Challenges and Opportunities of MicroRNAs in Lymphomas

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
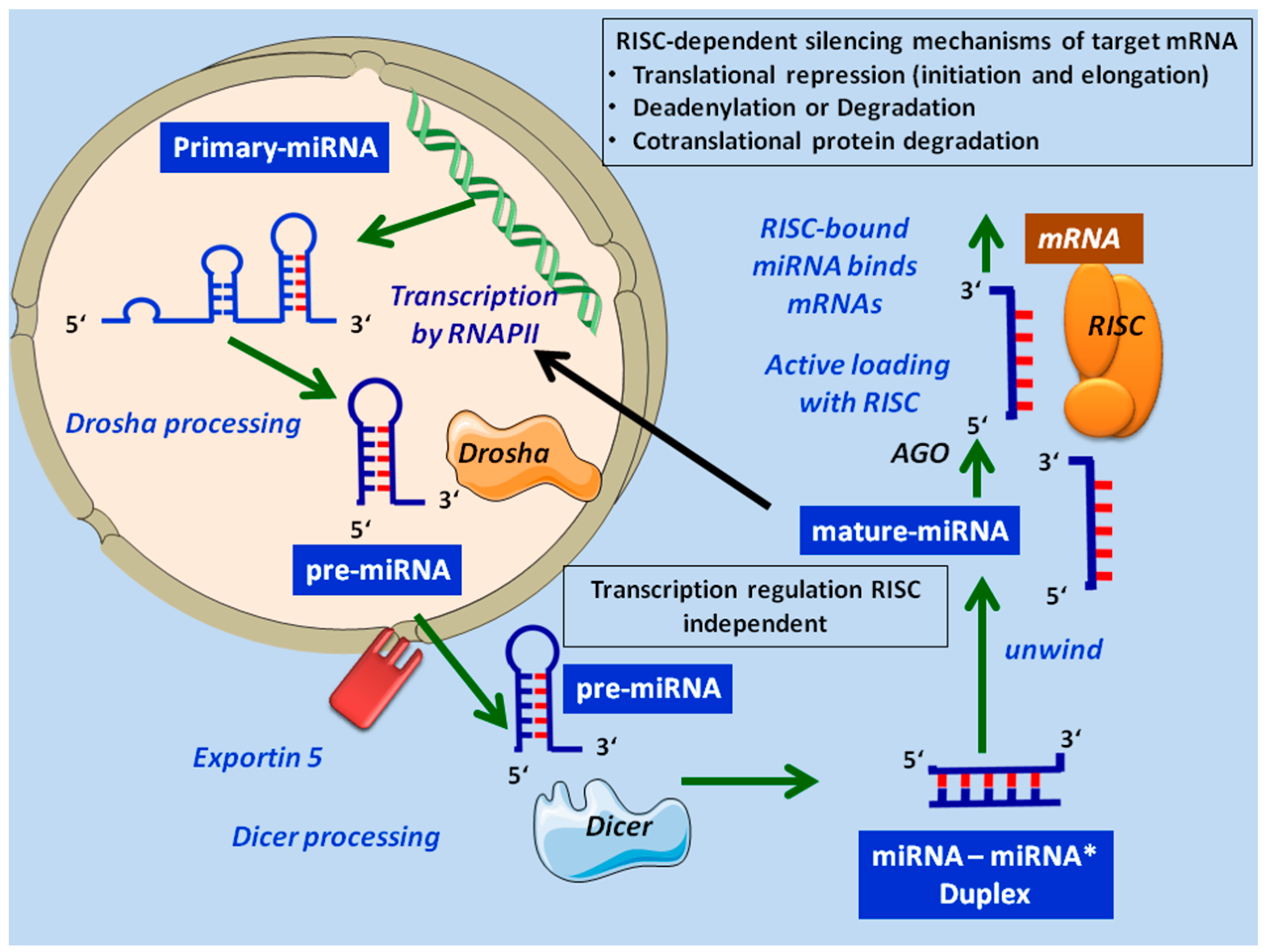
2. MicroRNA Biogenesis and Mechanism of MicroRNA Gene Regulation
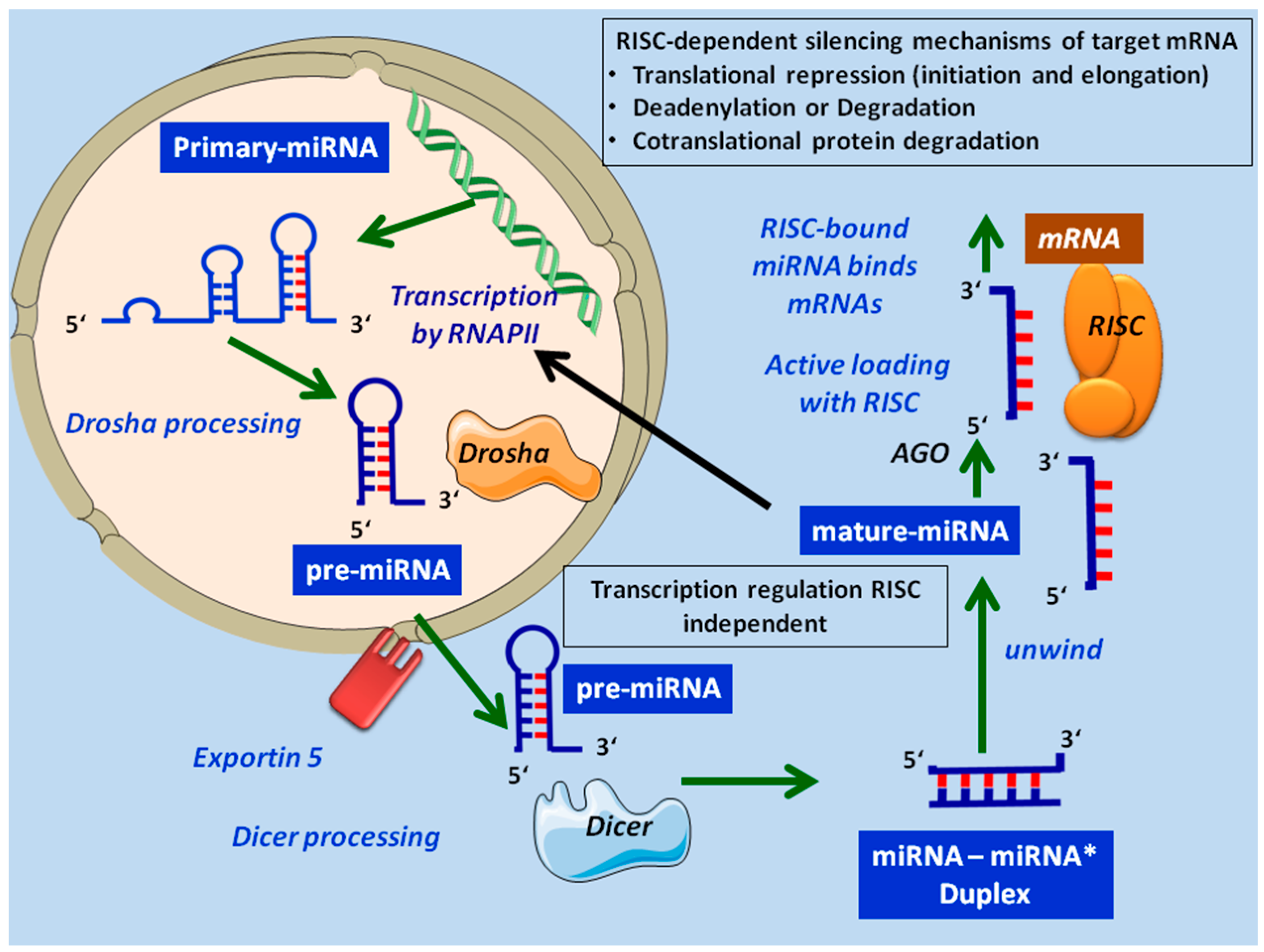
3. MicroRNAs as Regulators of Lymphoid Maturation
- Evaluation of global expression patterns of miRNAs in specific cell lineages, and comparison of these profiles in several stages of differentiation and/or in normal cells vs. lymphoma cells.
- In vitro and in vivo functional and mechanistic studies of miRNAs carried out by (a) replacing or knockdown of miRNAs or (b) silencing only specific single miRNA-mRNA target interactions through a mutation in complementary sites to the 3′-UTR or (c) using chemically-modified antisense oligonucleotides, termed antimiRs, which hold the mature miRNA in competition withtarget mRNAs leading to functional inhibition of the miRNA and repression of the direct targets.
