 3H, ArH), 7.10 (s, 3H, ArH), 7.54 (s, 3H, C=CH).
G1-N3 (21). Compound 20 (0.268 g, 0.288 mmol) was dissolved in DMF (10 mL). NaN3 (3.63 mmol) was added to the solution, and the solution was stirred at 50 °C for overnight. The solution was evaporated, and the residue was dissolved in CHCl3. The organic phase was washed with water for three times. The solution was dried with MgSO4, and MgSO4 was removed by filtration. The residue was purified by column chromatography (CHCl3–MeOH = 100:1) to yield white solid; Yield 0.260 g, 93.1%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.29 (s, 12H, CH2), 5.21 (s, 6H, CH2), 5.45 (s, 6H, CH2), 6.85 (s, 3H, ArH), 6.88 (s, 6H, ArH), 7.13 (s, 3H, ArH), 7.55 (s, 3H, C=CH). 1H-NMR (300 MHz, r.t., DMF-d7): δ/ppm 4.50 (s, 12H, CH2) 5.23 (s, 6H, CH2), 5.71 (s, 6H, CH2), 7.05 (s, 3H, ArH), 7.10 (s, 6H, ArH), 7.45 (s, 3H, ArH), 8.39 (s, 3H, C=CH). 13C-NMR (75 MHz, r.t., CDCl3): δ/ppm 53.4, 54.4, 62.1, 114.3, 120.6, 122.9, 127.6, 136.8, 137.7, 144.4, 158.8. ESI-MS: m/z 969.9.0 [M+H]+, 992.0 [M+Na]+.
pNP 6-Sulfo-GlcNAc hexamer(G1:22). Compounds 21 (6.15 mg, 6.34 μmol) and 10 were dissolved in DMF (400 μL), and an aqueous solution of sodium ascorbate (6.02 mg, 0.0304 mmol) and CuSO4 (2.40 mg, 0.0150 mmol) was added to the solution. The reaction mixture was stirred for 3 days. The solvent was evaporated and the residue was purified by reversed phase chromatography (H2O–MeOH = 2:1) to yield a white solid; Yield 7.70 mg, 28.2%. 1H-NMR (300 MHz, r.t., DMF-d7): δ/ppm 1.26–1.28 (m, 12H, CH2), 1.88 (s, 18H, OCOCH3), 1.96 (t, J = 7.5Hz, J = 7.5 Hz, 12H, CH2), 2.41 (t, J = 7.5 Hz, J = 7.8 Hz, 12H, CH2), 3.92 (dd, JH1-H2 = 9.5Hz, JH2-H3 = 9.0 Hz, 6H, H2), 4.03–4.06 (m, 6H, H5), 4.07–4.11 (m, 6H, H3), 4.19–4.26 (m, 6H, H4), 5.02 (d, JH1-H2 = 9.0 Hz, 6H, H1), 5.16 (s, 6H, CH2), 5.29 (broad, 6H, H6), 5.36 (broad, 6H, H6), 5.57 (s, 12H, CH2), 5.66 (s, 6H, CH2), 6.91 (s, 3H, ArH), 6.94 (d, 12H, Jo‑m = 9.0Hz, Ph-o), 7.03 (s, 6H, ArH), 7.44 (s, 3H, ArH), 7.58 (d, 12H, Jo-m = 9.0Hz, Ph-m), 7.94 (s, 6H, C=CH), 8.40 (s, 3H, C=CH). 13C-NMR (75 MHz, r.t., DMF-d7): δ/ppm 23.5, 28.2, 30.6, 31.0, 47.9, 58.4, 59.8, 61.8, 67.1, 71.7, 76.8, 80.4, 81.2, 106.0, 115.2, 119.8, 122.5, 126.0, 130.6, 133.6, 140.1, 143.1, 144.2, 159.4, 164.6, 175.5, 176.4. ESI-MS: m/z 822.9 [M+H+4Na]5+.
G2-Cl (23). G1-N3 (21, 0.0597 g, 0.0616 mmol) and 19 (0.114 g, 0.498 mmol) was dissolved in DMF (750 μL). CuI (21.0 mg, 0.110 mmol) was added to the solution and suspended. DIEA was added to the solution, and the solution was stirred for 24 h at room temperature. The reaction was confirmed by TLC (CHCl3–MeOH = 10:1), and the residue was purified by column chromatography (CHCl3–MeOH = 40:1) to yield a white solid; Yield 43.5 mg, 30.1%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.45 (s, 24H, CH2), 4.99 (s, 6H, CH2), 5.07 (s, 12H, CH2), 5.12 (s, 6H, CH2), 5.37 (s, 12H, CH2), 6.73 (s, 3H, ArH), 6.75 (s, 6H, ArH), 6.88 (s, 12H, ArH), 6.94 (s, 6H, ArH), 7.05 (s, 3H, ArH), 7.55 (s, 3H, C=CH), 7.62 (s, 6H, C=CH).
