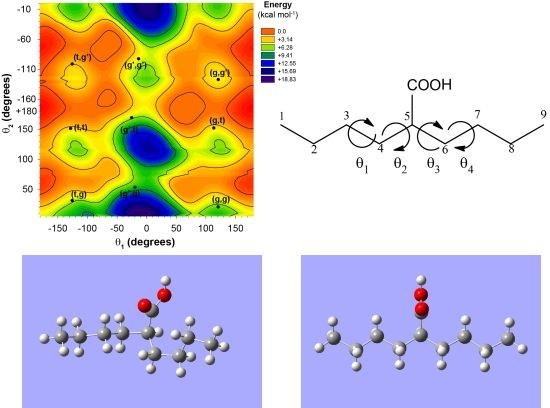
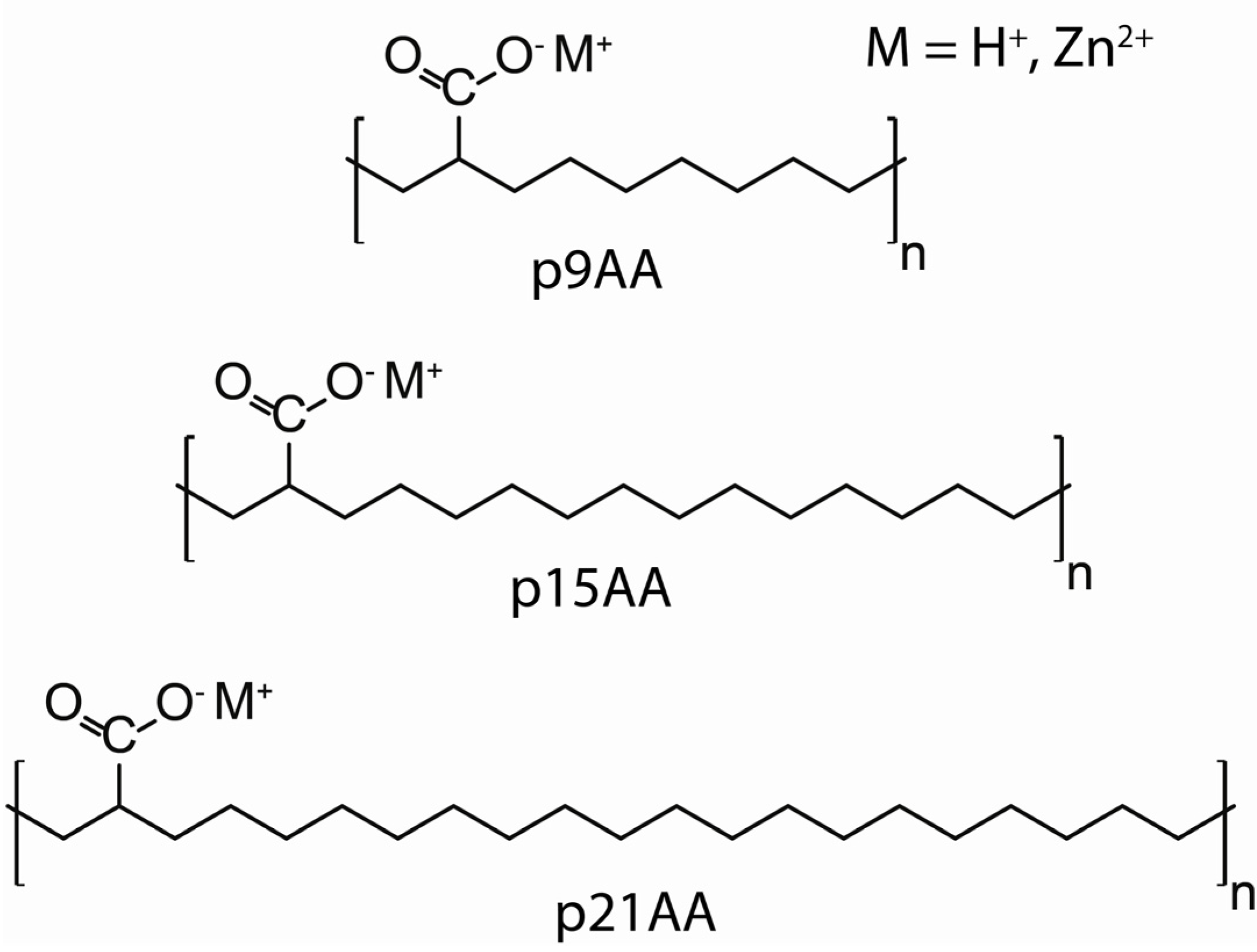
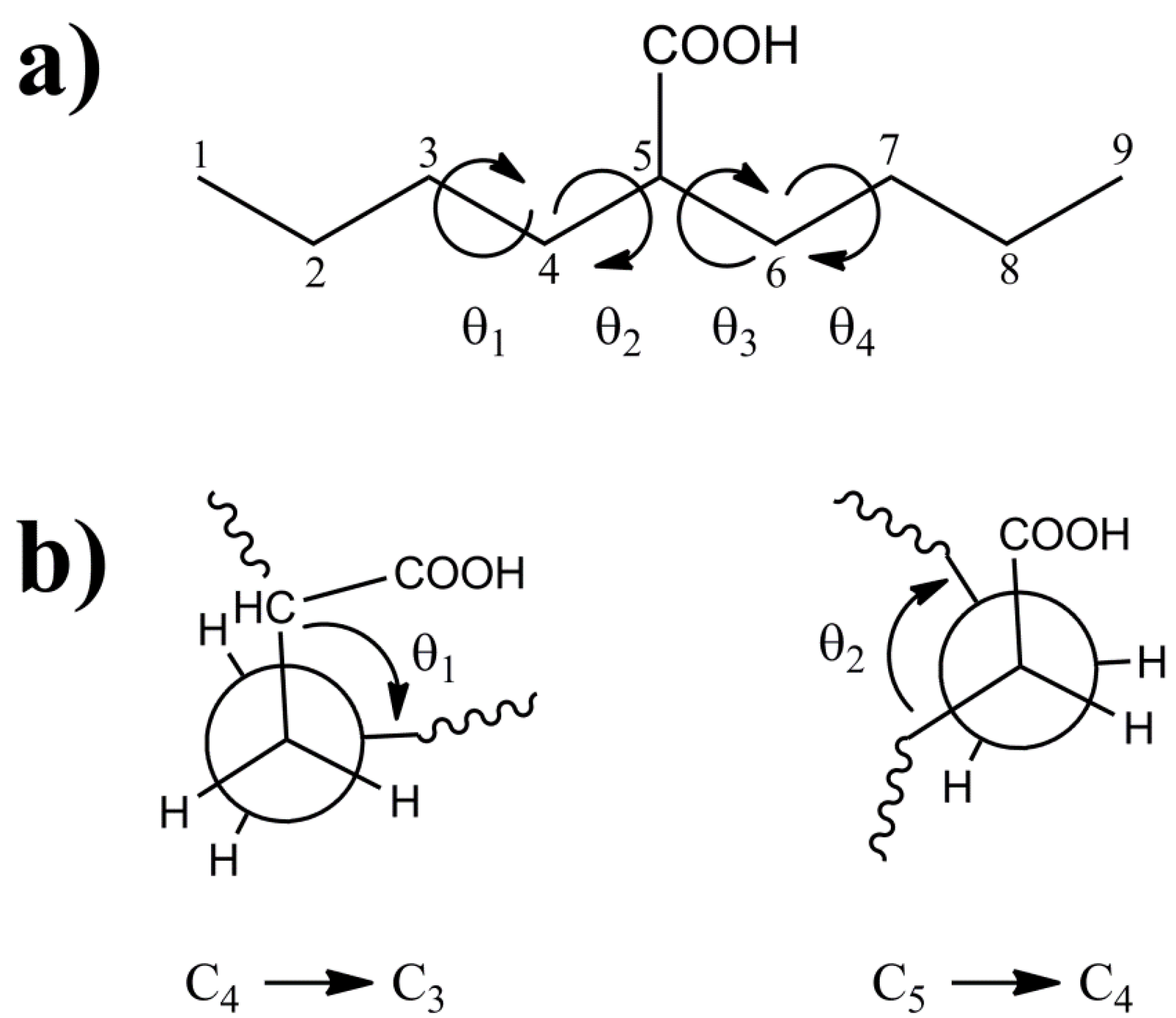
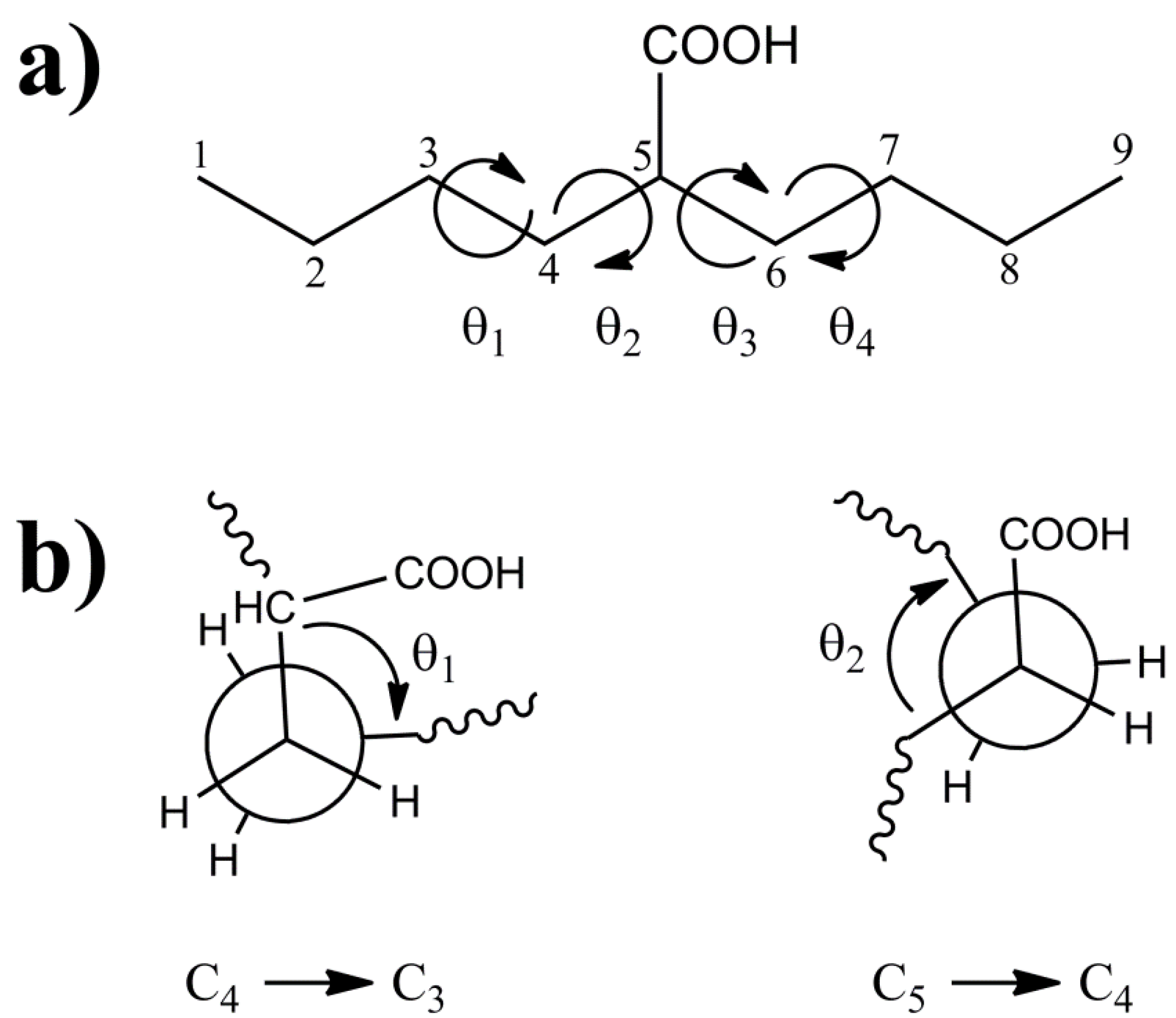
The 2-butylhexanoic acid molecular fragment (
Figure 1) was chosen as the model cluster for evaluation of the CH and CO
13C-NMR chemical shifts within p(E-AA) copolymers. The methine (CH) carbon, C5, is sufficiently removed from the end of this molecular cluster to minimize end-group effects [
9]. The dihedral angles (θ
1, θ
2, θ
3, θ
4) describing the local polymer chain conformations around the CH carbon ranging from −180° < θ
i ≤ 180° are shown in
Figure 1a. The rotational sense of the individual dihedral angles was always defined looking down the bond originating at the carbon closest to the central COOH pendant group (
i.e., along the C
4-C
3 for θ
1 or the C
5-C
4 bond for θ
2) as depicted in
Figure 1b. The rotations of dihedral angles near the ends of the chains were not considered to be important in describing the local conformation for the central carbon, such that these dihedral angles were not constrained and allowed to adopt a minimum energy conformation.
Figure 1.
(a) The model 2-butylhexanoic acid model cluster with the carbon numbering and torsional dihedral angles defined. (b) Projections along the C4-C3 and C5-C4 bonds defining the θ1 and θ2 dihedral angles.