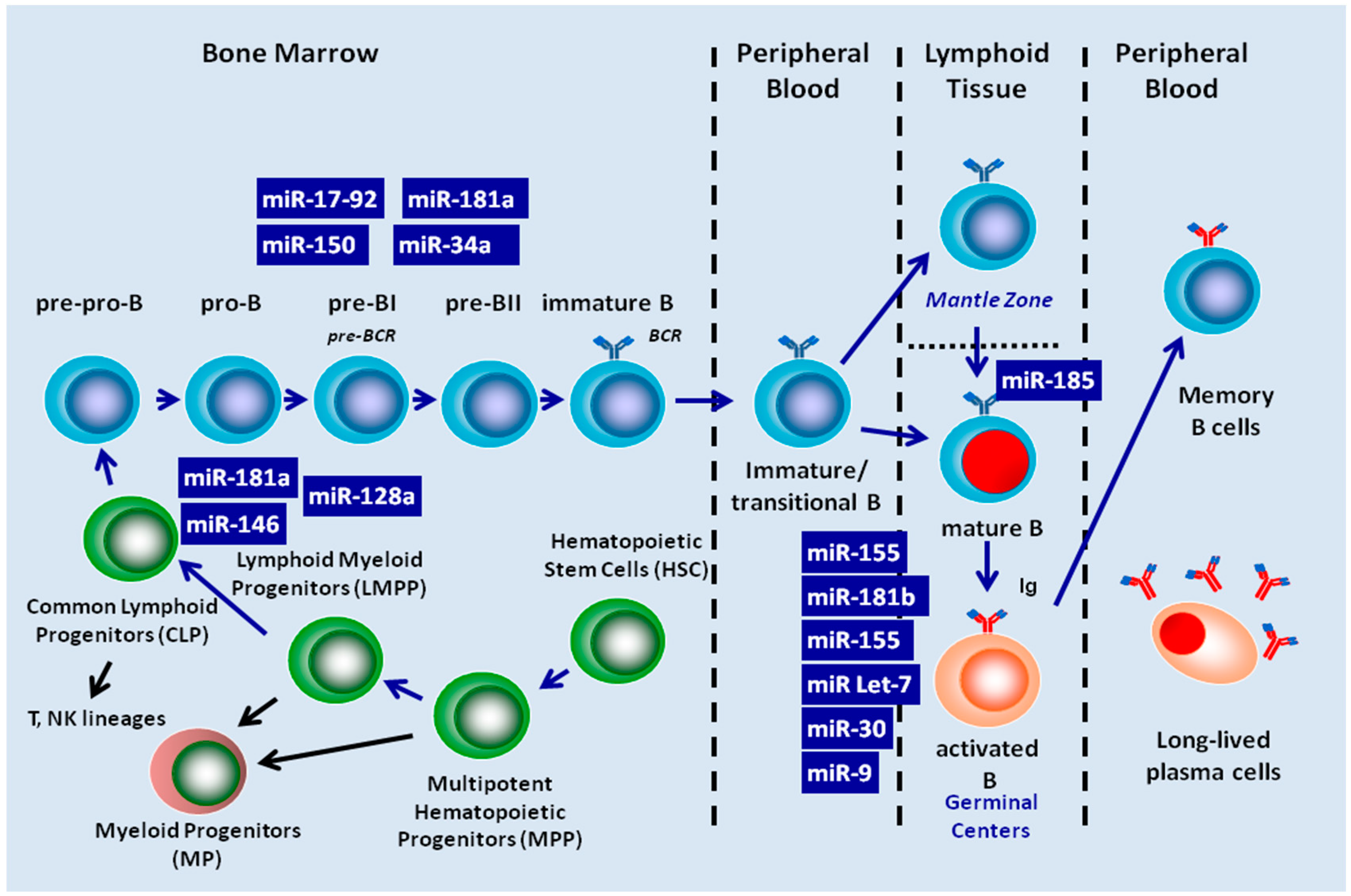
4. Role of miRNAs in B-Cell Maturation
4.1. miRNA Control of B-Cell Development in Bone Marrow

{kind=link}
{kind=link}
{kind=link}
| miRNAs | Direct/Indirect Target | Function/Ref. | Regulation | Lymphomas/miRNAs involved/Ref. |
|---|---|---|---|---|
| miR-17-92 |
| Control of transition from proB- to preB-cells. Enhancement of cell survival and inhibition of apoptosis by targeting PTEN and Bim, antagonizing with BCL2 [29,30]. Positive regulation by MYC and simultaneous repression of E2F1 expression by miR-17-5p and miR-20a: generation of MYC/miR-17-92/E2F1 circuit that accelerates the development and increases the aggressiveness of the tumor [31,32,33,34,35]. miR-19b represses apoptosis and promotes cell proliferation and angiogenesis by repressing PTEN expression and function, thus resulting a functional activation of Akt/mTOR pathway via PI3K pathway [36]. Induction of chemoresistance in MCL by activating the PI3K/AKT pathway trough targeting of PHLPP2 [37]. VEGF up-regulate the expression levels of only miR-18, miR-19 and miR-20 to participate in the control of angiogenic phenotypes [9]. Selective miR-17-92 biogenesis and likely post-transcriptional silencing mediated by each miRNA component within miR-17-92 may be regulated in a cell type- and context- dependent manner [9,38]. | ||
| miR-181a |
| Involvement in commitment to Lymphoid cell fate, B and T-cell differentiation, and specifically promotion of early B-cell development [19,45]. Block of human progenitor cell differentiation [46]. | ||
| miR-181b |
| Modulation of somatic hyper-mutation and class-switch recombination together with miR-155 but at different stage of B-cell activation [48,49]. | B-CLL [48] | |
| miR-150 |
| Control of transition from proB- to preB-cells [50,51]. Down-regulation of c-MYB, expression [18,51]: interaction important for oncogenesis and (or) tumor progression [52,53,54]. Regulation of the NK and iNKT cell development [55]. Reduction of phosphorylated AKT (ser473/4) levels and increasing Bim and TP53 by directly down-regulating the DKC1 and AKT2 expression [56]. |
| |
| miR-185 |
| BCR development. | ||
| miR-155 |
| Control of B-cell differentiation and GC reaction: activation and function of B cell in germinal centres and for T-cell dependent antibody responses [19,46,60,61,62] by negatively modulating somatic hypermutation and class-switch recombination through the targeting of AID [62] and PU.1 [63]. Down-regulation of HGAL expression, leading to a decreasing of RhoA activation and increasing of spontaneous and chemoattractant-induced lymphoma cell motility [64] Oncomir that regulates proliferation and enhances cell survival by:
|
| |
| miR-34a |
| Growth suppressive function and pro-apoptotic effect in pro-B cells of which it controls the transition to pre-B stage by targeting FOXP1 [77,78] know as B cell on cogene. [79] It is negatively regulated by c-MYC [80] and positively regulated by TP53 [80,81,82,83,84]. In turn, by inhibiting SIRT1, it actives TP53 resulting apoptotic effects mediated by TP53/SIRT1/miR-34a pathway. Epigenetically silenced in many lymphomas, mainly NK/T-NHL [55]. |
| |
| miR-9 |
| Regulation of B-cell terminal differentiation into plasma cells and memory B cells [85,86] It regulates E-cadherin [58] |
| |
| miR-30 |
| Regulation of B-cell differentiation by determining the ability of developing B cells to move to the GC [50] |
| |
| Let-7 |
| Regulation of B-cell terminal differentiation into plasma cells and memory B cells [85,86] | ||
| miR-29 |
| Down-regulates TCL1 and MCL1 expression [48,89] |
|

4.2. miRNA Regulate Mature B-Cell Activation and Functions
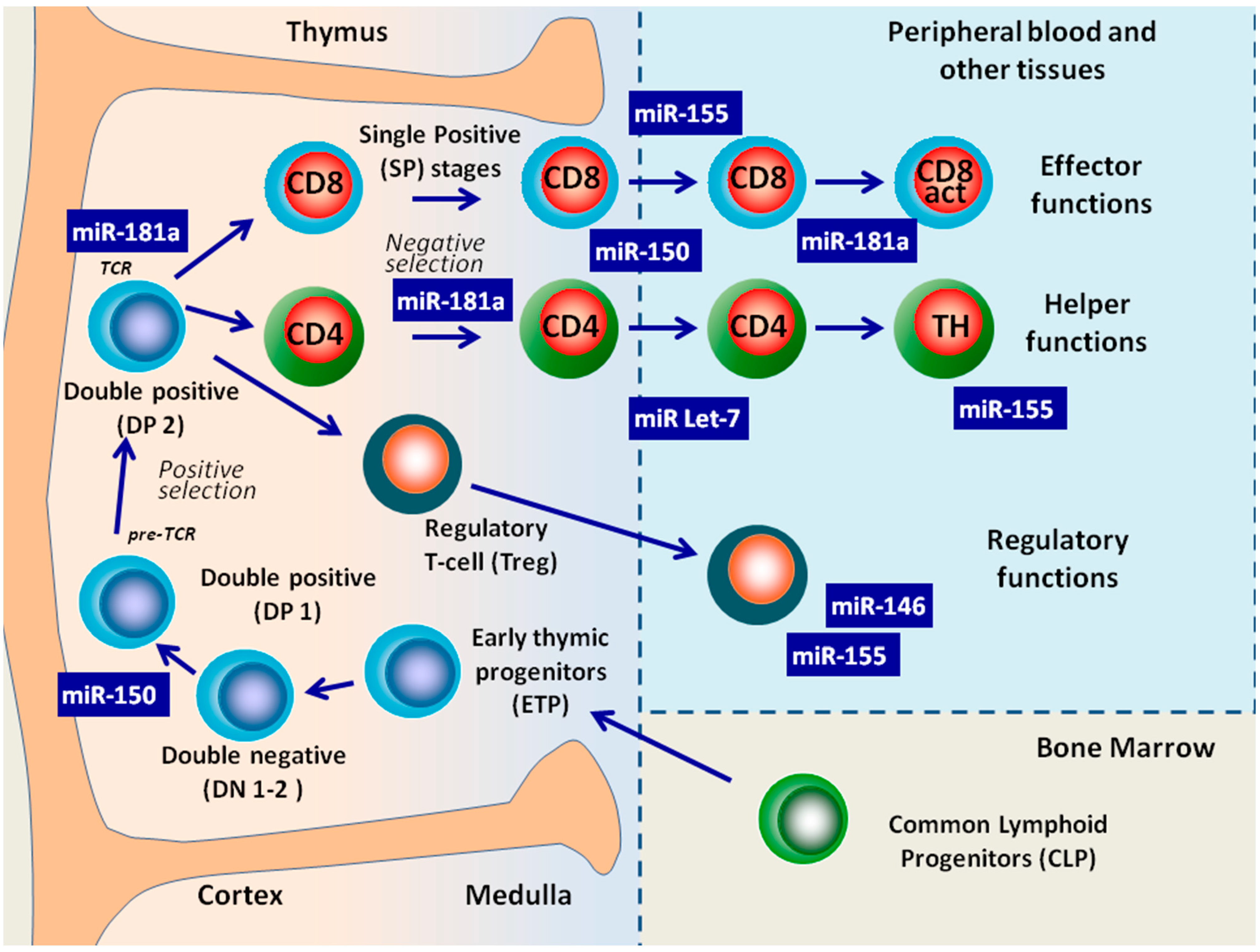
5. The Role of miRNAs in T-Cell Maturation
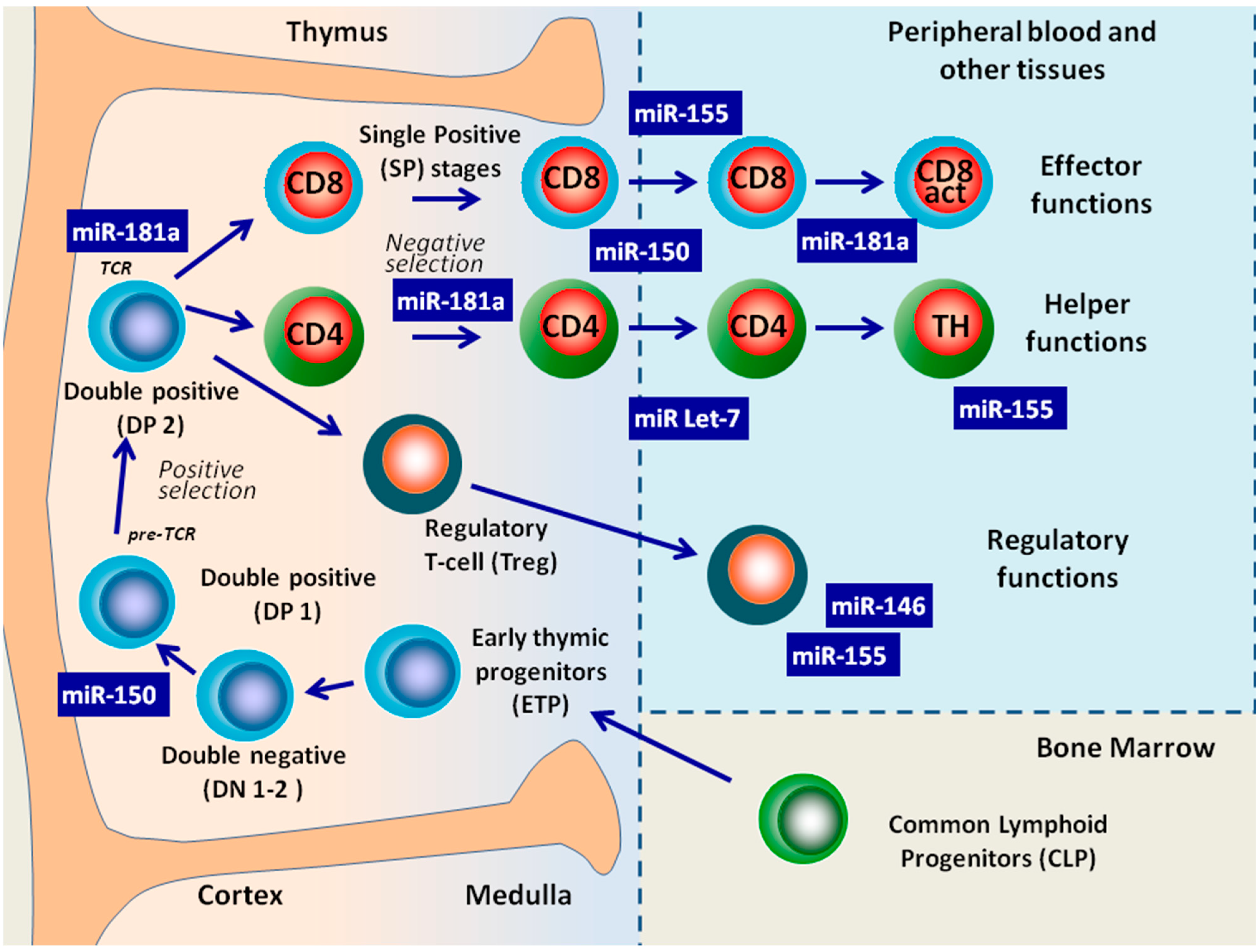
5.1. T-Cell Development in the Absence of Dicer
5.2. Individual MiRNAs Have the Potential to Regulate T-Cell Development and Functions
| miRNAs | Target | Function |
|---|---|---|
| miR-181a | CD69 BCL2 TCR-α (DUSP5, DSP6, SHP2, PTPN22) | Regulation of positive selection by governing the homeostasis of CD4/CD8 lymphocytes and modulation of T-cell sensitivity by increasing TCR signaling to peptide antigens through the down-regulation of multiple phosphatases [19,23,45,104,118,126]. Regulation of iNKT cell development through the modulation of TCR signaling threshold resulting the increase responsiveness of DP thymocytes to TCR signals [116]. |
| miR-17-92 | CREB1 PTEN Bim | Regulation of effector and memory CD8+ T-cell differentiation. Temporal expression is critical [129]. |
| miR-150 | NOTCH3 | Controls of T-cell differentiation [51]. Regulation of differentiation into the memory or effector phenotypes of T cells [59]. Regulation of the differentiation from DP into CD4+ and CD8+ T-cells [45,120,121,122]. Regulation of the intrathymic pre-T-cell receptor selection of T-cells [121,123] |
| miR-155 | SOCS1 | Regulation of differentiation into the memory or effector phenotypes of T cells [59]. Controls of T-cell differentiation: to favour Th1 responses partially by modulating cytokine production [60,61]. Control of proliferation and homeostasis of Treg cells by stabilizing the signal of FOXP3 through the targeting of SOCS1 [124]. |
| miR-146 | STAT1 | Regulation of the Treg suppressor function by modulating IFNγ responses through the targeting of STAT1 [45,128]: promotion of differentiation into Th1 cells rather than Th2 cells [45,50] |
6. miRNAs and Lymphoid Malignancies
6.1. miRNAs as Oncomirs in Lymphoid Malignancies
6.2. miRNAs as Dysregulated Tumor Suppressor Genes in Lymphoid Malignancies
| Ly Type | miRNA | Status | Func. | Target | BioM | Comment/Reference |
|---|---|---|---|---|---|---|
| HL | miR-135 | DR | JAK2 | The expression of miR-135a in cHL lymph nodes is down-regulated and correlates with clinical outcome. The miR-135a direct down-regulates the JAK2, thus affecting the expression of the antiapoptotic gene BCL-XL. In accordance with this the increased levels of miR-135a causes apoptosis and decreases cell growth [55] | ||
