
α-Linolenic Acid, A Nutraceutical with Pleiotropic Properties That Targets Endogenous Neuroprotective Pathways to Protect against Organophosphate Nerve Agent-Induced Neuropathology

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. α-Linolenic Acid—An Essential Nutraceutical
2. ALA Protects against Animal Models of NMDA Receptor-Mediated Excitotoxicity
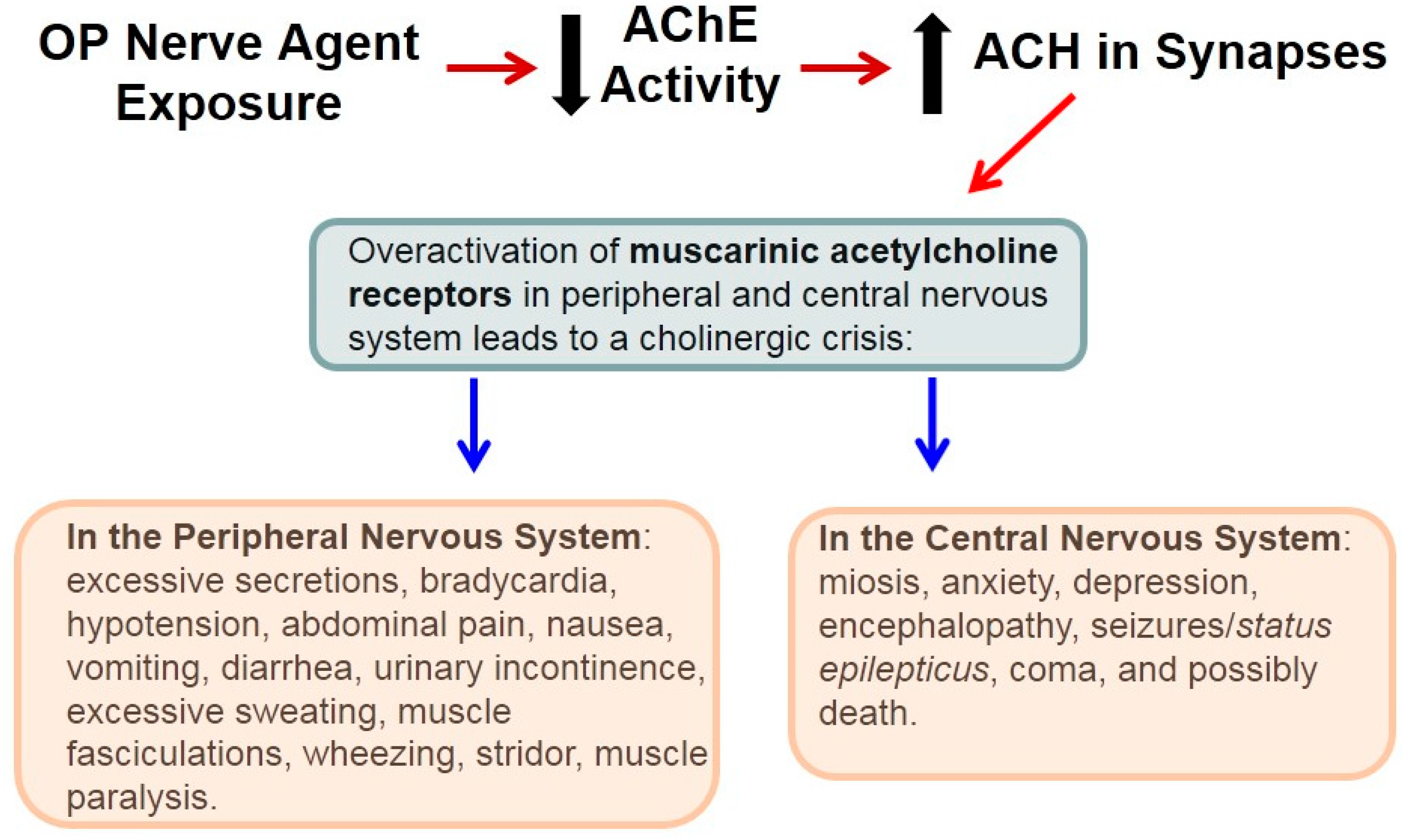
3. Organophosphate (OP) Nerve Agent-Induced Excitotoxicity and the Limited Availability of Neuroprotective Therapies

4. Lifesaving Treatment and OP Nerve Agents
5. Vulnerability to OP Nerve Agents Is Brain-Region Specific
6. The Nutraceutical α-Linolenic Acid Is a Potent Neuroprotective Agent against Soman-Induced Neuropathology

7. Cognitive Deficits and Neurodegeneration
8. The Nutraceutical ALA Improves Cognitive Function and Exerts an Anti-Depressant Effect after Soman Exposure in Vivo
9. The Amygdala and Hippocampus, Two Brain Regions Profoundly Damaged by Soman
10. OP Nerve Agents, Cognitive Deficits and Neurogenesis
11. ALA Induces Diverse and Mechanistically Distinct Endogenous Neuroprotective Pathways

12. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Tinoco, J. Dietary requirements and functions of α-linolenic acid in animals. Prog. Lipid Res. 1982, 21, 1–45. [Google Scholar] [CrossRef]
- Crupi, R.; Marino, A.; Cuzzocrea, S. N-3 Fatty acids: Role in neurogenesis and neuroplasticity. Curr. Med. Chem. 2013, 20, 2953–2963. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, N.; Nguemeni, C.; Debruyne, D.N.; Piens, M.; Wu, X.; Pan, H.; Hu, X.; Gandin, C.; Lipsky, R.H.; Plumier, J.C.; et al. Subchronic α-linolenic acid treatment enhances brain plasticity and exerts an antidepressant effect: A versatile potential therapy for stroke. Neuropsychopharmacology 2009, 34, 2548–2559. [Google Scholar] [CrossRef]
- Piermartiri, T.C.; Pan, H.; Chen, J.; McDonough, J.; Grunberg, N.; Apland, J.P.; Marini, A.M. α-Linolenic acid-induced increase in neurogenesis is a key factor in the improvement in the passive avoidance task after soman exposure. Neuromol. Med. 2015, 17, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Ganguli, S.; Menard, C.; Liede, A.C.; Hamadeh, M.J.; Chen, Z.Y.; Wolever, T.M.; Jenkins, D.J. High α-linolenic acid flaxseed (Linum usitatissimum): Some nutritional properties in humans. Br. J. Nutr. 1993, 69, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Nam, Y.A.; Kim, H.S.; Hayes, A.W.; Lee, B.M. α-Linolenic acid: Nutraceutical, pharmacological and toxicological evaluation. Food Chem. Toxicol. 2014, 70, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, M.; Pignatti, C.; Carpigiani, M.; Tarozzi, G.; Turchetto, E. Effect of C 18:3 (n-3) dietary supplementation on the fatty acid composition of the rat brain. Acta Vitaminol. Enzymol. 1984, 6, 151–156. [Google Scholar] [PubMed]
- Plourde, M.; Cunnane, S.C. Extremely limited synthesis of long chain polyunsaturates in adults: Implications for their dietary essentiality and use as supplements. Appl. Physiol. Nutr. Metab. 2007, 32, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Salem, N., Jr. Whole body distribution of deuterated linolenic and α-linolenic acids and their metabolites in the rat. J. Lipid Res. 2007, 48, 2709–2724. [Google Scholar] [CrossRef] [PubMed]
- Emken, E. Human studies using isotope labeled-fatty acids: Answered and unanswered questions. J. Oleo Sci. 2013, 62, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Anding, R.H.; Hwang, D.H. Effects of dietary linolenate on the fatty acid composition of brain lipids in rats. Lipids 1986, 21, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Saitoh, M.; Moriuchi, A.; Nomura, M.; Okuyama, H. Effect of dietary α-linolenate/linoleate balance on brain lipid compositions and learning ability of rats. J. Lipid Res. 1987, 28, 144–151. [Google Scholar] [PubMed]
- Bourre, J.M.; Francois, M.; Youyou, A.; Dumont, O.; Piciotti, M.; Pascal, G.; Durand, G. The effects of dietary α-linolenic acid on the composition of nerve membranes, enzymatic activity, amplitude of electrophysiological parameters, resistance to poisons and performance of learning tasks in rats. J. Nutr. 1989, 119, 1880–1892. [Google Scholar] [PubMed]
- Blondeau, N.; Lipsky, R.H.; Bourourou, M.; Duncan, M.W.; Gorelick, P.B.; Marini, A.M. α-linolenic acid: An omega-3 fatty acid with neuroprotectivge properties—Ready for use in the stroke clinic? BioMed. Res. Int. 2015, 2015, 519830. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Reilly, J.A.; Lysko, P.G.; Henneberry, R.C. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988, 451, 205–212. [Google Scholar] [CrossRef]
- Lipsky, R.H.; Xu, K.; Zhu, D.; Kelly, C.; Terhakopian, A.; Novelli, A.; Marini, A.M. NF-κB is a critical determinant for NMDA receptor-mediated neuroprotection. J. Neurochem. 2001, 78, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Blondeau, N.; Heurteaux, C.; Widmann, C.; Romey, G.; Lazdunski, M. Polyunsaturated fatty acids are potent neuroprotectors. EMBO J. 2000, 19, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, N.; Widmann, C.; Lazdunski, M.; Heurteaux, C. Activation of the nuclear factor-κB is a key event in brain tolerance. J. Neurosci. 2001, 21, 4668–4677. [Google Scholar] [PubMed]
- Pan, H.; Hu, X.Z.; Jacobowitz, D.M.; Chen, C.; McDonough, J.; van Shura, K.; Lyman, M.; Marini, A.M. A-linolenicacid is a potent neuroprotective agent against soman-induced neuropathology. Neurotoxicology 2012, 33, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Marini, A.M.; Jiang, X.; Wu, X.; Pan, H.; Guo, Z.; Mattson, M.P.; Blondeau, N.; Novelli, A.; Lipsky, R.H. Preconditioning and neurotrophins: A model for brain adaptation to seizures, ischemia and other stressful stimuli. Amino Acids 2007, 32, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Culmsee, C.; Yu, Z.; Camandola, S. Roles of nuclear factor κB in neuronal survival and plasticity. J. Neurochem. 2000, 74, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Meffert, M.K.; Baltimore, D. Physiological functions for brain NF-κB. Trends Neurosci. 2005, 28, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Meffert, M.K. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Boersma, M.C.; Dresselhaus, E.C.; de Biase, L.M.; Mihalas, A.B.; Bergles, D.E.; Meffert, M.K. A requirement for nuclear factor-κB in developmental and plasticity-associated synaptogenesis. J. Neurosci. 2011, 31, 5414–5425. [Google Scholar] [CrossRef] [PubMed]