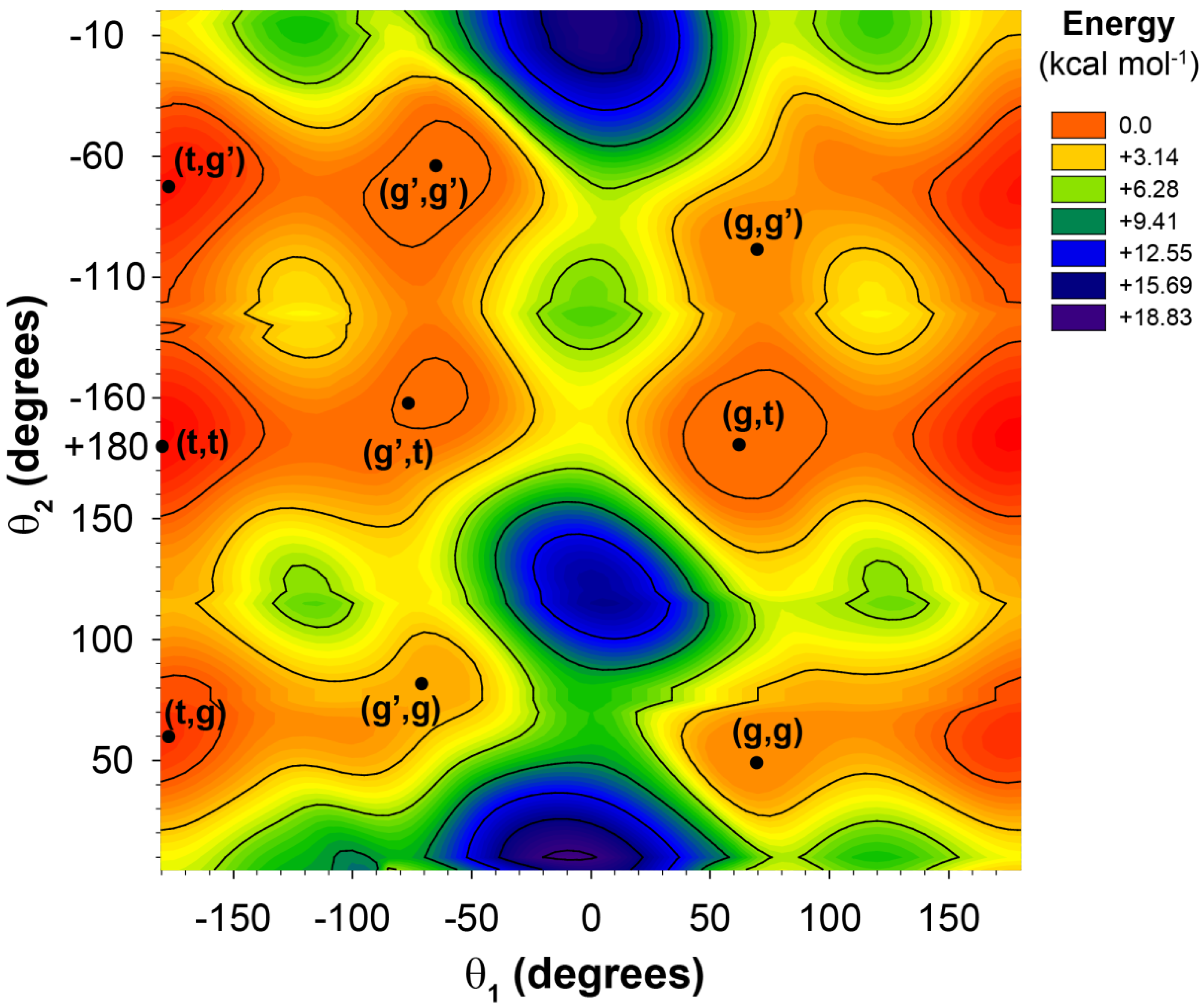
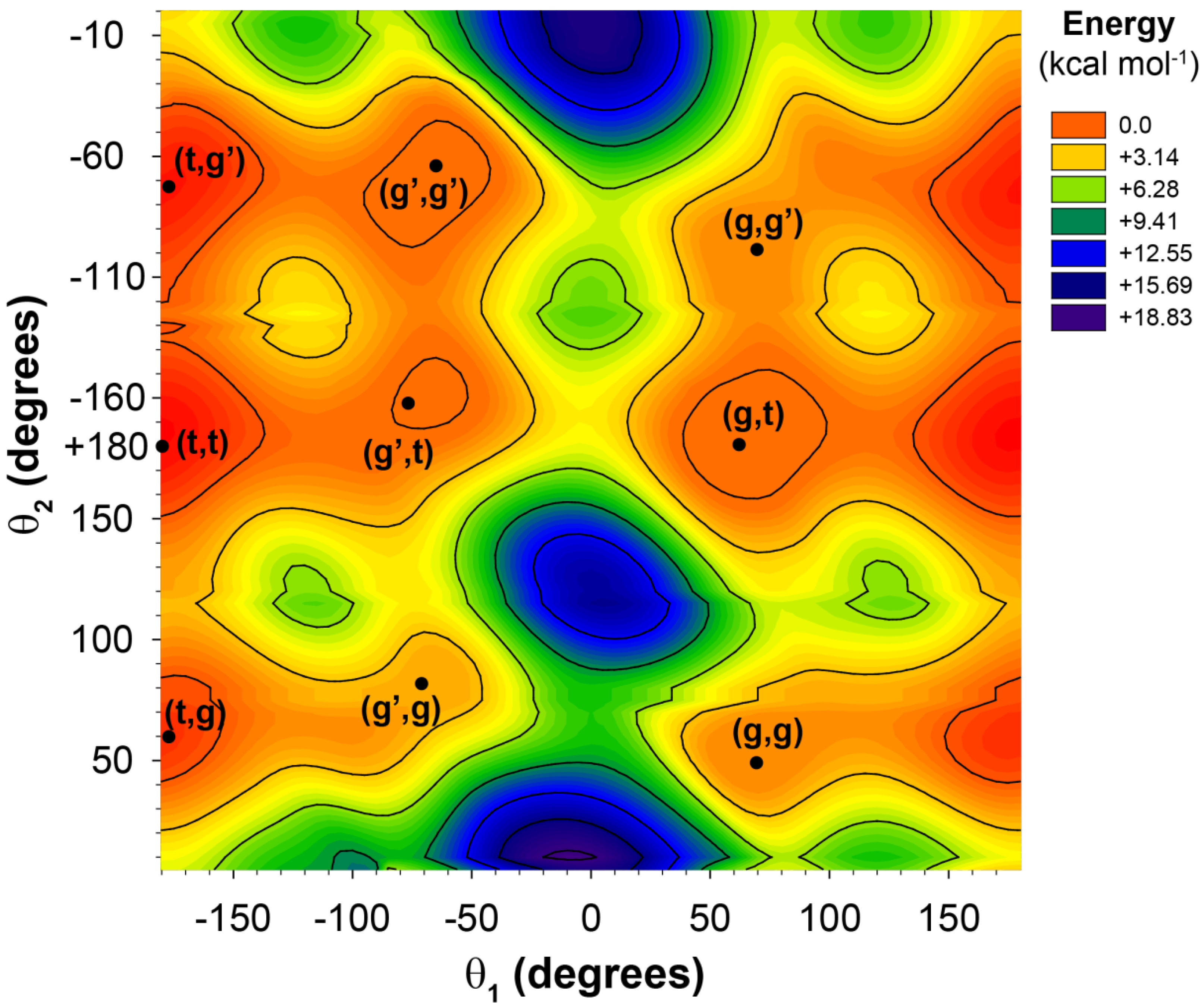
Figure 2.