| miR-155 | UP | OG | PU.1 | D | Specific biomarker of HL [43,71,72,143,144,145,146,147] | |
| let-7/miR-9 | UP | OG | PRDM1/blimp1 | D | [86,88] | |
| miR-17-92 | UP | OG | D | Compared to other B-cell lymphoma cell lines, overexpression of the miR-17-92 cluster members miR-17-5p, miR-19a, miR-19b, miR-20a, and miR-92, is prominent in HL [43] | ||
| miR-21 | UP | OG | PTEN | D, P, PR | Plasmatic levels are associated with Hasenclever scores ≥ 3 and returned to normal at remission [42,43]. Involved in expression in cHL pathogenesis and is associated with therapeutic resistance [184] | |
| miR-150 | DR | TSG | D | [43] | ||
| DBLCL | miR-155 | UP in ABC | OG | PU.1 INPP5D (SHIP1) SMAD5 | D, P | More aggressive subtypes [140] Higher in ABC-DBLCL than in GC-DBLCL; useful for differential diagnosis of ABC-DBLCL and as a P considering the poor prognosis of ABC as compared with GC-DBLCL subtypes [71,72,138]. Evaluation of serum levels: 83% sensitivity, 65% specificity [139]. Inverse correlation between NF-kB/miR-155 and PU.1/CD10 expression [140]. Stimulation by TNF-α increases miR-155 expression that induces aberrantly activation of PI3K/AKT pathway, one the most important unfavourable P, by directly targeting INPP5D [66,67,137,140,141]. By targeting SMAD5, it makes DBLCL cells resistant to the growth-inhibitory effects of both TGF-beta1 and BMP [58,64,65,70,100] |
| let-7f | UP | OG | D | [58] | ||
| let-7b | UP in ABC | OG | PRDM1/blimp1 | D | [88] | |
| miR-9 | UP in ABC | OG | PRDM1/blimp1 | D | [58,102] | |
| miR-15a | DR | TSG | BCL2 | D | [2] | |
| miR-17-92 (miR-17-5b, miR- 9b) | UP in GC | OG | BIM/PTEN | P | Consistent with more aggressive phenotype [31,40,41,87] MiR-19b promotes cell proliferation and angiogenesis, represses apoptosis | |
| miR-21 | UP in ABC | OG | BCL2 | D, P | Elevated levels (in serum or biopsy) are associated with a better RFS in de novo DBLCL [138] More highly expressed in the poor risk ABC than in the GC-DBLCL subtype and higher in disease stages III-IV compared to stages I-II [138,153,154]. Conversely in B-CLL [46,156,158] | |
| miR-30 | DR | TSG | BCL6 PRDM1 | [50,85] | ||
| miR-34a | DR | OG | ||||
| miR-150 | DR | TSG | c-Myb | [18,41] | ||
| MCL | miR-17-92 | UP | OG | E2F1 (c-MYC) PHLPP2 PTEN BIM | D, PR | Enhances resistance to chemo- [37] and radiotherapy via PI3K/AKT by targeting PTEN and PHLPP2 [152] [27,31,34,35,37,152] |
| miR-16-1 | 1. BS-del 2. DR | TSG | CCDN1 | P | 1. Truncation in CCDN1 mRNA alters its ability to be down-regulated by miR-16-1, resulting in MCL development and correlating with poor prognosis [174] 2. Myc represses miR-15/16-1 expression through recruitment of HDAC3 [175]. | |
| miR-181c | UP | [57,58] | ||||
| miR-155 | UP | OG | [185] | |||
| FL | miR-9 | UP | PRDM1/blimp1 | D | MiR-9 (-5p, -3p) is significantly upregulated; Activated by MYC. It regulates NF-kB and down-regulates PRMD1/BLIMP1 [41,58] | |
| let-7 | UP | TSG | PRDM1/blimp1 | D | Reduced PRDM1 levels are important in FL characterized by the tightly regulated expression of BCL6 and PRDM1 [86,87]. | |
| miR-155 | UP | [41] | ||||
| BL | let-7a, let-7c, let-7e, let-7-f | DR | PRDM1/blimp1 | Loss of the let-7 (a; c) participates to the genesis and maintenance of the lymphoma phenotype through c-MYC regulation [186] | ||
| miR-9 | UP | OG | ||||
| miR-17-92 | UP | miR-17-3p, miR-18a, miR-19a, miR-19b, miR-92 up-regulated in BL vs. NHL [58] | ||||
| miR-29 | DR/lost | TSG | TP53 TCL1 | D | MiR-29 family regulates TP53 [27,58] MiR-29b, regulates TCL-1 expression, whereby the aberrantly expression of TCL1 in BL has been proposed as a diagnostic marker [48,58] MiR-29 is negatively correlated with MCL-1. | |
| miR-34b | DR | Targeted by TP53 | ||||
| miR-150 | DR | TSG | [18,58] | |||
| miR-155 | DR/lost | MiR-155 is the most significantly lost miRNA in BL [72] followed by miR-29b and miR-146a [58], this making it one of the most suitable markers for differential diagnosis between BL vs. DBLCL [44,75]. | ||||
| miR-15a/miR-16-1 | DR/del. | TSG | BCL2 TP53 | D, P | Deleted (region 13q) or down-regulated in ≈ 68% of B-CLLs [133]. | |
| miR-15a/miR-16-1 | DR/del. | TSG | BCL2 TP53 | D, P | It is associated with pathogenesis and outcome of B-CLLs: monoallelic deletion slower growth kinetic than biallelic [164] due to the directly targeting of TP53 [166] and consistent with indolent B-CLL and more favourable prognosis than 17p13 or 11q23 deletions [90]. In addition targets BCL2, MCL1 and CDK6 [165] | |
| miR-17-92 | UP | OG | D | Specifically: miR-19a, miR-20a, miR-92 [43] | ||
| BL | miR-21 | UP | OG | ANP32A SMAR-CA4 PTEN | P | Dramatically overexpression without genomic loci amplification: dysregulation at post- or transcriptional level [137] High expression is a significantly unfavourable P independent of other clinic-pathological factors in B-CLL patients [156] Can trigger B-lymphomagenesis by targeting ANP32A, SMAR-CA4 and PTEN [157,158] |
| miR-29 | DR | TSG | TCL1 | P | Expression of members of miR-29 family could discriminate between good and bad prognosis CLL samples as results of TCL1 targeting. MiR-29b is down-regulated in aggressive and poor prognosis B-CLLs which are characterized by high levels of TCL1 [48,90]. MiR-29c is associated with TSF and OS [187]. | |
| miR-34 | DR | P | Aggressive B-CLL: in 11q deleted CLL with high levels of ZAP-70 [166]. | |||
| miR-181b | DR | TSG | TCL1 | P | Aggressive B-CLL. DR in poor prognosis.Down-regulation of TCL1 results the activation mTOR, NF- kB, Mdm2 and CyclinD1 pathways [178]. So its expression correlates with disease aggressiveness: high expression in aggressive B-CLL, and lower in indolent disease [48] | |
| miR-181a | DR | TSG | BCL2 | [45] | ||
| miR-150 | UP | TSG | c-MYB | [18,58] | ||
| miR-155 | UP | OG | P | Aggressive B-CLL. Increased expression for dysregulation at post- or transcriptional level. [71,90,137] | ||
| MZL | miR-9 | UP | OG | NF-κB | [41,44] | |
| miR-155 | UP | OG | [41,71] | |||