- Lang-Lazdunski, L.; Blondeau, N.; Jarretou, G.; Lazdunski, M.; Heurteaux, C. Linolenic acid prevents neuronal cell death and paraplegia after transient spinal cord ischemia in rats. J. Vasc. Surg. 2003, 38, 564–575. [Google Scholar] [CrossRef]
- Heurteaux, C.; Guy, N.; Laigle, C.; Blondeau, N.; Duprat, F.; Mazzuca, M.; Lang-Lazdunski, L.; Widmann, C.; Zanzouri, M.; Romey, G.; et al. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004, 23, 2684–2695. [Google Scholar] [CrossRef] [PubMed]
- Heurteaux, C.; Laigle, C.; Blondeau, N.; Jarretou, G.; Lazdunski, M. α-linolenic acid and riluzole treatment confer cerebral protection and improve survival after focal brain ischemia. Neuroscience 2006, 137, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Nguemeni, C.; Delplanque, B.; Rovère, C.; Simon-Rousseau, N.; Gandin, C.; Agnani, G.; Nahon, J.L.; Heurteaux, C.; Blondeau, N. Dietary supplementation of α-linolenic acid in an enriched rapeseed oil diet protects from stroke. Pharmacol. Res. 2010, 61, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Thiermann, H.; Szinicz, L.; Eyer, P.; Felgenhauer, N.; Zilker, T.; Worek, F. Lessons to be learnt from organophosphorus pesticide poisoning for the treatment of nerve agent poisoning. Toxicology 2007, 233, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Takasu, N.; Ishimatsu, S.; Miyanoki, S.; Mitsuhashi, A.; Kumada, K.; Tanaka, K.; Hinohara, S. Report on 640 victims of the Tokyo subway sarin attack. Ann. Emerg. Med. 1996, 28, 129–135. [Google Scholar] [CrossRef]
- Nozaki, H.; Aikawa, N.; Shinozawa, Y.; Hori, S.; Fujishima, S.; Takuma, K.; Sagoh, M. Sarin poisoning in Tokyo subway. Lancet 1995, 345, 980–981. [Google Scholar] [PubMed]
- Szinicz, L. History of chemical and biological warfare agents. Toxicology 2005, 214, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Syrian gas attack reinforces need for better anti-sarin drugs. Nat. Med. 2013, 19, 1194–1195. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.B.; Kryger, G.; Ordentlich, A.; Greenblatt, H.M.; Harel, M.; Raves, M.L.; Segall, Y.; Barak, D.; Shafferman, A.; Silman, I.; et al. Crystal structures of aged phosphonylated acetylcholinesterase: Nerve agent reaction products at the atomic level. Biochemistry 1999, 38, 7032–7039. [Google Scholar] [CrossRef] [PubMed]
- Wiener, S.W.; Hoffman, R.S. Nerve agents: A comprehensive review. J. Intensive Care Med. 2004, 19, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; de Gubareff, T.; Labruyere, J. Seizure-related brain damage induced by cholinergic agents. Nature 1983, 301, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Pazdernik, T.L.; Cross, R.S.; Giesler, M.; Samson, F.E.; McDonough, J., Jr. Delayed effect of soman: Brain glucose use and pathology. Neurotoxicology 1985, 6, 61–70. [Google Scholar] [PubMed]
- Shih, T.-M.; Duniho, S.M.; McDonough, J.H. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol. Appl. Pharmacol. 2003, 188, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Aroniadou-Anderjaska, V.; Figueiredo, T.H.; Apland, J.P.; Qashu, F.; Braga, M.F.M. Primary brain targets of nerve agents: The role of the amygdala in comparison to the hippocampus. Neurotoxicology 2009, 30, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.K.; Sheridan, R.D.; Green, A.C.; Scott, I.R.; Tattersall, J.E. A guinea pig hippocampal slice model of organophosphate-induced seizure activity. J. Pharmacol. Exp. Ther. 2004, 310, 678–686. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.H., Jr.; Shih, T.M. Pharmacological modulation of soman-induced seizures. Neurosci. Biobehav. Rev. 1993, 17, 203–215. [Google Scholar] [CrossRef]
- Lallement, G.; Carpentier, P.; Pernot-Marino, I.; Baubichon, D.; Collet, A.; Blanchet, G. Involvement of the different rat hippocampal glutamatergic receptors in development of seizures induced by soman: An autoradiographic study. Neurotoxicology 1991, 12, 655–664. [Google Scholar] [PubMed]
- Lallement, G.; Carpentier, P.; Collet, A.; Pernot-Marino, I.; Baubichon, D.; Blanchet, G. Effects of soman-induced seizures on different extracellular amino acid levels and on glutamate uptake in rat hippocampus. Brain Res. 1991, 563, 234–240. [Google Scholar] [CrossRef]
- Wade, J.V.; Samson, F.E.; Nelson, S.R.; Pazdernik, T.L. Changes in extracellular amino acids during soman- and kainic acid-induced seizures. J. Neurochem. 1987, 49, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Grasshoff, C.; Gillessen, T.; Thiermann, H.; Wagner, E.; Szinicz, L. The effect of acetylcholinesterase inhibition on depolarization-induced GABA release from rat striatal slices. Toxicology 2003, 1184, 149–156. [Google Scholar] [CrossRef]
- Santos, M.D.; Pereira, E.F.; Aracava, Y.; Castro, N.G.; Fawcett, W.P.; Randall, W.R.; Albuquerque, E.X. Low concentrations of pyridostigmine prevent soman-induced inhibition of GABAergic transmission in the central nervous system: Involvement of muscarinic receptors. J. Pharmacol. Exp. Ther. 2003, 304, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Solberg, Y.; Belkin, M. The role of excitotoxicity in organophosphorous nerve agents central poisoning. Trends Pharmacol. Sci. 1997, 18, 183–185. [Google Scholar] [CrossRef]
- Carpentier, P.; Lambrinidis, M.; Blanchet, G. Early dendritic changes in hippocampal pyramidal neurones (field CA1) of rats subjected to acute soman intoxication: A light microscopic study. Brain Res. 1991, 541, 293–299. [Google Scholar] [CrossRef]
- McDonough, H.J., Jr.; Shih, T.-M. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci. Biobehav. Rev. 1997, 21, 559–579. [Google Scholar] [CrossRef]