The potential energy surface for the 2-butylhexanoic acid model cluster as a function of the θ1 and θ2 dihedral angles.
2.1. Methine 13C-NMR Chemical Shifts
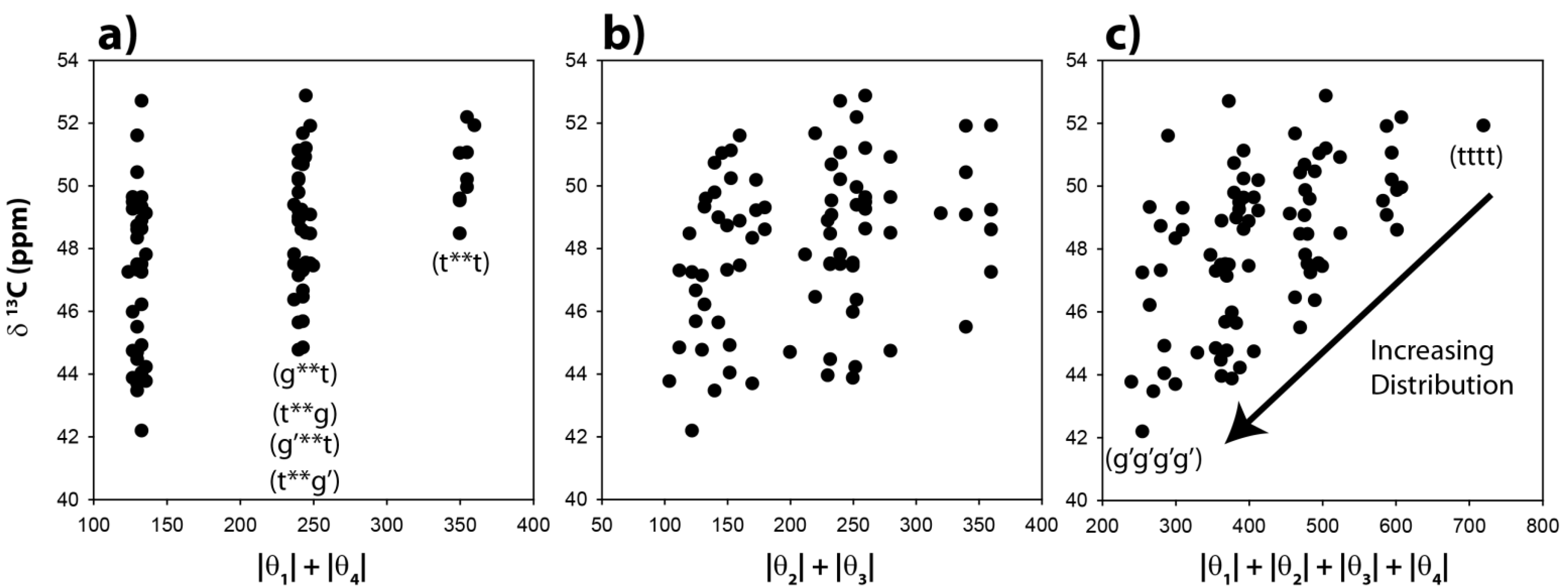
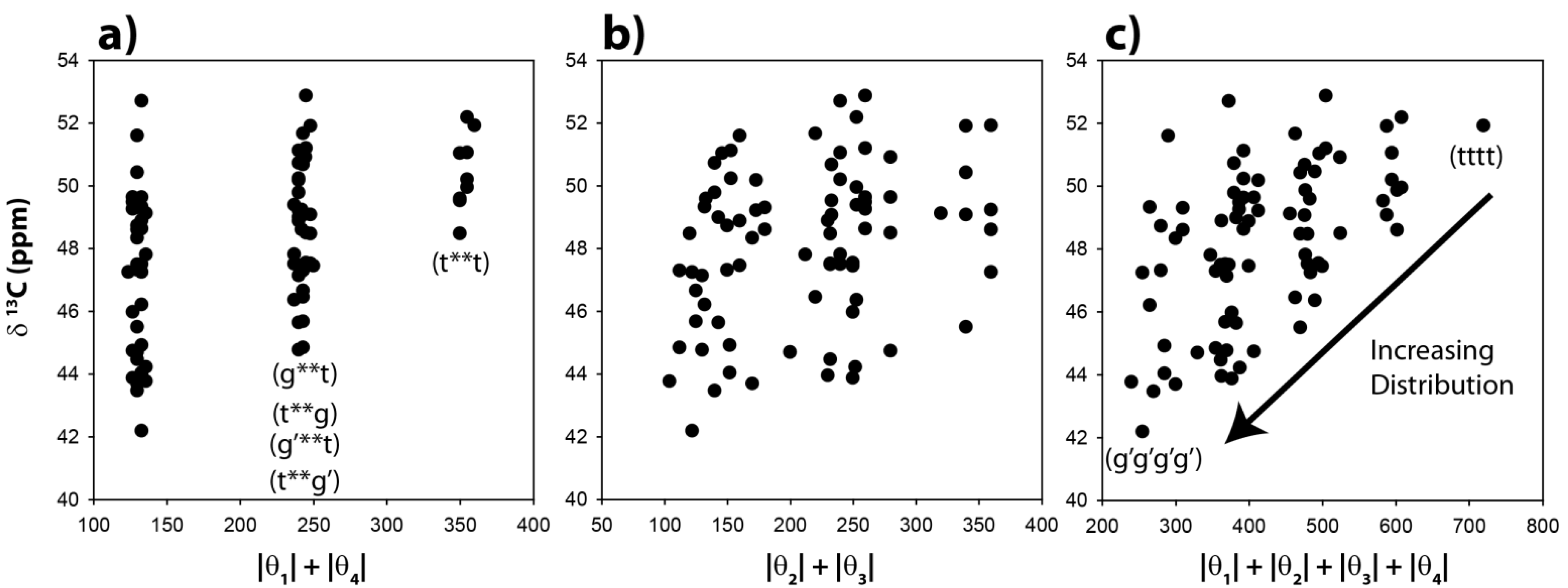
The
13C-NMR chemical shifts for the CH carbons in these 81 optimized clusters were evaluated and are shown as a function of the |θ|
1 + |θ|
4 angle summation in
Figure 3a. Variations of these dihedral angles describe the relative γ orientation of the CH carbon with respect to the remainder of the polymer chain on each side. For conformations where these two dihedral angles are both in a
trans conformation the |θ|
1 + |θ|
4 summation will be 360°, while those conformations involving both dihedrals in a
gauche orientation will have a summation of ~120°. It is immediately apparent that the calculated
13C-NMR chemical shifts show significant variations, and are not a simple function of the dihedral angles. For the θ
1, θ
4 gauche-gauche conformations (g**g, g**g’, g’**g, g’**g’, where * denotes the unspecified θ
2 and θ
3 dihedral angles) there is an almost 10 ppm chemical shift variation observed (vertical distribution). Not all of these chain conformations are energetically favorable, and would not be necessarily observable. For example, the relative energy of the gggg chain conformation is nearly 9 kcal higher than the tttt conformation. The all-
gauss conformations (gggg, gggg’, ggg’g’
etc.) where found to have very high relative energies, and are not expected to contribute significantly to the observed
13C-NMR spectra.