| miR-200a, miR-200b, miR-200c | UP | ZEB1 ZEB2 | miR-200 family inhibits the initiating step of metastasis, the EMT, by maintaining the epithelial phenotype through directly targeting the transcriptional repressors of E-cadherin, ZEB1 and ZEB2 [58] | |||
| miR-126 | DR | [58] | ||||
| NK/T Ly | miR-21 | UP | OG | PDCD4 PTEN | Overexpression may contribute to the typical aggressiveness of NK/T cell lymphoma by the strong repression of PDCD4 and PI3K/PTEN/AKT [159,160,161,162] | |
| miR-34a | DR | TSG | Hypermethylated in a tumor specific manner [55] | |||
| miR-150 | DR | TSG | DKC1 AKT2 | Potential causative event for the onset and progression of NK/T cell lymphoma since the transduction of miR-150 decreases cell proliferation and induces apoptosis into NK/T cell lymphoma lines [56] | ||
| ALCL | miR-29a | TSG | miR-29a targets MCL-1 that could promote tumor cell survival by inhibiting apoptosis. This down-modulation requires an active NPM-ALK Kinase since the absence of kinase results an increase of miR-29a expression. In ALCL cell lines and in a xenografted model the increased expression of miR-29a modulated apoptosis through inhibition of MCL-1 expression, with a concomitant tumor growth reduction suggesting a potential new tool to affect tumorigenesis in these lymphomas [89]. | |||
| ALCL | miR-16 | VEGF HIF-1a | Down-regulation of miR-16 increases both angiogenesis and tumor growth by up-regulating VEGF translation via the hypoxia-miR-16 pathway [176] | |||
| let-7 | UP | OG | PRDM1/blimp1 | [56] | ||
| B-NHLs | miR-15a/16-1 | DR/ | TSG | c-MYC-induced miR-15a/16-1 repression by HDAC may be a mechanism for malignant transformation in aggressive B-cell lymphoma [175]. Deletion in indolent disease. | ||
| miR-21 | UP | OG | ANP32A SMAR-CA4 PTEN | D, P | Key role in B lymphomagenesis [157,158] | |
| miR-17-92 | UP | OG | BIM PTEN E2F1 PHLPP2 | D, P, PR | Located in 13q31-q32 region frequently (65%) amplified in lymphomas, especially in aggressive B-NHLs.Oncogenic effects depend on the Bim, PTEN and E2F1 targeting [30,34]. | |
| miR-34a | UP | OG | SIRT1 | Confers drug resistance in B cells that overexpress MYC [79,81] | ||
| miR-155 | UP | OG | Distinguishes ABC-DLBCL from GC-DLBCL [71,138]. In more aggressive B-NHLs aberrant activation of NF-kB increases expression of miR-155, which then down-regulates PU.1, and consequently leads to reduced CD10 expression [140] |
6.3. miRNAs as Diagnostic or Prognostic Tool in Lymphoid Malignancies
| Lymphomas | Signatures | St. | Comments/Ref. |
|---|---|---|---|
| DBLCL | miR-210, miR-155, miR-106a, miR-17-5p | UP | Significantly higher expression in DBLCL than RLN [41] |
| miR-150, miR-145, miR-328, miR-139, miR-95, miR-99a, miR-10a, miR-149, miR-320, miR-151, let-7e (miR-17-3, miR-595, miR-663) | DR/lost | Significantly lower expression in DBLCL than RLN [41] miR-17-3, miR-595, miR-663 most significantly lost in DBLCL [58] | |
| FL | miR-9, miR-301, miR-213, miR-9*, miR-330, miR-106a, miR-338, miR-155, miR-210 | UP | Significantly higher expression in FL than RLN [41] |
| miR-320, miR-149, miR-139 | DR | Significantly lower expression in FL than RLN [41] | |
| DBLCL vs. FL | miR-150, miR-17-5p, miR-145, miR-328 vs. miR-9/9*, miR-301, miR-338 and miR-213 | Differentially expressed miRNAs in DBLCL and FL identify signatures respectively in DBLCL and FL [41] | |
| DBLCL/FL/RNL | mir-330, mir-17-5p, mir-106a and mir-210 | Correctly identifies 98% of DLBCL, FL and RLN [41] | |
| ABC-DBLCL vs. GC-DBLCL | miR-155, miR-21 and miR-221 | UP | More highly expressed in ABC- than GC-subtypes, distinguish between ABC- and GC-DBLCL cases (p < 0.05). MiR-21 expression is an independent prognostic indicator in de novo DLBCL (p < 0.05) [138] |
| DBLCL-dn vs. DBLCL-t | miR-27a, miR-19b, miR-25, miR-18a, miR-636, miR-92, miR-621, miR-526c, miR-766, miR-299-5p, miR-380-3p, miR-129, miR-588 | UP | More highly expressed (p < 0.05) in DLBCL de novo than DLBCL-t cases, correctly predict transformation >85% [87] |
| FL-t vs. FL-nt | miR-223, miR-217, miR-222, miR-221, let-7i, miR-7b | UP | Differentially expressed (p < 0.05) between FL-t and FL-nt, accurately (89%) predict transformation of FL cases [87] |
| DBLCL prognosis | miR-637, miR-608 and miR-302 | Poor prognosis [87] | |
| miR-330, miR-30e, miR-425, miR-27a, miR-24, miR-23a, miR-199b, miR-199a* and miR-100 | Better outcome [87] | ||
| miR-21, miR-127, miR-34a, miR-195, let-7g, miR-19a, miR-27a | Correlate with EFS and OS [41] | ||
| DBLCL drug sensitivity | miR-181a, miR-22 and miR-18a | Independent prognostic indicators of survival in R-CHOP treated DBLCL [190] | |
| B-CLL poor prognosis | miR-29 | ||
| miR-15a, miR-195, miR-221, miR-23b, miR-155, miR-24-1, miR-146, miR-16-1, miR-16-2 | UP | Significant relationship between the expression of 9 miRNAs and the time from diagnosis to beginning of chemotherapy [90] | |
| CLL vs. B-NHL | miR-182, miR-199a*(5p), let-7 family, miR-424, miR-10a, miR-7, mir-126, miR-218, MiR-197, miR-595 , miR-483 | DR/UP | MiR-197 the most highly expressed miRNA, miR-595 and miR-483 also upregulated [58] |
| BL vs. B-NHL | miR-17-3p, miR-18a, miR-19a, miR-19b, miR-92 | UP | Up/Down-regulated in BL vs. NHL [58] |
| let-7 family, miR-29 (a, b, c), miR-155, miR-146a | DR | ||
| HL | miR-17-92 cluster members, miR-16, miR-21, miR-24, and miR-155.miR-150 | UP DR | The HL-specific miRNAs up-regulated [43]. Only miR-150 is significantly down-regulated in HL compared with NHL [43]. |
| cHL vs. HL EBV+ | miR-96, miR-128a, miR-128b | DR | Selectively down-regulated in HL lymph nodes of EBV+ HL patients [42] |
| cHL prognosis | miR-135a | Expression of miR-135a in HL lymph nodes correlates with clinical outcome. Patients with low miR-135a expression had a higher probability of relapse and a shorter disease- free survival. [55] | |
| miR-21, miR-30e, miR-30d and miR-92b | To identify two different risk groups for 5-year FFS [184]. |
7. Novel Therapeutic Strategies by Modulation of miRNAs
- Inhibition of tumor-inducing miRNAs: miRNAs as therapeutic targets;
- MiRNA replacement by re-introducing miRNAs with tumor suppressor functions: miRNAs as therapeutic agents.