- Wood, S.J.; Tattersall, J.E. An improved brain slice model of nerve agent-induced seizure activity. J. Appl. Toxicol. 2001, 21 (Suppl. 1), S83–S86. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Hisaoka, T.; Naito, T.; Isonuma, H.; Okumura, S.; Miura, K.; Maekawa, H.; Ishimatsu, S.; Takasu, N.; Suzuki, K. Acute and chronic effects of sarin exposure from the Tokyo subway incident. Environ. Toxicol. Pharmacol. 2005, 19, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Araki, S.; Murata, K.; Nishikitani, M.; Okumura, T.; Ishimatsu, S.; Takasu, N.; White, R.F. Chronic neurobehavioral effects of Tokyo subway sarin poisoning in relation to posttraumatic stress disorder. Arch. Environ. Health 1998, 53, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Yamasue, H.; Abe, O.; Kasai, K.; Suga, M.; Iwanami, A.; Yamada, H.; Tochigi, M.; Ohtani, T.; Rogers, M.A.; Sasaki, T.; et al. Human brain structural change related to acute single exposure to sarin. Ann. Neurol. 2007, 61, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Haley, R.; Kurt, T. Neuropsychological correlates of Gulf War syndrome. Arch. Clin. Neuropsychol. 1997, 12, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Proctor, S.P.; Heaton, K.J.; Heeren, T.; White, R.F. Effects of sarin and cyclosarin exposure during the 1991 Gulf War on neurobehavioral functioning in US army veterans. Neurotoxicology 2006, 27, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.; Eisenkraft, A.; Finkelstein, A.; Schein, O.; Rotman, E.; Dushnitsky, T. A decade after the Tokyo sarin attack: A review of neurological follow-up of the victims. Mil. Med. 2007, 172, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, T.; Iwanami, A.; Kasai, K.; Yamasue, H.; Kato, T.; Sasaki, T.; Kato, N. Post-traumatic stress disorder symptoms in victims of Tokyo subway attack: A 5-year follow-up study. Psychiatry Clin. Neurosci. 2004, 58, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, K.; Nishiwaki, Y.; Maekawa, K.; Ogawa, Y.; Asukai, N.; Yoshimura, K.; Etoh, N.; Matsumoto, Y.; Kikuchi, Y.; Kumagai, N.; et al. Effects of sarin on the nervous system of subway workers seven years after the Tokyo subway sarin attack. J. Occup. Health 2005, 47, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.D.; Umbach, D.M.; Hoppin, J.A.; Richards, M.; Alavanja, M.C.; Blair, A.; Sandler, D.P.; Kamel, F. Pesticide exposure and depression among male private pesticide applicators in the agricultural health study. Environ. Health Perspect. 2014, 122, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.M.; Mergler, D.; Huel, G.; Cone, J.E. Psychological, psychosocial, psychophysiological sequelae in a community affected by a railroad chemical disaster. J. Trauma. Stress 1994, 7, 601–624. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanni, C., Jr. Domestic terrorism with chemical or biological agents: Psychiatric aspects. Am. J. Psychiatry 1999, 156, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.M.; Brewin, C.R.; Curran, H.V.; Furlong, C.E.; Abraham-Smith, K.M.; Harrison, V. Neuropsychological and psychiatric functioning in sheep farmers exposed to low levels of organophosphate pesticides. Neurotoxicol. Teratol. 2010, 12, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Toomey, R.; Alpern, R.; Vasterling, J.J.; Baker, D.G.; Reda, D.J.; Lyons, M.J.; Henderson, W.G.; Kang, H.K.; Eisen, S.A.; Murphy, F.M. Neuropsychological functioning of U.S. Gulf War veterans 10 years after the war. J. Int. Neuropsychol. Soc. 2009, 15, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Bowers, M.B.; Goodman, E.; Sim, V.M. Some behavioral changes in man following anti-cholinesterase administration. J. Nerv. Ment. Dis. 1964, 138, 383–389. [Google Scholar] [PubMed]
- Chao, L.L.; Rothlind, J.C.; Cardenas, V.A.; Meyerhoff, J.D.; Weiner, M.W. Effects of low-level exposure to sarin and cycloarin during the 1991 Gulf War on brain function and brain structure in US veterans. Neurotoxicology 2010, 31, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.L.; Abadjian, L.; Hlavin, J.; Meyerhoff, D.J.; Weiner, M.W. Effects of low-level sarin and cyclosarin exposure and Gulf War Illness on brain structure and function: A study at 4T. Neurotoxicology 2011, 32, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Brandeis, R.; Raveh, L.; Grunwald, J.; Cohen, E.; Ashani, Y. Prevention of soman-induced cognitive deficits by pretreatment with human butyrylcholinesterase in rats. Pharmacol. Biochem. Behav. 1993, 46, 889–896. [Google Scholar] [CrossRef]
- Filliat, P.; Baubichon, D.; Burckhart, M.F.; Pernot-Marino, I.; Foquin, A.; Masqueliez, C.; Perrichon, C.; Carpentier, P.; Lallement, G. Memory impairment after soman intoxication in rat: Correlation with central neuropathology. Improvement with anticholinergic and antiglutamatergic therapeutics. Neurotoxicology 1999, 20, 535–549. [Google Scholar] [PubMed]
- Filliat, P.; Coubard, S.; Pierard, C.; Liscia, P.; Beracochea, D.; Four, E.; Baubichon, D.; Masqueliez, C.; Lallement, G.; Collombet, J.M. Long-term behavioral consequences of soman poisoning in mice. Neurotoxicology 2007, 28, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.; Kunis, G.; Berkutzki, T.; Ronen, A.; Krivoy, A.; Yoles, E.; Last, D.; Mardor, Y.; van Shura, K.; McFarland, E.; et al. Immunomodulation by poly-YE reduces organophosphate-induced brain damage. Brain Behav. Immun. 2012, 26, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Joosen, M.J.; Jousma, E.; van den Boom, T.M.; Kuijpers, W.C.; Smit, A.B.; Lucassen, P.J.; van Helden, H.P. Long-term cognitive deficits accompanied by reduced neurogenesis after soman poisoning. Neurotoxicology 2009, 30, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Buccafusco, J.J.; Heithold, D.L.; Chon, S.H. Long-term behavioral and learning abnormalities produced by the irreversible cholinesterase inhibitor soman: Effect of a standard pretreatment regimen and clonidine. Toxicol. Lett. 1990, 52, 319–329. [Google Scholar] [CrossRef]