Obviously the γ-
gauche effect for the CH carbon, as controlled by the conformation of the θ
1 and θ
4 dihedral angles, is not the dominating (or only) factor dictating the
13C-NMR chemical shift, and that other structural components of the polymer chain are also influencing the chemical variations. These results are very different than those predicted for the CH
2 carbon in PE, where the γ-
gauche effect is distinct and on the order of Δδ ~ −4 ppm in going from all-
trans to all-
gauche in the γ carbon position [
12]. In addition, the broad distribution of predicted CH
13C-NMR chemical shifts was not observed experimentally, where relatively narrow line widths were observed for the CH carbon in all p(E-AA) copolymer materials [
14]. It is important to note that values of the θ
1 and θ
4 dihedral angles reveal a relatively narrow angle distribution (
Figure 3a, horizontal spread) demonstrating that the potential energy surface for this dihedral angle pair was not significantly perturbed by the presence of the COOH group attached to C
5.
The predicted CH
13C-NMR chemical shift as a function of the |θ|
2 + |θ|
3 dihedral angle summation is shown in
Figure 3b, and again reveals no correlation with these dihedral angles. It is clear that distinct β-orientational effects are minimal for the CH carbon. There is a significant variation (horizontal spread) in the θ
2 and θ
3 angles observed (
Figure 3b), demonstrating that the potential energy surface of these dihedral angles was perturbed from classic
trans-gauche values (180°, 60°) observed in un-substituted PE. The
13C NMR chemical shift as a function of all four dihedral angles is shown in
Figure 3c. No clear correlations are observed, but it can be argued that there is a decrease in the average chemical shift, and an increase in the distribution of chemical shifts with respect to the all
trans conformation (tttt) when higher percentages of the
gauche conformation are introduced into the polymer chain. These results demonstrate that while there is some influence of the