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Han, J.J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP- dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar]
- Suárez, Y.; Fernández-Hernando, C.; Yu, J.; Gerber, S.A.; Harrison, K.D.; Pober, J.S.; Iruela-Arispe, M.L.; Merkenschlager, M.; Sessa, W.C. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 14082–14087. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar]
- Lawrie, C.H. MicroRNAs and lymphomagenesis: A functional review. Br. J. Haematol. 2013, 160, 571–581. [Google Scholar]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar]
- Witwer, K.W.; Sisk, J.M.; Gama, L.; Clements, J.E. MicroRNA regulation of IFN-beta protein expression: Rapid and sensitive modulation of the innate immune response. J. Immunol. 2010, 184, 2369–2376. [Google Scholar]
- Zhou, X.; Jeker, L.T.; Fife, B.T.; Zhu, S.; Anderson, M.S.; McManus, M.T.; Bluestone, J.A. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med. 2008, 205, 1983–1991. [Google Scholar]
- Ha, T.Y. The Role of MicroRNAs in Regulatory T Cells and in the Immune Response. Immune Netw. 2011, 11, 11–41. [Google Scholar]
- Zhu, S.; Pan, W.; Qian, Y. MicroRNA in immunity and autoimmunity. J. Mol. Med. 2013, 91, 1039–1050. [Google Scholar]
- Xiao, C.; Calado, D.P.; Galler, G.; Thai, T.H.; Patterson, H.C.; Wang, J.; Rajewsky, N.; Bender, T.P.; Rajewsky, K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007, 131, 146–159. [Google Scholar]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar]
- Yang, J.S.; Lai, E.C. Alternative miRNA Biogenesis Pathways and the Interpretation of Core miRNA Pathway Mutants Molecular. Mol. Cell 2011, 43, 892–903. [Google Scholar]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar]
- Kanellopoulou, C.; Muljo, S.A.; Kung, A.L.; Ganesan, S.; Drapkin, R.; Jenuwein, T.; Livingston, D.M.; Rajewsky, K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005, 19, 489–501. [Google Scholar]
- Fabbri, M.; Croce, C.M.; Calin, G.A. MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk. Lymphoma 2009, 50, 160–170. [Google Scholar]
- Calvo, J.; BenYoucef, A.; Baijer, J.; Rouyez, M.C.; Pflumio, F. Assessment of Human Multi-Potent Hematopoietic Stem/Progenitor Cell Potential Using a Single In Vitro Screening System. PLoS One 2012, 7, e50495. [Google Scholar]
- Kawamoto, H. A close developmental relationship between the lymphoid and myeloid lineages. Trends Immunol. 2006, 27, 169–175. [Google Scholar]
- Doulatov, S.; Notta, F.; Eppert, K.; Nguyen, L.T.; Ohashi, P.S.; Dick, J.K. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat. Immunol. 2010, 11, 585–593. [Google Scholar]
- Di Lisio, L.; Martinez, N.; Montes-Moreno, S.; Piris-Villaespesa, M.; Sanchez-Beato, M.; Piris, M.A. The role of miRNAs in the pathogenesis and diagnosis of B-cell lymphomas. Blood 2012, 120, 1782–1790. [Google Scholar]
- Koralov, S.B.; Muljo, S.A.; Galler, G.R.; Krek, A.; Chakraborty, T.; Kanellopoulou, C.; Jensen, K.; Cobb, B.S.; Merkenschlager, M.; Rajewsky, N.; et al. Dicer Ablation Affects Antibody Diversity and Cell Survival in the B Lymphocyte Lineage. Cell 2008, 132, 860–874. [Google Scholar]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and auto-immunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- Leone, G.; DeGregori, J.; Sears, R.; Jakoi, L.; Nevins, J.R. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 1997, 387, 422–426. [Google Scholar]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar]
- Woods, K.; Thomson, J.M.; Hammond, S.M. Direct regulation of an oncogenic micro- RNA cluster by E2F transcription factors. J. Biol. Chem. 2007, 282, 2130–2134. [Google Scholar] [CrossRef]
- Tagawa, H.; Karube, K.; Tsuzuki, S.; Ohshima, K.; Seto, M. Synergistic action of the microRNA-17 polycistron and Myc in aggressive cancer development. Cancer Sci. 2007, 98, 1482–1490. [Google Scholar]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009, 23, 2839–2849. [Google Scholar]
- Rao, E.; Jiang, C.; Ji, M.; Huang, X.; Iqbal, J.; Lenz, G.; Wright, G.; Staudt, L.M.; Zhao, Y.; McKeithan, T.W.; et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia 2012, 26, 1064–1072. [Google Scholar]
- Guil, S.; Caceres, J.F. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat. Struct. Mol. Biol. 2007, 14, 591–596. [Google Scholar] [CrossRef]
- Sylvestre, Y.; de Guire, V.; Querido, E.; Mukhopadhyay, U.K.; Bourdeau, V.; Major, F.; Ferbeyre, G.; Chartrand, P. An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem. 2007, 282, 2135–2143. [Google Scholar]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar]
- Roehle, A.; Hoefig, K.P.; Repsilber, D.; Thorns, C.; Ziepert, M.; Wesche, K.O.; Thiere, M.; Loeffler, M.; Klapper, W.; Pfreundschuh, M.; et al. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. Br. J. Haematol. 2008, 142, 732–744. [Google Scholar]
- Navarro, A.; Gaya, A.; Martinez, A.; Urbano-Ispizua, A.; Pons, A.; Balague, O.; Gel, B.; Abrisqueta, P.; Lopez-Guillermo, A.; Artells, R.; et al. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood 2008, 111, 2825–2832. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Tan, L.P.; Harms, G.; Schakel, R.N.; de Jong, D.; Blokzijl, T.; Möller, P.; Poppema, S.; Kroesen, B.J.; van den Berg, A. Hodgkin Lymphoma Cell Lines Are Characterized by a Specific miRNA Expression Profile. Neoplasia 2009, 2, 167–176. [Google Scholar]
- Robertus, J.L.; Kluiver, J.; Weggemans, C.; Harms, G.; Reijmers, R.M.; Swart, Y.; Kok, K.; Rosati, S.; Schuuring, E.; van Imhoff, G.; et al. MiRNA Profiling in B Non-Hodgkin Lymphoma: A MYC-Related miRNA Profile Characterizes Burkitt Lymphoma. Br. J. Haematol. 2010, 6, 896–899. [Google Scholar]
- Neilson, J.R.; Zheng, G.X.; Burge, C.B.; Sharp, P.A. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev. 2007, 21, 578–589. [Google Scholar]
- Georgantas, R.W., III; Hildreth, R.; Morisot, S.; Alder, J.; Liu, C.G.; Heimfeld, S.; Cali, G.A.; Croce, C.M.; Civin, C.I. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: A circuit diagram of differentiation control. Proc. Natl. Acad. Sci. USA 2007, 104, 2750–2755. [Google Scholar]
- Cozzolino, A.M.; Pedace, L.; Castori, M.; de Simone, P.; Preziosi, N.; Sperduti, I.; Panetta, C.; Mogini, V.; de Bernardo, C.; Morrone, A.; et al. Analysis of the miR-34a locus in 62 patients with familial cutaneous melanoma negative for CDKN2A/CDK4 screening. Fam. Cancer 2012, 11, 201–208. [Google Scholar]
- Pekarsky, Y.; Santanam, U.; Cimmino, A.; Palamarchuk, A.; Efanov, A.; Maximov, V.; Volinia, S.; Alder, H.; Liu, C.G.; Rassenti, L.; Calin, G.A.; et al. TCL1 expression in CLL is regulated by miR-29 and miR-181. Cancer Res. 2006, 66, 11590–11593. [Google Scholar]
- Teng, G.; Hakimpour, P.; Landgraf, P.; Rice, A.; Tuschl, T.; Casellas, R.; Papavasiliou, F.N. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 2008, 28, 621–629. [Google Scholar] [CrossRef]
- Monticelli, S.; Ansel, K.M.; Xiao, C.; Socci, N.D.; Krichevsky, A.M.; Thai, T.H.; Rajewsky, N.; Marks, D.S.; Sander, C.; Rajewsky, K.; et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 2005, 6, R71:1–R71:15. [Google Scholar]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. MiR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar]
- Tan, L.P.; Wang, M.; Robertus, J.L.; Schakel, R.N.; Gibcus, J.H.; Diepstra, A.; Harms, G.; Peh, S.C.; Reijmers, R.M.; Pals, S.T. miRNA profiling of B-cell subsets: Specific miRNA profile for germinal center B cells with variation between centroblasts and centrocytes. Lab. Investig. 2009, 89, 708–716. [Google Scholar]
- Lin, Y.C.; Kuo, M.W.; Yu, J.; Kuo, H.H.; Lin, R.J.; Lo, W.L.; Yu, A.L. c-Myb is an evolutionary conserved miR-150 target and miR-150/c-Myb interaction is important for embryonic development. Mol. Biol. Evol. 2008, 10, 2189–2198. [Google Scholar]
- Lawrie, C.H.; Ballabio, E.; Dyar, O.J.; Jones, M.; Ventura, R.; Chi, J.; Tramonti, D.; Gooding, S.; Boultwood, J.; Wainscoat, J.S.; et al. MicroRNA expression in chronic lymphocytic leukaemia. Br. J. Haematol. 2009, 147, 398–402. [Google Scholar]
- Navarro, A.; Diaz, T.; Martinez, A.; Gaya, A.; Pons, A.; Gel, B.; Codony, C.; Ferrer, G.; Martinez, C.; Montserrat, E.; et al. Regulation of JAK2 by miR-135a: Prognostic impact in classic Hodgkin lymphoma. Blood 2009, 114, 2945–2951. [Google Scholar]
- Boi, M.; Rinaldi, A.; Kwee, I.; Bonetti, P.; Todaro, M.; Tabbò, F.; Piva, R.; Rancoita, P.M.V.; Matolcsy, A.; Timar, B.; et al. RDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood 2013, 122, 2683–2693. [Google Scholar] [CrossRef]
- Di Lisio, L.; Gomez-Lopez, G.; Sanchez-Beato, M.; Gomez-Abad, C.; Rodriguez, M.E.; Villuendas, R.; Ferreira, B.I.; Carro, A.; Rico, D.; Mollejo, M.; et al. Mantle Cell Lymphoma: Transcriptional Regulation by microRNAs. Leukemia 2010, 7, 1335–1342. [Google Scholar]
- Di Lisio, L.; Sánchez-Beato, M.; Gómez-López, G.M.; Rodríguez, E.; Montes-Moreno, S.; Mollejo, M.; Menárguez, J.; Martínez, M.A.; Alves, F.J.; Pisano, D.G.; et al. MicroRNA Signatures in B-Cell lymphomas. Blood Cancer J. 2012, 2. [Google Scholar] [CrossRef]
- Almanza, G.; Fernandez, A.; Volinia, S.; Cortez-Gonzalez, X.; Croce, C.M.; Zanetti, M. Selected microRNAs define cell fate determination of murine central memory CD8 T-cells. PLoS One 2010, 5, e11243. [Google Scholar]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef]
- Thai, T.H.; Calado, D.P.; Casola, S.; Ansel, K.M.; Xiao, C.; Xue, Y.; Murphy, A.; Frendewey, D.; Valenzuela, D.; Kutok, J.L.; et al. Regulation of the germinal center response by microRNA-155. Science 2007, 316, 604–608. [Google Scholar]
- Kuchen, S.; Resch, W.; Yamane, A.; Kuo, N.; Li, Z.; Chakraborty, T.; Wei, L.; Laurence, A.; Yasuda, T.; Peng, S.; et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity 2010, 32, 828–839. [Google Scholar]
- Vigorito, E.; Perks, K.L.; Abreu-Goodger, C.; Bunting, S.; Xiang, Z.; Kohlhaas, S.; Das, P.P.; Miska, E.A.; Rodriguez, A.; Bradley, A.; et al. MicroRNA-155 regulates the generation of immunoglobulin class- switched plasma cells. Immunity 2007, 27, 847–859. [Google Scholar] [CrossRef]