- Choi, E.K.; Park, D.; Yon, J.M.; Hur, G.H.; Ha, Y.C.; Che, J.H.; Kim, J.; Shin, S.; Jang, J.Y.; Hwang, S.Y.; et al. Protection by sustained release of physostigmine and procyclidine of soman poisoning in rats. Eur. J. Pharmacol. 2004, 505, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Piermartiri, T.C.; Chen, J.; McDonough, J.; Oppel, C.; Driwech, W.; Winter, K.; McFarland, E.; Black, K.; Figueiredo, T.; et al. Repeated administration of α-linolenic acid exerts neuroprotective efficacy, an anti-depressant effect and improves cognitive performance when given after soman exposure. Neurotoxicology 2015, 51, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Moffett, M.C.; Schultz, M.K.; Schwartz, J.E.; Stone, M.F.; Lumley, L.A. Impaired auditory and contextual fear conditioning in soman-exposed rats. Pharmacol. Biochem. Behav. 2011, 98, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Coubard, S.; Béracochéa, D.; Collombet, J.M.; Philippin, J.N.; Krazem, A.; Liscia, P.; Lallement, G.; Piérard, C. Long-term consequences of soman poisoning in mice: Part 2. Emotional behavior. Behav. Brain Res. 2008, 191, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.L.; Wright, L.K.; Connis, N.; Lumley, L.A. Characterizing the behavioral effects of nerve agent-induced seizure activity in rats: Increased startle reactivity and perseverative behavior. Pharmacol. Biochem. Behav. 2012, 100, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Prager, E.M.; Aroniadou-Anderjaska, V.; Almeida-Suhett, C.P.; Figueiredo, T.H.; Apland, J.P.; Rossetti, F.; Olsen, C.H.; Braga, M.F. The recovery of acetylcholinesterase activity and the progression of neuropathological and pathophysiological alterations in the rat basolateral amygdala after soman-induced status epilepticus: Relation to anxiety-like behavior. Neuropharmacology 2014, 81, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Lallement, G.; Denoyer, M.; Collet, A.; Pernot-Marino, I.; Baubichon, D.; Monmaur, P.; Blanchet, G. Changes in hippocampal acetylcholine and glutamate extracellular levels during soman-induced seizures; influence of septal cholinoceptive cells. Neurosci. Lett. 1992, 139, 104–107. [Google Scholar] [CrossRef]
- McDonough, J.H.; McMonagle, J.; Copeland, T.; Zoeffel, D.; Shih, T.-M. Comparative evaluation of benzodiazepines for control of soman-induced seizures. Arch. Toxicol. 1999, 73, 473–478. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.H.; Dochterman, L.W.; Smith, C.D.; Shih, T.-M. Protection against nerve agent-induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology 1995, 15, 123–132. [Google Scholar]
- McDonough, J.H.; Zoeffel, L.D.; McMonagle, J.; Copeland, T.L.; Smith, C.D.; Shih, T.-M. Anticonvulsant treatment of nerve agent seizures anticholinergics versus diazepam in soman-intoxicated guinea pigs. Epilepsy Res. 2000, 38, 1–14. [Google Scholar] [CrossRef]
- Clement, J.G.; Broxup, B. Efficacy of diazepam and avizafone against soman-induced neuropathology in brain of rats. Neurotoxicology 1993, 14, 485–504. [Google Scholar] [PubMed]
- Apland, J.P.; Aroniadou-Anderjaska, V.; Figueiredo, T.H.; Rossetti, F.; Miller, S.L.; Braga, M.F. The limitations of diazepam as a treatment for nerve agent-induced seizures and neuropathology in rats: Comparison with UBP302. J. Pharmacol. Exp. Ther. 2014, 351, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, F.; de Araujo Furtado, M.; Pak, T.; Bailey, K.; Shields, M.; Chanda, S.; Addis, M.; Robertson, B.D.; Moffett, M.; Lumley, L.A.; et al. Combined diazepam and HDAC inhibitor treatment protects against seizures and neuronal damage caused by soman exposure. Neurotoxicology 2012, 33, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.W.; Jaax, N.K.; Graham, J.S.; McLeod, C.G., Jr. Cardiomyopathy in soman and sarin intoxicated rats. Toxicol. Lett. 1987, 36, 243–249. [Google Scholar] [CrossRef]
- De Araujo Furtado, M.; Lumley, L.A.; Robison, C.; Tong, L.C.; Lichtenstein, S.; Yourick, D.L. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia 2010, 51, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Apland, J.P.; Aroniadou-Anderjaska, V.; Braga, M.F. Soman induces ictogenesis in the amygdala and interictal activity in the hippocampus that are blocked by a GluR5 kainate receptor antagonist in vitro. Neuroscience 2009, 159, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Baille, V.; Clarke, P.G.; Brochier, G.; Dorandeu, F.; Verna, J.M.; Four, E.; Lallement, G.; Carpentier, P. Soman-induced convulsions: The neuropathology revisited. Toxicology 2005, 215, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.G.; Shinmei, S.S.; Cai, B. Seizure-induced neuronal necrosis: Implications for programmed cell death mechanisms. Epilepsia 2000, 41 (Suppl. 6), S9–S13. [Google Scholar] [CrossRef] [PubMed]
- Lallement, G.; Delamanche, I.S.; Pernot-Marino, I.; Baubichon, D.; Denoyer, M.; Carpentier, P.; Blanchet, G. Neuroprotective activity of glutamate receptor antagonists against soman-induced hippocampal damage: Quantification with an omega 3 site ligand. Brain Res. 1993, 618, 227–237. [Google Scholar] [CrossRef]
- Raveh, L.; Chapman, S.; Cohen, G.; Alkalay, D.; Gilat, E.; Rabinovitz, I.; Weissman, B.A. The involvement of the NMDA receptor complex in the protective effect of anticholinergic drugs against soman poisoning. Neurotoxicology 1999, 20, 551–559. [Google Scholar] [PubMed]
- Dematteis, M.; Mallaret, M.; Baubichon, D.; Pernot-Marino, I.; Lallement, G. Evaluation of dextromethorphan and dextrorphan as a preventive treatment of soman toxicity in mice. Neurosci. Lett. 1997, 234, 91–94. [Google Scholar] [CrossRef]
- White, H.S.; Alex, A.B.; Pollock, A.; Hen, N.; Shekh-Ahmad, T.; Wilcox, K.S.; McDonough, J.H.; Stables, J.P.; Kaufmann, D.; Yagen, B.; et al. A new derivative of valproic acid amide possesses a broad-spectrum antiseizure profile and unique activity against status epilepticus and organophosphate neuronal damage. Epilepsia 2012, 53, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Shekh-Ahmad, T.; Hen, N.; McDonough, J.H.; Yagen, B.; Bialer, M. Valnoctamide and sec-butyl-propylacetamide (SPD) for acute seizures and status epilepticus. Epilepsia 2013, 54 (Suppl. 6), 99–102. [Google Scholar] [CrossRef] [PubMed]
- Gunosewoyo, H.; Tipparaju, S.K.; Pieroni, M.; Wang, Y.; Doctor, B.P.; Nambiar, M.P.; Kozikowski, A.P. Structural analogs of huperzine A improve survival in guinea pigs exposed to soman. Bioorg. Med. Chem. Lett. 2013, 23, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Myhrer, T.; Enger, S.; Mariussen, E.; Aas, P. Two medical therapies very effective shortly after high levels of soman poisoning in rats, but only one with universal utility. Toxicology 2013, 314, 221–228. [Google Scholar] [CrossRef]