G2-N3 (24). 23 (0.0435 g, 0.0186 mmol) was dissolved in DMF (10 mL). NaN3 (0.0201 g, 0.334 mmol) was added to the solution, and stirred at 50 °C for overnight. The solution was evaporated and the residue was dissolved in CHCl3. The organic phase was washed with water three times. The organic phase was dried over MgSO4, and MgSO4 was filtrated. The residue was purified by column chromatography (CHCl3–MeOH = 40:1) to yield a white solid; Yield 0.253 g, 56.3%. 1H-NMR (300 MHz, r.t., CDCl3): δ/ppm 4.27 (s, 24H, CH2), 4.5.04 (s, 6H, CH2), 5.16 (s, 12H, CH2), 5.17 (s, 6H, CH2), 5.40 (s, 12H, CH2), 6.76 (s, 3H, ArH), 6.78 (s, 6H, ArH), 6.83 (s, 6H, ArH), 6.86 (s, 12H, ArH), 7.08 (s, 3H, ArH), 7.54 (s, 3H, C=CH), 7.61 (s, 6H, C=CH). 1H-NMR (300 MHz, r.t., DMF-d7): δ/ppm 4.48 (s, 24H, CH2), 5.15 (s, 6H, CH2), 5.24 (s, 12H, CH2), 5.66 (s, 6H, CH2), 5.69 (s, 12H, CH2), 7.03 (s, 6H, ArH), 7.06 (s, 9H, ArH), 7.09 (s, 12H, ArH), 7.44 (s, 3H, ArH), 8.35 (s, 3H, C=CH), 8.41 (s, 6H, C=CH). 13C-NMR (75 MHz, r.t., CDCl3): δ/ppm 53.3, 53.6, 54.3, 61.7, 62.0, 114.2, 114.6, 120.0, 120.6, 123.2, 123.5, 127.7, 136.7, 137.2, 137.7, 143.6, 144.1, 158.7, 159.0. MALDI-TOF-MS: m/z 2447.2 [M+Na]+.
pNP 6-Sulfo-GlcNAc dodecamer (G2:25). Compounds 24 (8.00 mg, 3.30 μmol) and 10 were dissolved in DMF (350 μL). The aqueous solution (83 μL) of sodium ascorbate (6.28 mg, 31.7 mmol) and CuSO4 (2.52 mg, 0.0158 mmol) was added to the solution. The reaction mixture was stirred for 3 days. The reaction was confirmed by TLC (H2O–MeOH = 3:1). The solution was evaporated and the residue was purified by reversed phase column chromatography (H2O–MeOH = 3:1) to yield a white solid; Yield 6.30 mg, 21.1%. 1H-NMR (500 MHz, r.t., DMF-d7): δ/ppm 1.26–1.28 (m, 24H, CH2), 1.88 (s, 36H, OCOCH3), 1.94–199 (m, 24H, CH2), 2.40 (t, J = 7.5 Hz, J = 7.0 Hz, 24H, CH2), 3.78 (broad, 12H, H5), 3.87–3.90 (m, 12H, H2), 4.06–4.10 (m, 12H, H3), 4.23–4.26 m, 12H, H4), 4.25 (m, 6H, H4), 5.01 (d, JH1-H2 = 8.5 Hz, 12H, H1), 5.16–5.18 (m, 12H, H6), 5.22–5.24 (broad, 6H, CH2), 5.31–5.34 (m, 6H, H6), 5.36 (broad, 6H, H6′), 5.53 (s, 24H, CH2), 5.56 (s, 12H, CH2), 5.58 (s, 12H, CH2), 5.61–5.66 (broad, 6H, CH2), 6.88 (s, 6H, ArH), 6.96–6.98 (overlap, 24H, Ph-o), 6.96–6.98 (overlap, 9H, ArH), 7.03(s, 12H, ArH), 7.44 (broad, 3H, ArH), 7.58 (d, 2H, Jo-m = 8.1 Hz, Ph-m), 7.94 (s, 12H, C=CH), 8.35 (s, 3H, C=CH), 8.35 (s, 6H, C=CH). 13C-NMR (75 MHz, r.t., DMF-d7): δ/ppm 22.6, 28.2, 30.6, 31.1, 58.4, 61.3, 61.8, 67.1, 71.7, 76.8, 80.3, 81.2, 82.3, 91.5, 106.0, 115.4, 120.0, 122.5, 126.0, 127.8, 130.0, 140.1, 143.2, 143.4, 144.2, 148.7, 155.4, 156.6, 156.6, 164.6, 164.7, 176.4, 181.0. ESI-MS m/z 1238.0 [M+H+6Na]7+.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}