- Natkunam, Y.; Lossos, I.S.; Taidi, B.; Zhao, S.; Lu, X.; Ding, F.; Hammer, A.S.; Marafioti, T.; Byrne, G.E.; Levy, S., Jr.; et al. Expression of the human germinal center-associated lymphoma (HGAL) protein, a new marker of germinal center B-cell derivation. Blood 2005, 105, 3979–3986. [Google Scholar]
- Rai, D.; Kim, S.W.; McKeller, M.R.; Dahia, P.L.; Aguiar, R.C. Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3111–3116. [Google Scholar]
- Rai, D.; Karanti, S.; Jung, I.; Dahia, P.L.; Aguiar, R.C. Coordinated expression of microRNA-155 and predicted target genes in diffuse large B-cell lymphoma. Cancer Genet. Cytogenet. 2008, 181, 8–15. [Google Scholar]
- Pedersen, I.M.; Otero, D.; Kao, E.; Miletic, A.V.; Hother, C.; Ralfkiaer, E.; Rickert, R.C.; Gronbaek, K.; David, M. Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol. Med. 2009, 1, 288–295. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Los-sos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar]
- Fruman, D.A. Phosphoinositide 3-kinase and its targets in B− cell and T− cell signaling. Curr. Opin. Immunol. 2004, 16, 314–320. [Google Scholar]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar]
- Kluiver, J.; Poppema, S.; de Jong, D.; Blokzijl, T.; Harms, G.; Jacobs, S.; Kroesen, B.J.; van den Berg, A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J. Pathol. 2005, 207, 243–249. [Google Scholar]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, 1862–1879. [Google Scholar]
- Dorsett, Y.; McBride, K.M.; Jankovic, M.; Gazumyan, A.; Thai, T.H.; Robbiani, D.F.; di Virgilio, M.; Reina San-Martin, B.; Heidkamp, G.; Schwickert, T.A.; et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 2008, 28, 630–638. [Google Scholar] [CrossRef]
- Kluiver, J.; Haralambieva, E.; de Jong, D.; Blokzijl, T.; Jacobs, S.; Kroesen, B.J.; Poppema, S.; van den Berg, A. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosom. Cancer 2006, 45, 147–153. [Google Scholar]
- Calin, G.A.; Cimmino, A.; Fabbri, M.; Ferracin, M.; Wojcik, S.E.; Shimizu, M.; Taccioli, C.; Zanesi, N.; Garzon, R.; Aqeilan, R.I.; et al. Mi-15a and miR-16–1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 5166–5171. [Google Scholar]
- Rao, D.S.; O’Connell, R.M.; Chaudhuri, A.A.; Garcia-Flores, Y.; Geiger, T.L.; Baltimore, D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010, 33, 48–59. [Google Scholar] [CrossRef]
- Hu, H.; Wang, B.; Borde, M.; Nardone, J.; Maika, S.; Allred, L.; Tucker, P.W.; Rao, A. Foxp1 is an essential transcriptional regulator of B cell development. Nat. Immunol. 2006, 7, 819–826. [Google Scholar]
- Sotillo, E.; Laver, T.; Mellert, H.; Schelter, J.M.; Cleary, M.A.; McMahon, S.; Thomas-Tikhonenko, A. Myc overexpression brings out unexpected antiapoptotic effects of miR-34a. Oncogene 2011, 30, 2587–2594. [Google Scholar]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C.; et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar]
- Merkel, O.; Asslaber, D.; Piñón, J.D.; Egle, A.; Greil, R. Interdependent regulation of p53 and miR-34a in chronic lymphocytic leukemia. Cell Cycle 2010, 9, 2764–2768. [Google Scholar]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Lin, J.; Lwin, T.; Zhao, J.J.; Tam, W.; Choi, Y.S.; Moscinski, L.C.; Dalton, W.S.; Sotomayor, E.M.; Wright, K.L.; Tao, J. Follicular dendritic cell-induced microRNA-mediated upregulation of PRDM1 and down-regulation of BCL-6 in non-Hodgkin’s B-cell lymphomas. Leukemia 2011, 25, 145–152. [Google Scholar]
- Nie, K.; Gomez, M.; Landgraf, P.; Garcia, J.F.; Liu, Y.; Tan, L.H.; Chadburn, A.; Tuschl, T.; Knowles, D.M.; Tam, W. MicroRNA-mediated down-regulation of PRDM1/Blimp-1 in Hodgkin/Reed-Sternberg cells: A potential pathogenetic lesion in Hodgkin lymphomas. Am. J. Pathol. 2008, 173, 242–252. [Google Scholar] [CrossRef]
- Lawrie, C.H.; Chi, J.; Taylor, S.; Tramonti, D.; Ballabio, E.; Palazzo, S.; Saunders, N.J.; Pezzella, F.; Boultwood, J.; Wainscoat, J.S.; et al. Expression of microRNAs in diffuse large B cell lymphoma is associated with immunophenotype, survival and transformation from follicular lymphoma. J. Cell. Mol. Med. 2009, 13, 1248–1260. [Google Scholar]
- Nie, K.; Zhang, T.; Allawi, H.; Gomez, M.; Liu, Y.; Chadburn, A.; Wang, Y.L.; Knowles, D.M.; Tam, W. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: A potential role of the microRNA let-7. Am. J. Pathol. 2010, 177, 1470–1479. [Google Scholar]
- Desjobert, C.; Renalier, M.H.; Bergalet, J.; Dejean, E.; Joseph, N.; Kruczynski, A.; Soulier, J.; Espinos, E.; Meggetto, F.; Cavaillé, J.; et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 2011, 117, 6627–6637. [Google Scholar]
- Calin, GA.; Ferracin, M.; Cimmino, A.; di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar]
- Santanam, U.; Zanesi, N.; Efanov, A.; Costinean, S.; Palamarchuk, A.; Hagan, J.P.; Volinia, S.; Alder, H.; Rassenti, L.; Kipps, T.; et al. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 12210–12215. [Google Scholar] [CrossRef]
- Havelange, V.; Garzon, R. MicroRNAs: Emerging Key Regulators of Hematopoiesis. Am. J. Hematol. 2010, 85, 935–942. [Google Scholar]
- Thomas, M.D.; Kremer, C.S.; Ravichandran, K.S.; Bender, T.P. c-Myb is critical for B cell development and maintenance of follicular B cells. Immunity 2005, 23, 275–286. [Google Scholar]
- Merkerova, M.; Belickova, M.; Bruchova, H. Differential expression of microRNAs in hematopoietic cell lineages. Eur. J. Haematol. 2008, 81, 304–310. [Google Scholar] [CrossRef]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17–92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009, 28, 2719–2732. [Google Scholar]
- Craig, V.J.; Cogliatti, S.B.; Imig, J.; Renner, C.; Neuenschwander, S.; Rehrauer, H.; Schlapbach, R.; Dirnhofer, S.; Tzankov, A.; Muller, A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 2011, 117, 6227–6236. [Google Scholar] [CrossRef]
- Belver, L.; de Yebenes, V.G.; Ramiro, A.R. MicroRNAs prevent the generation of autoreactive antibodies. Immunity 2010, 33, 713–722. [Google Scholar]
- Tam, W.; Ben-Yehuda, D.; Hayward, W.S. Bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell. Biol. 1997, 17, 1490–1502. [Google Scholar]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar]
- Dagan, L.N.; Jiang, X.; Bhatt, S.; Cubedo, E.; Rajewsky, K.; Lossos, I.S. miR-155 regulates HGAL expression and increases lymphoma cell motility. Blood 2012, 119, 513–520. [Google Scholar]
- Lu, X.; Chen, J.; Malumbres, R.; Cubedo Gil, E.; Helfman, D.M.; Lossos, I.S. HGAL, a lymphoma prognostic biomarker, interacts with the cytoskeleton and mediates the effects of IL-6 on cell migration. Blood 2007, 110, 4268–4277. [Google Scholar]
- Pasqualucci, L.; Compagno, M.; Houldsworth, J.; Monti, S.; Grunn, A.; Nandula, S.V.; Aster, J.C.; Murty, V.V.; Shipp, M.A.; Dalla-Favera, R. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J. Exp. Med. 2006, 203, 311–317. [Google Scholar]
- Tam, W.; Gomez, M.; Chadburn, A.; Lee, J.W.; Chan, W.C.; Knowles, D.M. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 2006, 107, 4090–4100. [Google Scholar]
- Liu, J.; Wu, C.P.; Lu, B.F.; Jiang, J.T. Mechanism of T cell regulation by microRNAs. Cancer Biol. Med. 2013, 10, 131–137. [Google Scholar]
- Wu, H.; Neilson, J.R.; Kumar, P.; Manocha, M.; Shankar, P.; Sharp, P.A.; Manjunath, N. MiRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2007, 2, e1020. [Google Scholar]
- Cobb, B.S.; Nesterova, T.B.; Thompson, E.; Hertweck, A.; O’Connor, E.; Godwin, J.; Wilson, C.B.; Brockdorff, N.; Fisher, A.G.; Smale, S.T.; et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J. Exp. Med. 2005, 201, 1367–1373. [Google Scholar]
- Wolfer, A.; Bakker, T.; Wilson, A.; Nicolas, M.; Ioannidis, V.; Littman, D.R.; Lee, P.P.; Wilson, C.B.; Held, W.; MacDonald, H.R.; et al. Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8 T cell development. Nat. Immunol. 2001, 2, 235–241. [Google Scholar]
- Muljo, S.A.; Ansel, K.M.; Kanellopoulou, C.; Livingston, D.M.; Rao, A.; Rajewsky, K.; Aberrant, T. Cell differentiation in the absence of Dicer. J. Exp. Med. 2005, 202, 261–269. [Google Scholar]
- Tian, L.; de Hertogh, G.; Fedeli, M.; Staats, K.A.; Schonefeldt, S.; Humblet-Baron, S.; Liston, A. Loss of T cell microRNA provides systemic protection against autoimmune pathology in mice. J. Autoimmun. 2012, 38, 39–48. [Google Scholar]
- Paul, W.E.; Seder, R.A. Lymphocyte responses and cytokines. Cell 1994, 76, 241–251. [Google Scholar]
- Cobb, B.S.; Hertweck, A.; Smith, J.; O’Connor, E.; Graf, D.; Cook, T.; Smale, S.T.; Sakaguchi, S.; Livesey, F.J.; Fisher, A.G.; et al. A role for Dicer in immune regulation. J. Exp. Med. 2006, 203, 2519–2527. [Google Scholar]
- Chong, M.M.; Rasmussen, J.P.; Rudensky, A.Y.; Littman, D.R. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J. Exp. Med. 2008, 205, 2005–2017. [Google Scholar]
- Liston, A.; Lu, L.F.; O’Carroll, D.; Tarakhovsky, A.; Rudensky, A.Y. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J. Exp. Med. 2008, 205, 1993–2004. [Google Scholar]
- Zhou, L.; Seo, K.H.; Wong, H.K.; Mi, Q.S. MicroRNAs and immune regulatory T cells. Int. Immunopharmacol. 2009, 9, 524–527. [Google Scholar]
- Nakayama, T.; Kasprowicz, D.J.; Yamashita, M.; Schubert, L.A.; Gillard, G.; Kimura, M.; Didierlaurent, A.; Koseki, H.; Ziegler, S.F. The generation of mature, single-positive thymocytes in vivo is dysregulated by CD69 blockade or overexpression. J. Immunol. 2002, 168, 87–94. [Google Scholar]
- Ziętara, N.; Łyszkiewicz, M.; Witzlau, K.; Naumann, R.; Hurwitz, R.; Langemeier, J.; Bohne, J.; Sandrock, I.; Ballmaier, M.; Weiss, S.; et al. Critical role for miR-181a/b-1 in agonist selection of invariant natural killer T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7407–7412. [Google Scholar]
- Henao-Mejia, J.; Williams, A.; Goff, L.A.; Staron, M.; Licona-Limón, P.; Kaech, S.M.; Nakayama, M.; Rinn, J.L.; Flavell, R.A. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity 2013, 38, 984–997. [Google Scholar]
- Li, Q.J.; Chau, J.; Ebert, P.J.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. MiR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 2007, 129, 147–161. [Google Scholar]