- Schmued, L.C.; Stowers, C.C.; Scallet, A.C.; Xu, L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005, 1035, 24–31. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Crook, J.E.; Meschia, J.F.; Brott, T.G.; Dickson, D.W.; McKinney, M. Aging blunts ischemic-preconditioning-induced neuroprotection following transient global ischemia in rats. Curr. Neurovasc. Res. 2005, 2, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, J.F.; Schmued, L.C. Fluoro-Ruby labeling prior to an amphetamine neurotoxic insult shows a definitive massive loss of dopaminergic terminals and axons in the caudate-putamen. Brain Res. 2006, 1075, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, J.F.; Ali, S. High doses of methamphetamine that cause disruption of the blood-brain barrier in limbic regions produce extensive neuronal degeneration in mouse hippocampus. Synapse 2006, 60, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Park, J.E.; Kang, L.; Ko, S.Y.; Jung, K.H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 1118, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, T.H.; Aroniadou-Anderjaska, V.; Qashu, F.; Apland, J.P.; Pidoplichko, V.; Stevens, D.; Ferrara, T.M.; Braga, M.F. Neuroprotective efficacy of caramiphen against soman and mechanisms of its action. Br. J. Pharmacol. 2011, 164, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Schmued, L.C.; Sarkar, S.; Paule, M.G.; Raymick, B. One-step labeling of degenerative neurons in unfixed brain tissue samples using Fluoro-Jade C. J. Neurosci. Methods 2012, 208, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Haley, R.; Kurt, T. Self-reported exposure to neurotoxic chemical combinations in the Gulf War: A cross-sectional epidemiologic study. JAMA 1997, 277, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, Y.; Maekawa, K.; Ogawa, Y.; Asukai, N.; Minami, M.; Omae, K.; Sarin Health Effects Study Group. Effects of sarin on the nervous system in rescue team staff members and police officers 3 years after the Tokyo subway sarin attack. Environ. Health Perspect. 2001, 109, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Baze, W.B. Soman-induced morphological changes: An overview in the non-human primate. J. Appl. Toxicol. 1993, 13, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.; Delamanche, I.S.; le Bert, M.; Blanchet, G.; Bouchaud, C. Seizure-related opening of the blood-brain barrier induced by soman: Possible correlation with the acute neuropathology observed in poisoned rats. Neurotoxicology 1990, 11, 493–508. [Google Scholar] [PubMed]
- Prager, E.M.; Aroniadou-Anderjaska, V.; Almeida-Suhett, C.P.; Figueiredo, T.H.; Apland, J.P.; Braga, M.F. Acetylcholinesterase inhibition in the basolateral amygdala plays a key role in the induction of status epilepticus after soman exposure. Neurotoxicology 2013, 38, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Myers, T.M.; Langston, J.L. Diet composition exacerbates or attenuates soman toxicity in rats: Implied metabolic control of nerve agent toxicity. Neurotoxicology 2011, 32, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Philippens, I.H.; Melchers, B.P.; de Groot, D.M.; Wolthuis, O.L. Behavioral performance, brain histology, and EEG sequela after immediate combined atropine/diazepam treatment of soman-intoxicated rats. Pharmacol. Biochem. Behav. 1992, 42, 711–719. [Google Scholar] [CrossRef]
- Pernot, F.; Carpentier, P.; Baille, V.; Testylier, G.; Beaup, C.; Foquin, A.; Filliat, P.; Liscia, P.; Coutan, M.; Piérard, C.; et al. Intrahippocampal cholinesterase inhibition induces epileptogenesis in mice without evidence of neurodegenerative events. Neuroscience 2009, 162, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Rodriguiz, R.M.; Wetsel, W.C. Assessments of cognitive deficits in mutuant mice. In Animal Models of Cognitive Impairment; Levin, E.D., Buccafusco, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Cryan, J.F.; Markou, A.; Lucki, I. Assessing antidepressant activity in rodents: Recent developments and future needs. Trends Pharmacol. Sci. 2002, 23, 238–245. [Google Scholar] [CrossRef]
- Porsolt, R.D.; Bertin, A.; Jalfre, M. Behavioral despair in mice: A primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 1977, 229, 327–336. [Google Scholar] [PubMed]
- Toomey, R.; Kang, H.K.; Karlinsky, J.; Baker, D.G.; Vasterling, J.J.; Alpern, R.; Reda, D.J.; Henderson, W.G.; Murphy, F.M.; Eisen, S.A. Mental health of US Gulf War veterans 10 years after the war. Br. J. Psychiatry 2007, 190, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Bazylewicz-Walczak, B.; Majczakowa, W.; Szymczak, M. Behavioral effects of occupational exposure to organophosphorous pesticides in female greenhouse planting workers. Neurotoxicology 1999, 20, 819–826. [Google Scholar] [PubMed]
- Campbell, S.; MacQueen, G. An update on regional brain volume differences associated with mood disorders. Curr. Opin. Psychiatry 2006, 19, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.; Mackay, D.; Landau, S.; Kerwin, R.; Everall, I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch. Gen. Psychiatry 2001, 58, 543–553. [Google Scholar] [CrossRef]
- Tizabi, Y.; Hauser, S.R.; Tyler, K.Y.; Getachew, B.; Madani, R.; Sharma, Y.; Manaye, K.F. Effects of nicotine on depressive-like behavior and hippocampal volume of female WKY rats. Prog. Neuropsycholopharmacol. Biol. Psychiatry 2010, 34, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Heaton, K.J.; Palumbo, C.L.; Proctor, S.P.; Killiany, R.J.; Yurgelun-Todd, D.A.; White, R.F. Quantitative magnetic resonance brain imaging in US army veterans of the 1991 Gulf War potentially exposed to sarin and cyclosarin. Neurotoxicology 2007, 28, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.L.; Kriger, S.; Buckley, S.; Ng, P.; Mueller, S.G. Effects of low-level sarin and cyclosarin exposure on hippocampal subfields in Gulf War Veterans. Neurotoxicology 2014, 44, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. Is impaired neurogenesis relevant to the affective symptoms of depression? Biol. Psychiatry 2004, 56, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, D.; Yasui, Y.; Yamada, K.; Ohara, N.; Okuyama, H. Biochemical responses to dietary α-linolenic acid restriction proceed differently among brain regions in mice. Biomed. Res. 2011, 32, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Mirzaei, F.; O’Reilly, E.J.; Pan, A.; Willett, W.C.; Kawachi, I.; Koenen, K.; Ascherio, A. Dietary intake of n-3 and n-6 fatty acids and the risk of clinical depression in women: A 10-years prospective follow-up study. Am. J. Clin. Nutr. 2011, 93, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Hibbeln, J.R. Fish consumption and major depression. Lancet 1998, 351, 1213. [Google Scholar] [CrossRef]