γ-gauche on the CH
13C NMR chemical shift, there are clearly other structural perturbations influencing the shift.
Figure 3.
Predicted 13C-NMR chemical shift variation of the methine (CH) carbon as a function of (a) the |θ|1 + |θ|4 dihedral angle summation, (b) the |θ|2 + |θ|3 dihedral angle summation, and (c) the |θ|1 + |θ|2 + |θ|3 + |θ|4 dihedral angle summation.
Figure 3.
Predicted 13C-NMR chemical shift variation of the methine (CH) carbon as a function of (a) the |θ|1 + |θ|4 dihedral angle summation, (b) the |θ|2 + |θ|3 dihedral angle summation, and (c) the |θ|1 + |θ|2 + |θ|3 + |θ|4 dihedral angle summation.
In previous
ab initio investigation of
13C-NMR chemical shifts in “crowded” polymer systems such as PIB, there were deviations from the ideal tetrahedral coordination underlying the geometric assumptions in formulating the γ-
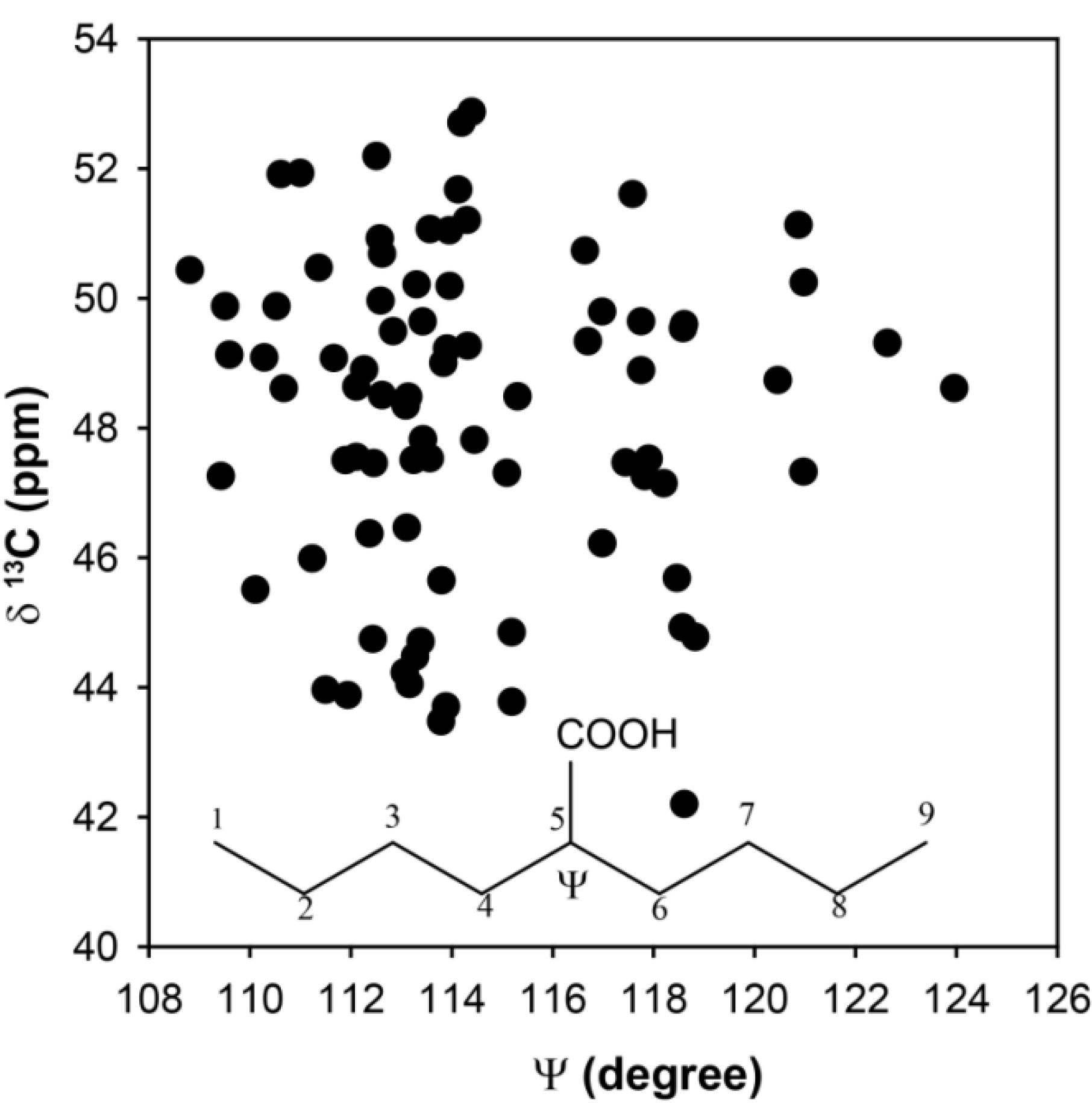
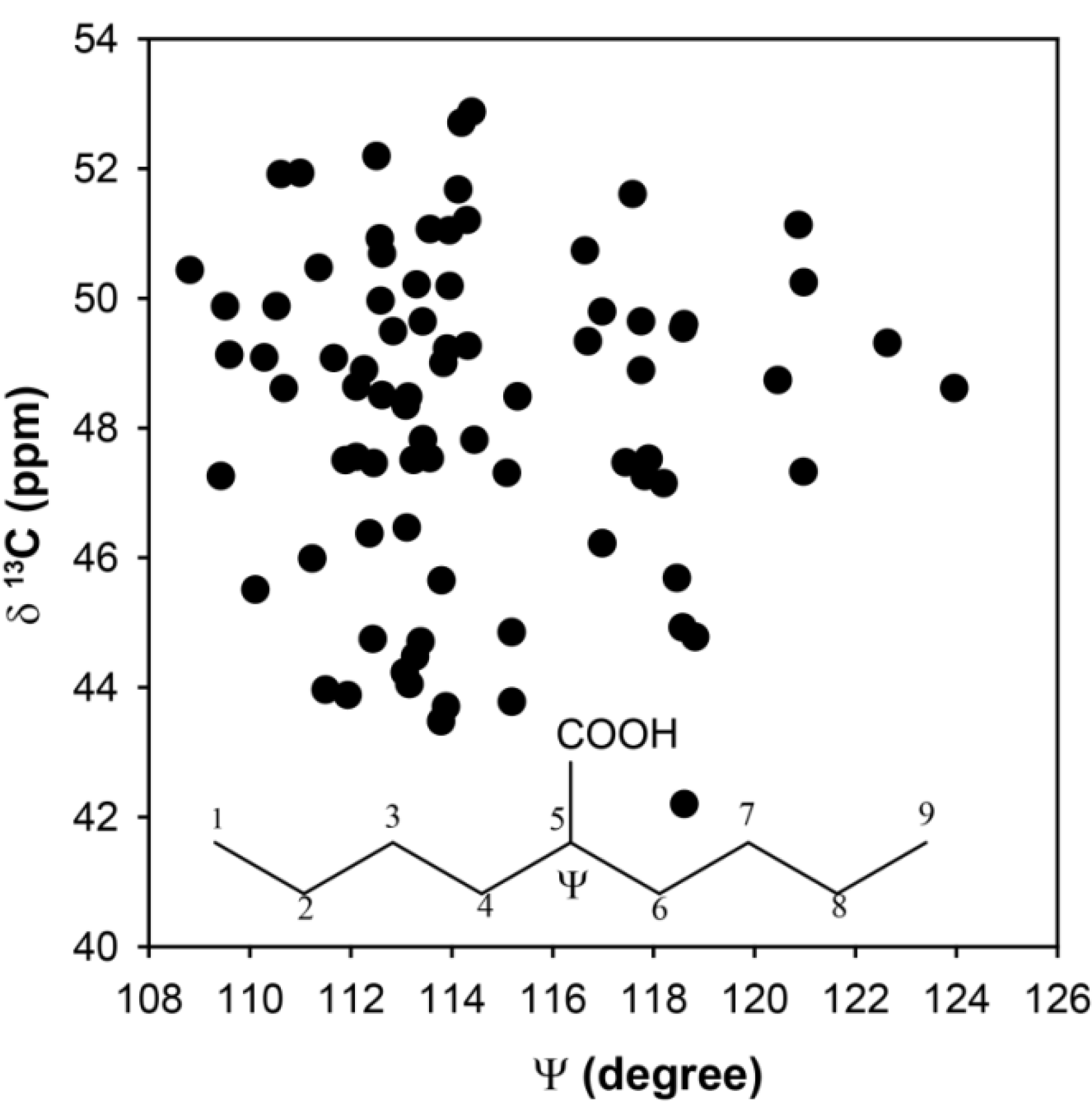
gauche effect. As the size of the substituent was increased, the distortions of the chain tetrahedral angles near the substituent were perturbed. In the current study, the tetrahedral angles were not constrained such that these types of structural perturbations could occur in the selected clusters. To explore if tetrahedral distortions might account for the observed chemical shift distribution, the CH
13C-NMR chemical shift as a function of the C
4-C
5-C
6 bond angle Ψ are shown in
Figure 4. While there is a range of dihedrals observed, the distribution of the chemical shifts is again significant (Δδ ~ 10 ppm), but does not appear to have a simple correlation with bond angle distortions. Based on these
ab initio results there does not appear to be a single structural parameter that can be extracted from the experimentally determined
13C-NMR chemical shifts in the p(E-AA) copolymers.
Figure 4.
Predicted 13C-NMR chemical shift variation for the methine (CH) carbon as a function of the bond angle Ψ.
Figure 4.
Predicted 13C-NMR chemical shift variation for the methine (CH) carbon as a function of the bond angle Ψ.