- Belvera, L.; Papavasilioub, N.F.; Ramiro, A.R. MicroRNA control of lymphocyte differentiation and function. Curr. Opin. Immunol. 2011, 23, 368–373. [Google Scholar]
- Ghisi, M.; Corradin, A.; Basso, K.; Frasson, C.; Sera-fin, V.; Mukherjee, S.; Mussolin, L.; Ruggero, K.; Bonanno, L.; Guffanti, A.; et al. Modulation of microRNA expression in human T-cell development: Targeting of NOTCH3 by miR-150. Blood 2011, 117, 7053–7062. [Google Scholar]
- Bellavia, D.; Campese, A.F.; Vacca, A.; Gulino, A.; Screpanti, I. Notch3, another Notch in T-cell development. Semin. Immunol. 2003, 15, 107–112. [Google Scholar]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; di Mario, G.; et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar]
- Campese, A.F.; Bellavia, D.; Gulino, A.; Screpanti, I. Notch signaling at the crossroads of T-cell devel- opment and leukemogenesis. Semin. Cell. Dev. Biol. 2003, 14, 151–157. [Google Scholar]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar]
- Li, L.; Zhang, J.; Diao, W.; Wang, D.; Wei, Y.; Zhang, C.; Zen, K. MicroRNA-155 and MicroRNA-21 Promote the Expansion of Functional Myeloid-Derived Suppressor Cells. J. Immunol. 2014, 192, 1034–1043. [Google Scholar]
- Das, L.M.; Torres-Castillo, M.D.; Gill, T.; Levine, A.D. TGF-beta conditions intestinal T cells to express increased levels of miR-155, associated with down-regulation of IL-2 and itk mRNA. Mucosal Immunol. 2013, 6, 167–176. [Google Scholar]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar]
- Wu, T.; Wieland, A.; Araki, K.; Davis, C.W.; Ye, L.; Hale, J.S.; Ahmed, R. Temporal expression of microRNA cluster miR-17–92 regulates effector and memory CD8+ T-cell differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 9965–9970. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragilesites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Osada, H.; Takahashi, T. MicroRNAs in biological processes and carcinogenesis. Carcinogenesis 2007, 28, 2–12. [Google Scholar]
- Huppi, K.; Volfovsky, N.; Mackiewicz, M.; Runfola, T.; Jones, T.L.; Martin, S.E.; Stephens, R.; Caplen, N.J. MicroRNAs and genomic instability. Semin. Cancer Biol. 2007, 17, 65–73. [Google Scholar]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebet, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Calin, C.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar]
- Fulci, V.; Chiaretti, S.; Goldoni, M.; Azzalin, G.; Carucci, N.; Tavolaro, S.; Castellano, L.; Magrelli, A.; Citarella, F.; Messina, M.; et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 2007, 109, 4944–4951. [Google Scholar]
- Lawrie, C.H.; Soneji, S.; Marafioti, T.; Cooper, C.D.; Palazzo, S.; Paterson, J.C.; Cattan, H.; Enver, T.; Mager, R.; Boultwood, J.; et al. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int. J. Cancer 2007, 121, 1156–1161. [Google Scholar]
- Fang, C.; Zhu, D.X.; Dong, H.J.; Zhou, Z.J.; Wang, Y.H.; Liu, L.; Fan, L.; Miao, K.R.; Liu, P.; Xu, W.; et al. Serum microRNAs are promising novel biomarkers for diffuse large B cell lymphoma. Ann. Hematol. 2012, 91, 553–559. [Google Scholar]
- Thompson, R.C.; Herscovitch, M.; Zhao, I.; Ford, T.J.; Gilmore, T.D. NF-kappaB down-regulates expression of the B-lymphoma marker CD10 through a miR-155/PU. 1 pathway. J. Biol. Chem. 2011, 286, 1675–1682. [Google Scholar]
- Costinean, S.; Sandhu, S.K.; Pedersen, I.M.; Tili, E.; Trotta, R.; Perrotti, D.; Ciarlariello, D.; Neviani, P.; Harb, J.; Kauffman, L.R.; et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of micro-MiR-155 transgenic mice. Blood 2009, 114, 1374–1382. [Google Scholar]
- Huang, X.; Shen, Y.; Liu, M.; Bi, C.; Jiang, C.; Iqbal, J.; McKeithan, T.W.; Chan, W.C.; Ding, S.J.; Fu, K. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am. J. Pathol. 2012, 181, 26–33. [Google Scholar]
- Haasch, D.; Chen, Y.W.; Reilly, R.M.; Chiou, X.G.; Koterski, S.; Smith, M.L.; Kroeger, P.; McWeeny, K.; Halbert, D.N.; Mollison, K.W.; et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell. Immunol. 2002, 217, 78–86. [Google Scholar]
- Meltzer, P.S. Cancer genomics: Small RNAs with big impacts. Nature 2005, 435, 745–746. [Google Scholar]
- Van den Berg, A.; Kroesen, B.J.; Kooistra, K.; de Jong, D.; Briggs, J.; Blokzijl, T.; Jacobs, S.; Kluiver, J.; Diepstra, A.; Maggio, E.; et al. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosom. Cancer 2003, 37, 20–28. [Google Scholar]
- Tam, W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene 2001, 274, 157–167. [Google Scholar]
- Tam, W.; Hughes, S.H.; Hayward, W.S.; Besmer, P. Avian bic, a gene isolated from a common retroviral site in avian leukosis virus-induced lymphomas that encodes a noncoding RNA, cooperates with c-myc in lymphomagenesis and erythroleukemogenesis. J. Virol. 2002, 76, 4275–4286. [Google Scholar]
- Metzler, M.; Wilda, M.; Busch, K.; Viehmann, S.; Borkhardt, A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt Lymphoma. Genes Chromosom. Cancer 2004, 39, 167–169. [Google Scholar]
- Gatto, G.; Rossi, A.; Rossi, D.; Kroening, S.; Bonatti, S.; Mallardo, M. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB path- way. Nucleic Acids Res. 2008, 36, 6608–6619. [Google Scholar]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar]
- Zhao, Y.; Xu, H.; Yao, Y.; Smith, L.P.; Kgosana, L.; Green, J.; Petherbridge, L.; Baigent, S.J.; Nair, V. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek’s disease lymphomas. PLoS Pathog. 2011, 7, e1001305. [Google Scholar]
- Jiang, P.; Rao, E.Y.; Meng, N.; Zhao, Y.; Wang, J.J. MicroRNA-17–92 significantly enhances radioresistance in human mantle cell lymphoma cells. Radiat. Oncol. 2010, 5, 100. [Google Scholar]
- Lawrie, C.H.; Gal, S.; Dunlop, H.M.; Pushkaran, B.; Liggins, A.P.; Pulford, K.; Banham, A.H.; Pezzella, F.; Boultwood, J.; Wainscoat, J.S.; et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2008, 141, 672–675. [Google Scholar]
- Chen, W.; Wang, H.; Chen, H.; Liu, S.; Lu, H.; Kong, D.; Huang, X.; Kong, Q.; Lu, Z. Clinical significance and detection of microRNA-21 in serum of patients with diffuse large B-cell lymphoma in Chinese Population. Eur. J. Haematol. 2014, 92, 407–412. [Google Scholar]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar]
- Rossi, S.; Shimizu, M.; Barbarotto, E.; Nicoloso, M.S.; Dimitri, F.; Sampath, D.; Fabbri, M.; Lerner, S.; Barron, L.L.; Rassenti, L.Z.; et al. MicroRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood 2010, 116, 945–952. [Google Scholar]
- Medina, P.P.; Nolde, M.; Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86–90. [Google Scholar]
- Schramedei, K.; Morbt, N.; Pfeifer, G.; Lauter, J.; Rosolowski, M.; Tomm, J.M.; von Bergen, M.; Horn, F.; Brocke-Heidrich, K. MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4. Oncogene 2011, 30, 2975–2985. [Google Scholar]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar]
- Yamanaka, Y.; Tagawa, H.; Takahashi, N.; Watanabe, A.; Guo, Y.M.; Iwamoto, K.; Yamashita, J.; Saitoh, H.; Kameoka, Y.; Shimizu, N.; et al. Aberrant over-expression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood 2009, 114, 3265–3275. [Google Scholar]
- Cmarik, J.L.; Min, H.; Hegamyer, G.; Zhan, S.; Kulesz-Martin, M.; Yoshinaga, H.; Matsuhashi, S.; Colburn, N.H. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc. Natl. Acad. Sci. USA 1999, 96, 14037–14042. [Google Scholar]
- Leupold, J.H.; Yang, H.S.; Colburn, N.H.; Asangani, I.; Post, S.; Allgayer, H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene 2007, 26, 4550–4562. [Google Scholar]
- Bullrich, F.; Fujii, H.; Calin, G.; Mabuchi, H.; Negrini, M.; Pekarsky, Y.; Rassenti, L.; Alder, H.; Reed, J.C.; Keating, M.J.; et al. Characterization of the 13q14 tumor suppressor locus in CLL: Identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001, 61, 6640–6648. [Google Scholar]
- Pfeifer, D.; Pantic, M.; Skatulla, I.; Rawluk, J.; Kreutz, C.; Martens, U.M.; Fisch, P.; Timmer, J.; Veel-ken, H. Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays. Blood 2007, 109, 1202–1210. [Google Scholar]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar]
- Fabbri, M.; Bottoni, A.; Shimizu, M.; Spizzo, R.; Nicoloso, M.S.; Rossi, S.; Barbarotto, E.; Cimmino, A.; Adair, B.; Wojcik, S.E.; et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. J. Am. Med. Assoc. 2011, 305, 59–67. [Google Scholar]
- Kitada, S.; Andersen, J.; Akar, S.; Zapata, J.M.; Takayama, S.; Krajewski, S.; Wang, H.G.; Zhang, X.; Bullrich, F.; Croce, C.M.; et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: Correlations with in vitro and in vivo chemoresponses. Blood 1998, 91, 3379–3389. [Google Scholar]
- Adachi, M.; Tefferi, A.; Greipp, P.R.; Kipps, T.J.; Tsujimoto, Y. Preferential linkage of Bcl-2 to immunoglobulin light chain gene in chronic lymphocytic leukemia. J. Exp. Med. 1990, 171, 559–564. [Google Scholar]
- Ouillette, P.; Erba, H.; Kujawski, L.; Kaminski, M.; Shedden, K.; Malek, S.N. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res. 2008, 68, 1012–1021. [Google Scholar]
- Liu, Y.; Corcoran, M.; Rasool, O.; Ivanova, G.; Ibbotson, R.; Grander, D.; Iyengar, A.; Baranova, A.; Kashuba, V.; Merup, M.; et al. Cloning of two candidate tumor suppressor genes within a 10 kb region on chromosome 13q14, frequently deleted in chronic lymphocytic leukemia. Oncogene 1997, 15, 2463–2473. [Google Scholar]
- Lia, M.; Carette, A.; Tang, H.; Shen, Q.; Mo, T.; Bhagat, G.; Dalla-Favera, R.; Klein, U. Functional dissection of the chromosome 13q14 tumor-suppressor locus using transgenic mouse lines. Blood 2012, 119, 2981–2990. [Google Scholar]
- Klein, U.; Lia, M.; Crespo, M.; Siegel, R.; Shen, Q.; Mo, T.; Ambesi-Impiombato, A.; Califano, A.; Migliazza, A.; Bhagat, G.; et al. The DLEU2/miR-15a/16–1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010, 17, 28–40. [Google Scholar]
- Brown, J.R.; Lawrence, M.S.; Hanna, M.; Tesar, B.; Stojanov, P.; Vartanov, A.R.; Fernandes, S.M.; Sougnez, C.; MacConaill, L.; Pochet, N.; et al. Novel germline genetic variants associated with familial chronic lymphocytic leukemia (CLL). In Proceedings of the 53rd ASH Annual Meeting and Exposition, San Diego, CA, USA, 10–13 December 2011. 118, Abstract 465.