- Peet, M.; Murphy, B.; Shay, J.; Horrobin, D. Depletion of omega-3 fatty acid levels in red blood cell membranes of depressive patients. Biol. Psychiatry 1998, 43, 315–319. [Google Scholar] [CrossRef]
- Peet, M.; Stokes, C. Omega-3 fatty acids in the treatment of psychiatric disorders. Drugs 2005, 65, 1041–1059. [Google Scholar] [CrossRef]
- Venna, V.R.; Deplanque, D.; Allet, C.; Belarbi, K.; Hamdane, M.; Bordet, R. PUFA induce antidepressant-like effects in parallel to structural and molecular changes in the hippocampus. Psychoneuroendocrinology 2009, 34, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, F.; Galfalvy, H.; Chen, S.; Huang, Y.Y.; Cooper, T.B.; Burke, A.K.; Oquendo, M.A.; Mann, J.J.; Sublette, M.E. DNA methylation perturbations in genes involved in polyunsaturated Fatty Acid biosynthesis associated with depression and suicide risk. Front Neurol. 2015, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [PubMed]
- Aloyz, R.; Fawcett, J.P.; Kaplan, D.R.; Murphy, R.A.; Miller, F.D. Activity-dependent activation of TrkB neurotrophin receptors in the adult CNS. Learn Mem 1999, 6, 216–231. [Google Scholar] [PubMed]
- Thoenen, H. Neurotrophins and neuronal plasticity. Science 1995, 270, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Katoh-Semba, R.; Asano, T.; Ueda, H.; Morishita, R.; Takeuchi, I.K.; Inaguma, Y.; Kato, K. Riluzole enhances expression of brain-derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. FASEB J. 2002, 16, 1328–1330. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Shirayama, Y.; Chen, A.C.; Nakagawa, S.; Russell, D.S.; Duman, R.S. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 2002, 22, 3251–3261. [Google Scholar] [PubMed]
- Deltheil, T.; Guiard, B.P.; Cerdan, J.; David, D.J.; Tanaka, K.F.; Repérant, C.; Guilloux, J.P.; Coudoré, F.; Hen, R.; Gardier, A.M. Behavioral and serotonergic consequences of decreasing or increasing hippocampus brain-derived neurotrophic factor protein levels in mice. Neuropharmacology 2008, 55, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Võikar, V.; Rantamäki, T. Role of neurotrophic factors in depression. Curr. Opin. Pharmacol. 2007, 7, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, J.S.; Castrén, E. Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front. Behav. Neurosci. 2014, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.D.; Duman, R.S. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology 2010, 35, 2378–2391. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Barrot, M.; Autry, A.E.; Theobald, D.; Monteggia, L.M. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol. Psychiatry 2008, 63, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Saarelainen, T.; Hendolin, P.; Lucas, G.; Koponen, E.; Sairanen, M.; MacDonald, E.; Agerman, K.; Haapasalo, A.; Nawa, H.; Aloyz, R.; et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 2003, 23, 349–357. [Google Scholar] [PubMed]
- Phelps, E.A. Human emotion and memory: Interactions of the amygdala and hippocampal complex. Curr. Opin. Neurobiol. 2004, 14, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.L.; Wickelgren, W.O. Hippocampal ablation and passive avoidance. Science 1962, 138, 1104–1106. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, R. The hippocampus and contextual retrieval of information from memory: A theory. Behav. Biol. 1974, 12, 421–444. [Google Scholar] [CrossRef]
- Riekkinen, P., Jr.; Riekkinen, M.; Sirviö, J. Cholinergic drugs regulate passive avoidance performance via the amygdala. J. Pharmacol. Exp. Ther. 1993, 267, 1484–1492. [Google Scholar] [PubMed]
- Ghiasvand, M.; Rezayof, A.; Zarrindast, M.R.; Ahmadi, S. Activation of cannabinoid CB1 receptors in the central amygdala impairs inhibitory avoidance memory consolidation via NMDA receptors. Neurobiol. Learn Mem 2011, 96, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.; László, K.; Lukács, E.; Lénárd, L. Intraamygdaloid microinjection of acylated-ghrelin influences passive avoidance learning. Behav. Brain Res. 2009, 202, 308–311. [Google Scholar] [CrossRef] [PubMed]
- László, K.; Tóth, K.; Kertes, E.; Péczely, L.; Ollmann, T.; Madarassy-Szücs, A.; Lénárd, L. The role of neurotensin in passive avoidance learning in the rat central nucleus of amygdala. Behav. Brain Res. 2012, 226, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.J.; Tilson, H.A.; DeHaven, D.L.; Mailman, R.B.; Fisher, A.; Hanin, I. AF64A, a cholinergic neurotoxin, selectively depletes acetylcholine in hippocampus and cortex, and produces long-term passive avoidance and radial-arm maze deficits in the rat. Brain Res. 1984, 21, 91–102. [Google Scholar] [CrossRef]
- Dobryakova, Y.V.; Gurskaya, O.Y.; Markevich, V.A. Administration of nicotinic receptor antagonists during the period of memory consolidation affects passive avoidance learning and modulates synaptic efficiency in the CA1 region in vivo. Neuroscience 2015, 284, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Maia, G.H.; Quesado, J.L.; Soares, J.I.; do Carmo, J.M.; Andrade, P.A.; Andrade, J.P.; Lukoyanov, N.V. Loss of hippocampal neurons after kainate treatment correlates with behavioral deficits. PLoS ONE 2014, 9, e84722. [Google Scholar] [CrossRef] [PubMed]