These CH chemical shift correlations (
Figure 3) along with the experimentally narrow CH resonance argue that only a limited number of local chain conformations occur around the central CH species in these p(E-AA) copolymers and corresponding Zn-modified ionomers. Other possibilities include the partitioning of CH defect between the crystalline and amorphous phases within the p(E-AA) copolymers, or that local dynamics are averaging the
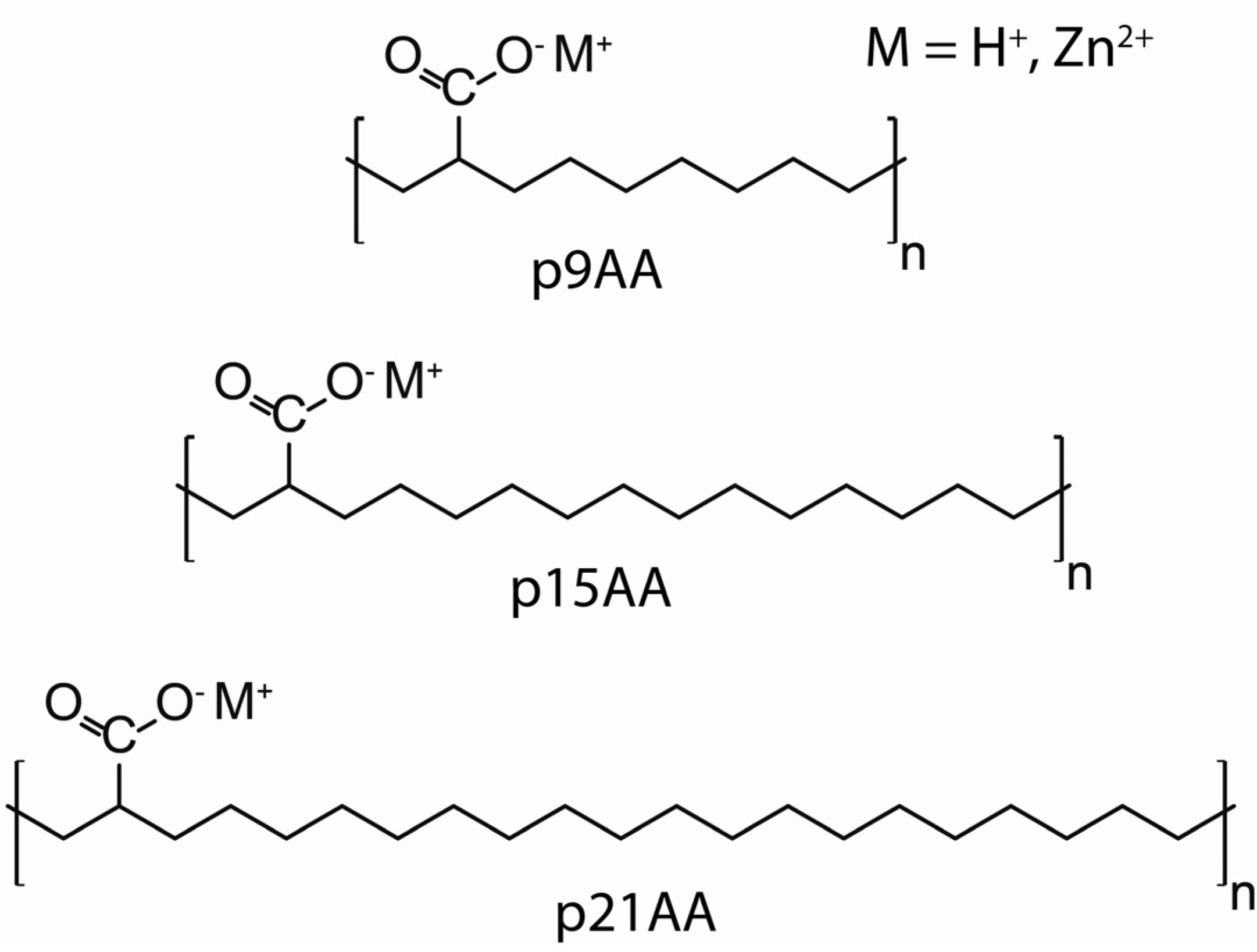
13C chemical shift over multiple conformations. The p21AA sample contained ~ 44% crystalline
trans conformation phase, and the remainder of the material was in the amorphous
trans/
gauche or
gauche defect phase, while the p9AA sample is entirely in an amorphous
trans/gauche phase. For the p21AA polymer only a single CH resonance was observed, inconsistent with the CH defect being present in both the crystalline and amorphous phases (
i.e., partitioned into a single phase). On the other hand, the reduction in the CH
13C- NMR chemical shift between the p21AA and p9AA copolymer is consistent with the increase in
gauche conformations with decreasing chain length. As noted above, the narrow CH line width experimentally observed is at odds with the large chemical shift distributions predicted in
Figure 3. The reduced line width may reflect chemical shift averaging due to chain dynamics. A Boltzmann average of the predicted CH
13C-NMR chemical shift for the 81 different structures investigated is δ = +50.4 ppm, which is lower than the δ = +51.9 ppm predicted for the all-
trans conformation (Δδ ~ −1.5 ppm). This chemical shift difference is similar to the change observed experimentally. Chain dynamics have been observed within these p(E-AA) copolymers [
14] and in precise methyl-substituted PE [
15], based on experimental observation of CH
2 chemical shift anisotropy (CSA) tensor averaging.
2.2. Carbonyl 13C-NMR Chemical Shifts
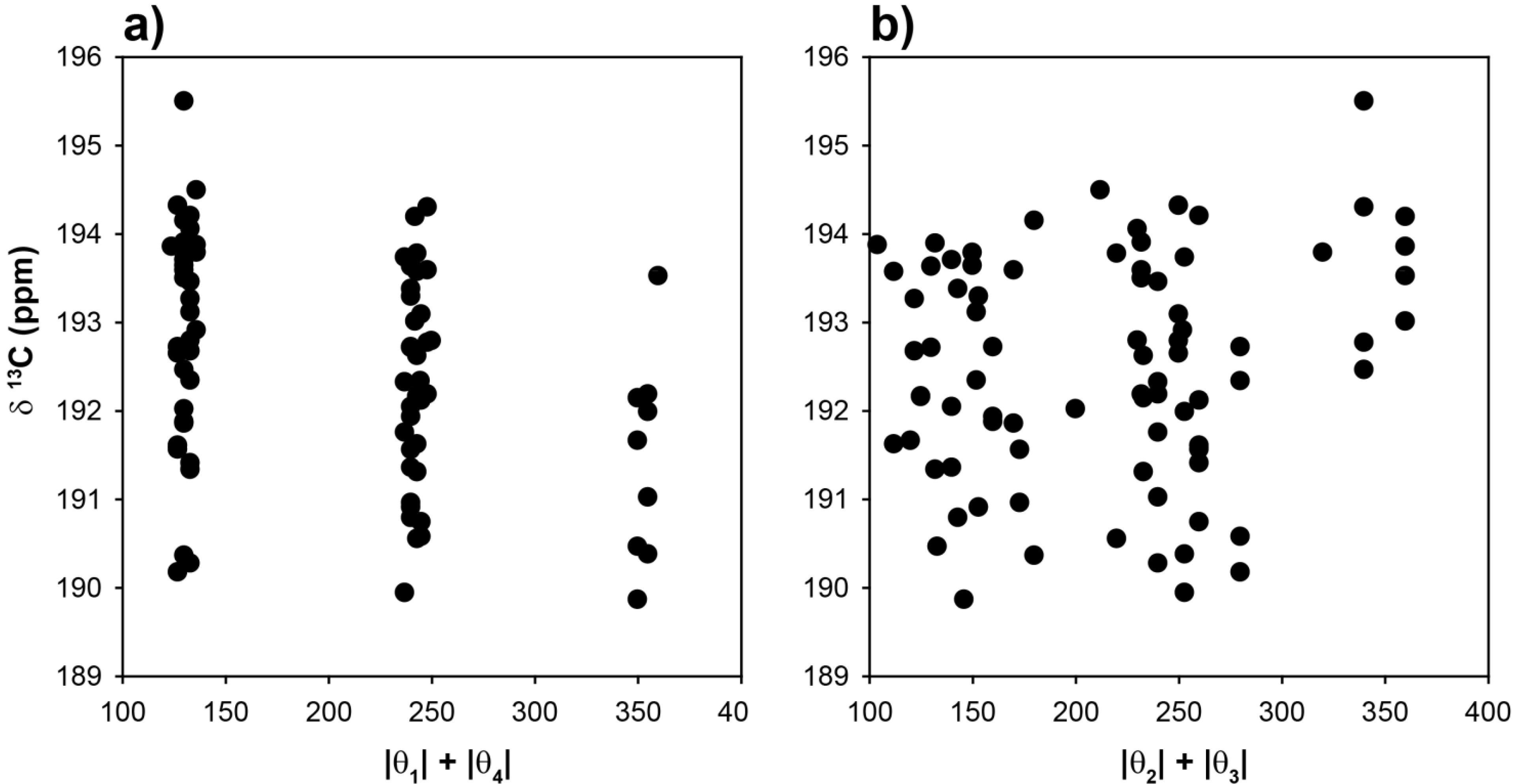
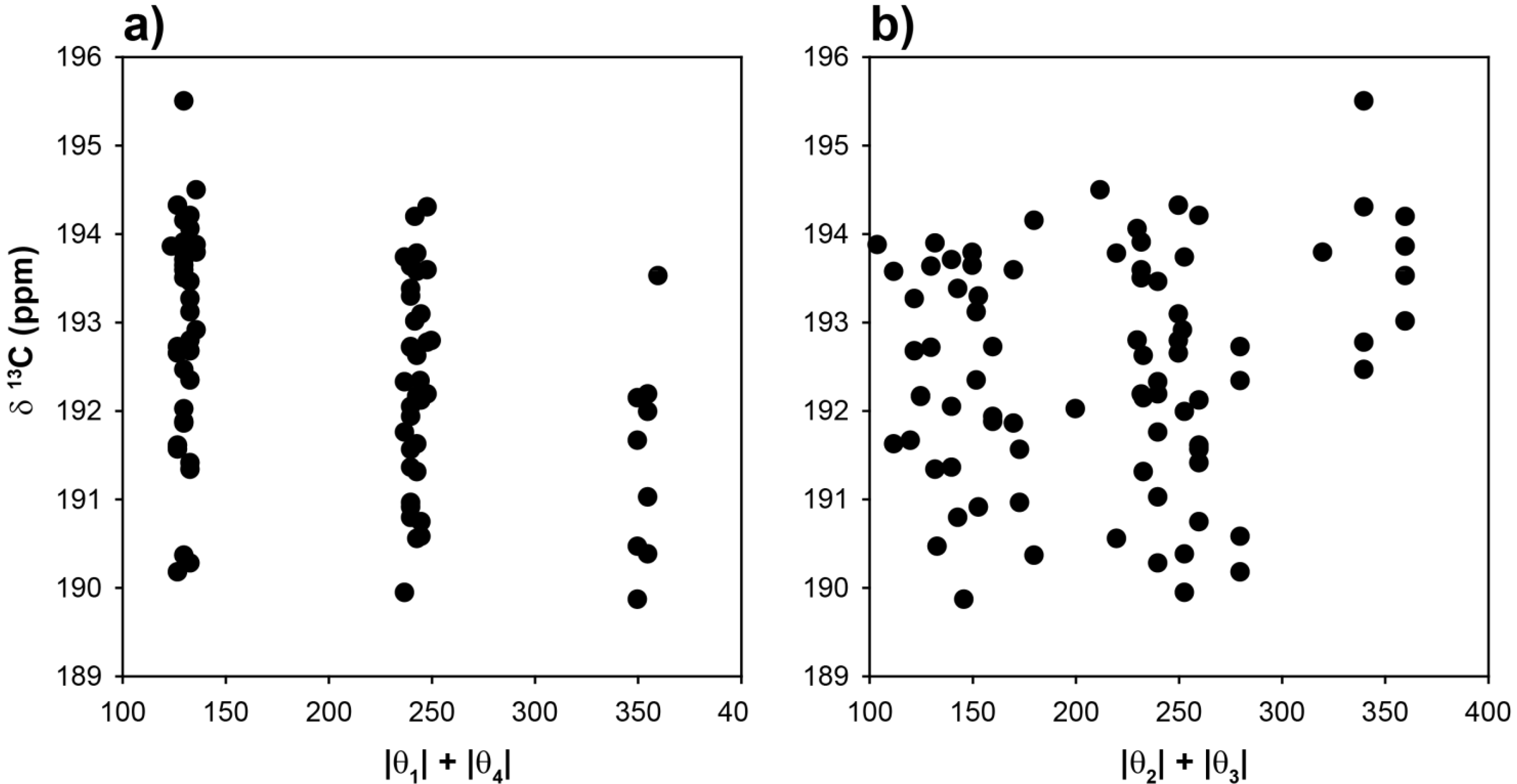
To explore the role of chain conformations on the carbonyl
13C-NMR spectra, the COOH chemical shifts for the 81 different clusters were calculated and are shown in