- Chen, R.W.; Bemis, L.T.; Amato, C.M.; Myint, H.; Tran, H.; Birks, D.K.; Eckhardt, S.G.; Robinson, W.A. Truncation in CCND1 mRNA alters miR-16–1 regulation in mantle cell lymphoma. Blood 2008, 112, 822–829. [Google Scholar]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16–1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar]
- Dejean, E.; Renalier, M.H.; Foisseau, M.; Agirre, X.; Joseph, N.; de Paiva, G.R.; Meggetto, F. Hypoxia-microRNA-16 downregulation induces VEGF expression in anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphomas. Leukemia 2011, 25, 1882–1890. [Google Scholar]
- Bertoni, F.; Rinaldi, A.; Zucca, E.; Cavalli, F. Review Update on the molecular biology of mantle cell lymphoma. Hematol. Oncol. 2006, 24, 22–27. [Google Scholar]
- Chan, T.O.; Rittenhouse, S.E.; Tsichlis, P.N. AKT/PKB and other D3 phosphoinositide-regulated kinases: Kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biol.-Chem. 1999, 68, 965–1014. [Google Scholar]
- Herling, M.; Patel, K.A.; Khalili, J.; Schlette, E.; Kobayashi, R.; Medeiros, L.J.; Jones, D. TCL1 shows a regulated expression pattern in chronic lymphocytic leukemia that correlates with molecular subtypes and proliferative state. Leukemia 2006, 20, 280–285. [Google Scholar]
- Said, J.W.; Hoyer, K.K.; French, S.W.; Rosenfelt, L.; Garcia-Lloret, M.; Koh, P.J.; Cheng, T.C.; Sulur, G.G.; Pinkus, G.S.; Kuehl, W.M.; et al. TCL1 oncogene expression in B cell subsets from lymphoid hyperplasia and distinct classes of B cell lymphoma. Lab. Investig. 2001, 81, 555–564. [Google Scholar]
- Yan, X.J.; Albesiano, E.; Zanesi, N.; Yancopoulos, S.; Sawyer, A.; Romano, E.; Petlickovski, A.; Efremov, D.G.; Croce, C.M.; Chiorazzi, N. B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 11713–11718. [Google Scholar]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar]
- Chim, C.S.; Wong, K.Y.; Qi, Y.; Loong, F.; Lam, W.L.; Wong, L.G.; Jin, D.Y.; Costello, J.F.; Liang, R. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 2010, 31, 745–750. [Google Scholar]
- Sánchez-Espiridión, B.; Martin-Moreno, A.M.; Montalbán, C.; Figueroa, V.; Vega, F.; Younes, A.; Medeiros, L.J.; Alvés, F.J.; Canales, M.; Estévez, M.; et al. MicroRNA signatures and treatment response in patients with advanced classical Hodgkin lymphoma. Br. J. Haematol. 2013, 162, 336–347. [Google Scholar]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of MYC-Mediated miR-29 by HDAC3 and EZH2 as a Therapeutic Target of Histone Modification in Aggressive B Cell Lymphoma. Cancer Cell 2012, 4, 506–523. [Google Scholar]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar]
- Stamatopoulos, B.; Meuleman, N.; Haibe-Kains, B.; Saussoy, P.; van den Neste, E.; Michaux, L.; Heimann, P.; Martiat, P.; Bron, D.; Lagneaux, L. MicroRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood 2009, 113, 5237–5245. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, bio-genesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Kipps, T.J. Chronic lymphocytic leukemia and related diseases. In Williams Hematology; Beutler, E., Lichtman, M.A., Coller, B.S., Kipps, T.J., Seligson, U., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 1163–1194. [Google Scholar]
- Alencar, A.J.; Malumbres, R.; Kozloski, G.A.; Advani, R.; Talreja, N.; Chinichian, S.; Briones, J.; Natkunam, Y.; Sehn, L.H.; Gascoyne, R.D.; et al. MicroRNAs are independent predictors of outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Clin. Cancer Res. 2011, 15, 4125–4135. [Google Scholar]
- Jones, K.; Nourse, J.P.; Keane, C.; Bhatnagar, A.; Ganghi, M.K. Plasma microRNA are disease response biomarkers in classical Hodgkin lymphoma. Clin. Cancer Res. 2014, 20, 253–264. [Google Scholar]
- Westin, J.R.; Fayad, L.E. Beyond R-CHOP and the IPI in large-cell lymphoma: Molecular markers as an opportunity for stratification. Curr. Hematol. Malig. Rep. 2009, 4, 218–224. [Google Scholar]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31. [Google Scholar] [CrossRef]
- Fabani, M.M.; Abreu-Goodger, C.; Williams, D.; Lyons, P.A.; Torres, A.G.; Smith, K.G.; Enright, A.J.; Gait, M.J.; Vigorito, E. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010, 38, 4466–4475. [Google Scholar]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar]
- Fontana, L.; Fiori, M.E.; Albini, S.; Cifaldi, L.; Giovinazzi, S.; Forloni, M.; Boldrini, R.; Donfrancesco, A.; Federici, V.; Giacomini, P.; et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and Bim. PLoS One 2008, 3, e2236. [Google Scholar]
- Pereira, D.M.; Rodrigues, P.M.; Borralho, P.M.; Rodrigues, C.M. Delivering the promise of miRNA cancer therapeutics. Drug Discov. Today 2013, 18, 282–289. [Google Scholar]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 1695–1704. [Google Scholar]
- Craig, V.J.; Tzankov, A.; Flori, M.; Schmid, C.A.; Bader, A.G.; Muller, A. Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia 2012, 26, 2421–2424. [Google Scholar]
- Zhao, J.; Kelnar, K.; Bader, A.G. In-Depth Analysis Shows Synergy between Erlotinib and miR-34a. PLoS One 2014, 9, e89105. [Google Scholar]
- Bader, A.G. MiR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3. [Google Scholar] [CrossRef]
- Roman-Gomez, J.; Agirre, X.; Jimenez-Velasco, A.; Arqueros, V.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Martin-Subero, I.; Garate, L.; Cordeu, L.; San José-Eneriz, E.; et al. Epigenetic regulation of microRNAs in acute lympho-blastic leukemia. J. Clin. Oncol. 2009, 27, 1316–1322. [Google Scholar]
- Agirre, X.; Vilas-Zornoza, A.; Jimenez-Velasco, A.; Martin-Subero, J.I.; Cordeu, L.; Garate, L.; San Jose-Eneriz, E.; Abizanda, G.; Rodriguez-Otero, P.; Fortes, P.; et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009, 69, 4443–4453. [Google Scholar]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of micro- RNA-127 with down-regulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar]
- Parekh, S.; Polo, J.M.; Shaknovich, R.; Juszczynski, P.; Lev, P.; Ranuncolo, S.M.; Melnick, A. BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood 2007, 110, 2067–2074. [Google Scholar]
- Hummel, R.; Hussey, D.J.; Haier, J. MicroRNAs: Predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur. J. Cancer 2010, 46, 298–311. [Google Scholar]
- Chun-Zhi, Z.; Lei, H.; An-Ling, Z.; Yan-Chao, F.; Xiao, Y.; Guang-Xiu, W.; Zhi-Fan, J.; Pei-Yu, P.; Qing-Yu, Z.; Chun-Sheng, K. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer 2010, 10. [Google Scholar] [CrossRef]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Tullio, G.; De Fazio, V.; Sgherza, N.; Minoia, C.; Serratì, S.; Merchionne, F.; Loseto, G.; Iacobazzi, A.; Rana, A.; Petrillo, P.; et al. Challenges and Opportunities of MicroRNAs in Lymphomas. Molecules 2014, 19, 14723-14781. https://doi.org/10.3390/molecules190914723
De Tullio G, De Fazio V, Sgherza N, Minoia C, Serratì S, Merchionne F, Loseto G, Iacobazzi A, Rana A, Petrillo P, et al. Challenges and Opportunities of MicroRNAs in Lymphomas. Molecules. 2014; 19(9):14723-14781. https://doi.org/10.3390/molecules190914723
Chicago/Turabian StyleDe Tullio, Giacoma, Vincenza De Fazio, Nicola Sgherza, Carla Minoia, Simona Serratì, Francesca Merchionne, Giacomo Loseto, Angela Iacobazzi, Antonello Rana, Patrizia Petrillo, and et al. 2014. "Challenges and Opportunities of MicroRNAs in Lymphomas" Molecules 19, no. 9: 14723-14781. https://doi.org/10.3390/molecules190914723
APA StyleDe Tullio, G., De Fazio, V., Sgherza, N., Minoia, C., Serratì, S., Merchionne, F., Loseto, G., Iacobazzi, A., Rana, A., Petrillo, P., Silvestris, N., Iacopino, P., & Guarini, A. (2014). Challenges and Opportunities of MicroRNAs in Lymphomas. Molecules, 19(9), 14723-14781. https://doi.org/10.3390/molecules190914723