- Collombet, J.M.; Piérard, C.; Béracochéa, D.; Coubard, S.; Burckhart, M.F.; Four, E.; Masqueliez, C.; Baubichon, D.; Lallement, G. Long-term consequences of soman poisoning in mice Part 1. Neuropathology and neuronal regeneration in the amygdala. Behav. Brain Res. 2008, 191, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Collombet, J.M.; Carpentier, P.; Baille, V.; Four, E.; Bernabe, D.; Burckhart, M.-F.; Masqueliez, C.; Baubichon, D.; Lallement, G. Neuronal regeneration partially compensates the delayed neuronal cell death observed in the hippocampal CA1 field of soman-poisoned mice. Neurotoxicology 2006, 27, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Collombet, J.M.; Four, E.; Bernabé, D.; Masqueliez, C.; Burckhart, M.F.; Baille, V.; Baubichon, D.; Lallement, G. Soman poisoning increases neural progenitor proliferation and induces long-term glial activation in mouse brain. Toxicology 2005, 208, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Saxe, M.D.; Battaglia, F.; Wang, J.W.; Malleret, G.; David, D.J.; Monckton, J.E.; Garcia, A.D.; Sofroniew, M.V.; Kandel, E.R.; Santarelli, L.; et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2006, 103, 17501–17506. [Google Scholar] [CrossRef] [PubMed]
- Gheusi, G.; Cremer, H.; McLean, H.; Chazal, G.; Vincent, J.D.; Lledo, P.M. Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc. Natl. Acad. Sci. USA 2000, 97, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Cameron, H.A.; McKay, R.D. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol. 2001, 435, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Kempermann, G.; Palmer, T.D.; Peterson, D.A.; Ray, J. Multipotent progenitor cells in the adult dentate gyrus. J. Neurobiol. 1998, 36, 249–266. [Google Scholar] [CrossRef]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Schinder, A.F.; Christie, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Merz, K.; Herold, S.; Lie, D.C. CREB in adult neurogenesis—Master and partner in the development of adult-born neurons? Eur. J. Neurosci. 2011, 33, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, M.; Lucas, G.; Ernfors, P.; Castrén, M.; Castrén, E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 2005, 25, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Platel, J.C.; Stamboulian, S.; Nguyen, I.; Bordey, A. Neurotransmitter signaling in postnatal neurogenesis: The first leg. Brain Res. Rev. 2010, 63, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. More hippocampal neurons in adult mice living in an enriched environment. Nature 1997, 386, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Leuner, B.; Gould, E.; Shors, T.J. Is there a link between adult neurogenesis and learning? Hippocampus 2006, 16, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Sakamoto, M.; Ohtsuka, T.; Takao, K.; Miyakawa, T.; Yamaguchi, M.; Mori, K.; Ikeda, T.; Itohara, S.; Kageyama, R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008, 11, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Warner-Schmidt, J.L.; Madsen, T.M.; Duman, R.S. Electroconvulsive seizure restores neurogenesis and hippocampus-dependent fear memory after disruption by irradiation. Eur. J. Neurosci. 2008, 27, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Winocur, G.; Wojtowicz, J.M.; Sekeres, M.; Snyder, J.S.; Wang, S. Inhibition of neurogenesis interferes with hippocampus-dependent memory function. Hippocampus 2006, 16, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Denny, C.A.; Burghardt, N.S.; Schachter, D.M.; Hen, R.; Drew, M.R. 4- to 6-week-old adult-born hippocampal neurons influence novelty-evoked exploration and contextual fear conditioning. Hippocampus 2012, 22, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.S.; Hong, N.S.; McDonald, R.J.; Wojtowicz, J.M. A role for adult neurogenesis in spatial long-term memory. Neuroscience 2005, 130, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Shors, T.J.; Miesegaes, G.; Beylin, A.; Zhao, M.; Rydel, T.; Gould, E. Neurogenesis in the adult is involved in the formation of trace memories. Nature 2001, 410, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Clelland, C.D.; Choi, M.; Romberg, C.; Clemenson, G.D., Jr.; Fragniere, A.; Tyers, P.; Jessberger, S.; Saksida, L.M.; Barker, R.A.; Gage, F.H.; et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 2009, 325, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Petrik, D.; Lagace, D.C.; Eisch, A.J. The neurogenesis hypothesis of affective and anxiety disorders: Are we mistaking the scaffolding for the building? Neuropharmacology 2012, 62, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.B.; Park, H.R.; Jang, Y.J.; Choi, S.Y.; Son, T.G.; Lee, J. Baicalein attenuates impaired hippocampal neurogenesis and the neurocognitive deficits induced by gamma-ray radiation. Br. J. Pharmacol. 2013, 168, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Pereira, I.; Canals, S.; Carrion, A.M. Adult newborn neurons are involved in learning acquisition and long-term memory formation: The distinct demands on temporal neurogenesis of different cognitive tasks. Hippocampus 2015, 25, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.G.; Kim, D.H.; Park, S.J.; Kim, J.M.; Cai, M.; Liu, X.; Lee, C.H.; Ryu, J.H. The memory-enhancing effects of Kami-ondam-tang in mice. J. Ethnopharmacol. 2011, 137, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Shim, K.J.; Choi, M.J.; Park, S.Y.; Choi, B.J.; Chang, M.S.; Park, S.K. Novel effects of Nelumbo nucifera rhizome extract on memory and neurogenesis in the dentate gyrus of the rat hippocampus. Neurosci. Lett. 2008, 443, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.M.; Kim, D.H.; Park, S.J.; Liu, X.; Cai, M.; Hong, J.G.; Park, J.H.; Ryu, J.H. Effects of Sun ginseng on memory enhancement and hippocampal neurogenesis. Phytother. Res. 2013, 27, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.J.; Park, S.J.; Woo, H.; Lee, H.E.; Kim, H.J.; Kwon, G.; Gao, Q.; Jang, D.S.; Ryu, J.H. Effects of allantoin on cognitive function and hippocampal neurogenesis. Food Chem. Toxicol. 2014, 64, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Akers, K.G.; Martinez-Canabal, A.; Restivo, L.; Yiu, A.P.; de Cristofaro, A.; Hsiang, H.L.; Wheeler, A.L.; Guskjolen, A.; Niibori, Y.; Shoji, H.; et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 2014, 344, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.T.; Sun, C.L.; Wo, P.Y.; Yen, H.H.; Tang, T.H.; Ng, M.C.; Huang, M.L.; Yang, Y.L. Hippocampal neurogenesis after traumatic brain injury is mediated by vascular endothelial growth factor receptor-2 and the Raf/MEK/ERK cascade. J. Neurotrauma 2011, 28, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Ponnusamy, K.; Song, M.R.; Ming, G.L.; Song, H. Molecular genetic analysis of FGFR1 signalling reveals distinct roles of MAPK and PLCγ1 activation for self-renewal of adult neural stem cells. Mol. Brain 2009, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar] [CrossRef] [PubMed]