Figure 5 as a function of the |θ
1| + |θ
4| and |θ
2| + |θ
3| dihedral angle summations. In this case the dihedral angles θ
2 and θ
3 would control the
γ-gauche effect. Similar to the methine chemical shifts there is a very large variation of ~15 ppm observed for the COOH chemical shift. This is significantly larger than the experimentally observed variation in the copolymer (~+0.7 ppm), or the Zn-neutralized ionomer (~ +2 to +4 ppm). These results (
Figure 5) demonstrate that COOH NMR chemical shift correlations with dihedral angles describing the different chain conformations were not beneficial in determining the local chain conformation. In addition, there was no correlation between the COOH chemical shift and the tetrahedral angle Ψ (data not shown). It was noted that the absolute value of the COOH chemical shifts were very sensitive to the basis set used in structural optimization, but the overall Δδ range as a function of angle was consistent between different levels of theory. This basis set effect reflects the need to adequately model the electron density in the carbonyl region, and required both diffuse and polarized basis functions. These results support the earlier conclusion that for the experimentally observed carbonyl
13C-NMR chemical shifts, the γ-
gauche effects was not observed. It is proposed that the experimental COOH NMR chemical shifts are governed by intermolecular effects such as hydrogen bonding and neutralization. Based on these correlations (
Figure 5), the lack of large dispersions in the COOH experimental line width (broadened line shape) argues against multiple different chain conformations in both the p(E-AA) copolymers and the Z,-modified ionomers.
Figure 5.
Predicted 13C-NMR chemical shift variation of the carbonyl (COOH) carbon as a function of (a) the |θ|1 + |θ|4 dihedral angle summation and, (b) the |θ|2 + |θ|3 dihedral angle summation.
Figure 5.
Predicted 13C-NMR chemical shift variation of the carbonyl (COOH) carbon as a function of (a) the |θ|1 + |θ|4 dihedral angle summation and, (b) the |θ|2 + |θ|3 dihedral angle summation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}