- Wahane, S.D.; Hellbach, N.; Prentzell, M.T.; Weise, S.C.; Vezzali, R.; Kreutz, C.; Timmer, J.; Krieglstein, K.; Thedieck, K.; Vogel, T. PI3K-p110-α-subtype signalling mediates survival, proliferation and neurogenesis of cortical progenitor cells via activation of mTORC2. J. Neurochem. 2014, 130, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Fournier, N.M.; Lee, B.; Banasr, M.; Elsayed, M.; Duman, R.S. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and PI3K/Akt-dependent signaling. Neuropharmacology 2012, 63, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Chandran, A.; Iyo, A.H.; Jernigan, C.S.; Legutko, B.; Austin, M.C.; Karolewicz, B. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 40, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J., III; Govindarajan, A.; Jung, H.Y.; Kang, H.; Tonegawa, S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 2004, 116, 467–479. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Aimone, J.B.; Wiles, J.; Gage, F.H. Potential role for adult neurogenesis in the encoding of time in new memories. Nat. Neurosci. 2006, 9, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Czéh, B.; Michaelis, T.; Watanabe, T.; Frahm, J.; de Biurrun, G.; van Kampen, M.; Bartolomucci, A.; Fuchs, E. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. USA 2001, 98, 12796–12801. [Google Scholar] [CrossRef] [PubMed]
- Lesage, F.; Lazdunski, M. Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiol. Renal Physiol. 2000, 279, F793–F801. [Google Scholar] [PubMed]
- Zhao, G.; Etherton, T.D.; Martin, K.R.; Gillies, P.J.; West, S.G.; Kris-Etherton, P.M. Dietary α-linolenic acid inhibits proinflammatory cytokine production by peripheral blood monomuclear cells in hypercholesterolemic subjects. Am. J. Clin. Nutr. 2007, 85, 385–391. [Google Scholar] [PubMed]
- Mutoh, T.; Hamano, T.; Tokuda, A.; Kuriyama, M. Unglycosylated Trk protein does not co-localize nor associate with ganlioside GM1 in stable clone of PC12 cells overexpressing Trk (PCtrk cells). Glycoconj. J. 2000, 17, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.; Wacha, S.; Caroni, P. Spatial and temporal control of signaling through lipid rafts. Curr. Opin. Neurobiol. 2004, 15, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Besshoh, S.; Chen, S.; Brown, I.R.; Gurd, J.W. Developmental changes in the association of NMDA receptors with lipid rafts. J. Neurosci. Res. 2007, 85, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Capone, C.; Frigerio, S.; Fumagalli, S.; Gelati, M.; Principato, M.C.; Storini, C.; Montinaro, M.; Kraftsik, R.; de Curtis, M.; Parati, E.; et al. Neurosphere-derived cells exert a neuroprotective action by changing the ischemic microenvironment. PLoS ONE 2007, 2, e373. [Google Scholar] [CrossRef] [PubMed]
- Maysami, S.; Lan, J.Q.; Minami, M.; Simon, R.P. Proliferating progenitor cells: A required cellular element for induction of ischemic tolerance in the brain. J. Cereb. Blood Flow Metab. 2008, 28, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Honmou, O.; Harada, K.; Houkin, K.; Hamada, H.; Kocsis, J.D. I.V. infusion of brain-derived neurotrophic factor gene-modified human mesenchymal stem cells protects against injury in a cerebral ischemia model in adult rat. Neuroscience 2005, 136, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Pallini, R.; Vitiani, L.R.; Bez, A.; Casalbore, P.; Facchiano, F.; di Giorgi Gerevini, V.; Falchetti, M.L.; Fernandez, E.; Maira, G.; Peschle, C.; et al. Homologous transplantation of neural stem cells to the injured spinal cord of mice. Neurosurgery 2005, 57, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef]
- Marini, A.M.; Rabin, S.J.; Lipsky, R.H.; Mocchetti, I. Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J. Biol. Chem. 1998, 273, 29394–29399. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Araya, C.; Palma, V.; Bronfman, M. PPARβ/δ and PPARγ maintain undifferentiated phenotypes of mouse adult neural precursor cells from the subventricular zone. Front Cell Neurosci. 2015, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Robbins, M.E. PPARα ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-κB and AP-1 pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Nevin, D.K.; Mulas, G.; Casu, M.A.; Diana, A.; Rossi, D.; Sacchetti, G.; Carta, A.R. Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARγ agonist in vitro and in MPTP-treated mice. Neuroscience 2015, 302, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piermartiri, T.; Pan, H.; Figueiredo, T.H.; Marini, A.M. α-Linolenic Acid, A Nutraceutical with Pleiotropic Properties That Targets Endogenous Neuroprotective Pathways to Protect against Organophosphate Nerve Agent-Induced Neuropathology. Molecules 2015, 20, 20355-20380. https://doi.org/10.3390/molecules201119698
Piermartiri T, Pan H, Figueiredo TH, Marini AM. α-Linolenic Acid, A Nutraceutical with Pleiotropic Properties That Targets Endogenous Neuroprotective Pathways to Protect against Organophosphate Nerve Agent-Induced Neuropathology. Molecules. 2015; 20(11):20355-20380. https://doi.org/10.3390/molecules201119698
Chicago/Turabian StylePiermartiri, Tetsade, Hongna Pan, Taiza H. Figueiredo, and Ann M. Marini. 2015. "α-Linolenic Acid, A Nutraceutical with Pleiotropic Properties That Targets Endogenous Neuroprotective Pathways to Protect against Organophosphate Nerve Agent-Induced Neuropathology" Molecules 20, no. 11: 20355-20380. https://doi.org/10.3390/molecules201119698
APA StylePiermartiri, T., Pan, H., Figueiredo, T. H., & Marini, A. M. (2015). α-Linolenic Acid, A Nutraceutical with Pleiotropic Properties That Targets Endogenous Neuroprotective Pathways to Protect against Organophosphate Nerve Agent-Induced Neuropathology. Molecules, 20(11), 20355-20380. https://doi.org/10.3390/molecules201119698