
Xerogel-Sequestered Silanated Organochalcogenide Catalysts for Bromination with Hydrogen Peroxide and Sodium Bromide

Abstract
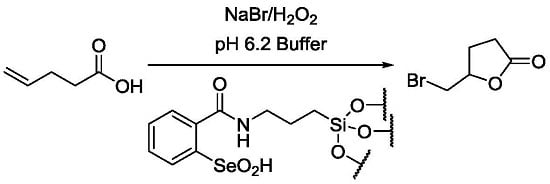
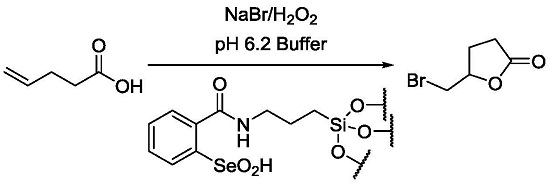
:
1. Introduction
2. Results and Discussion
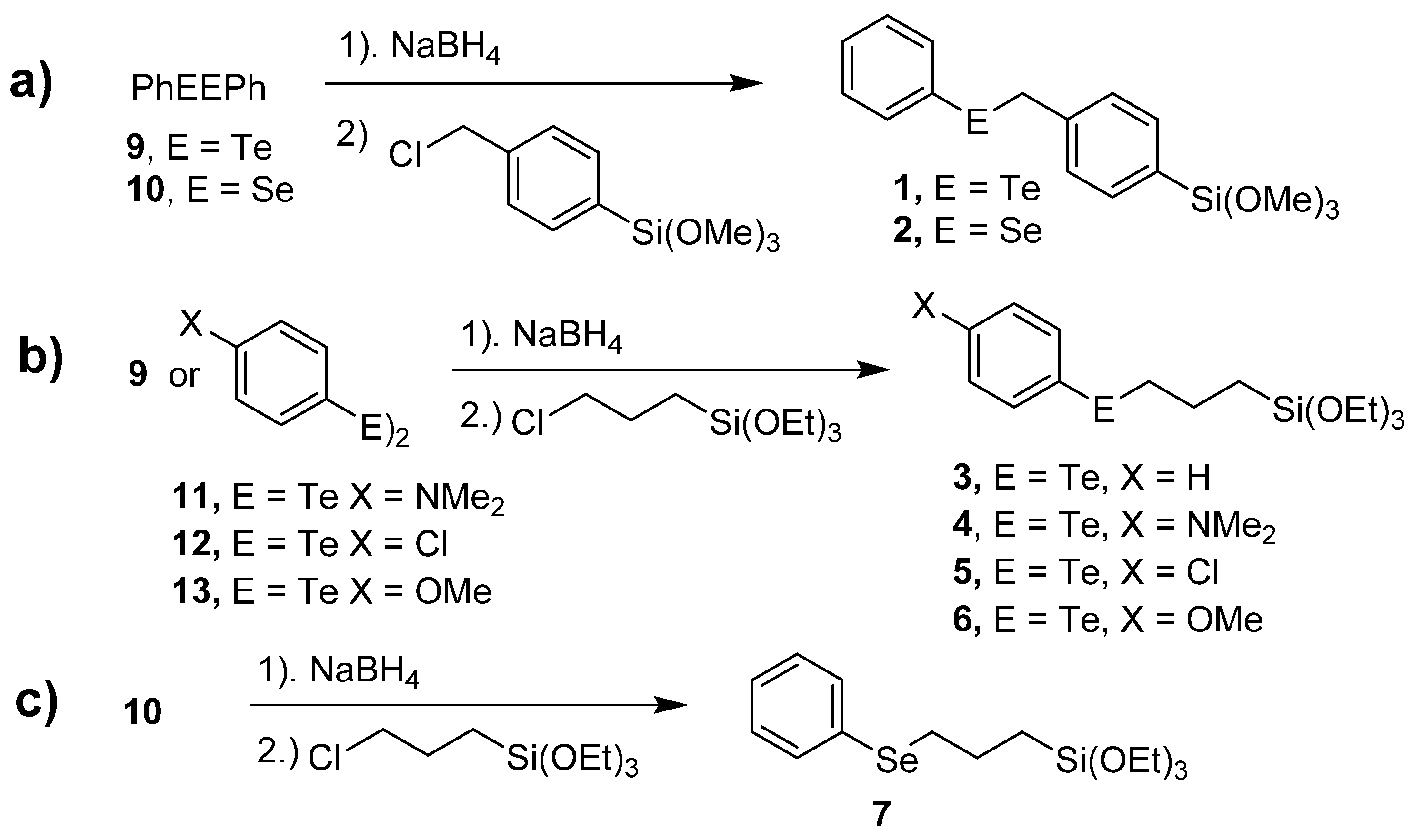
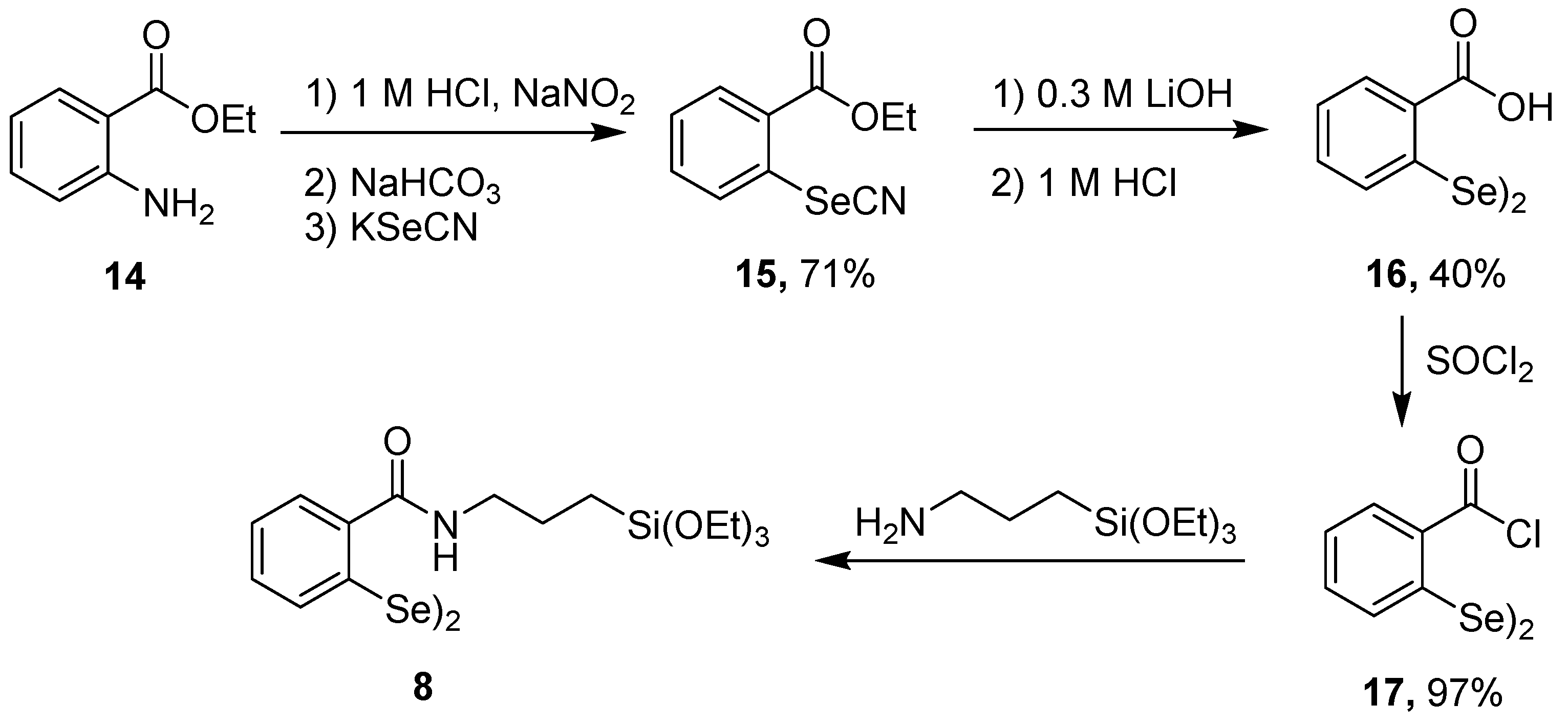
2.1. Synthesis of Chalcogenide Catalysts and Catalyst Precursors



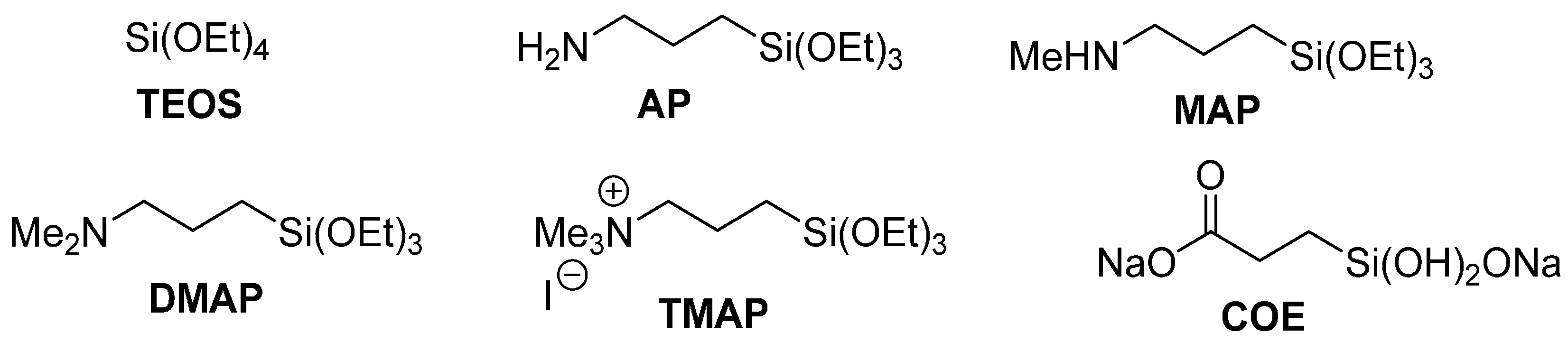
2.2. Preparation of Xerogel Monoliths with Chalcogenide Catalysts/Catalyst Precursors
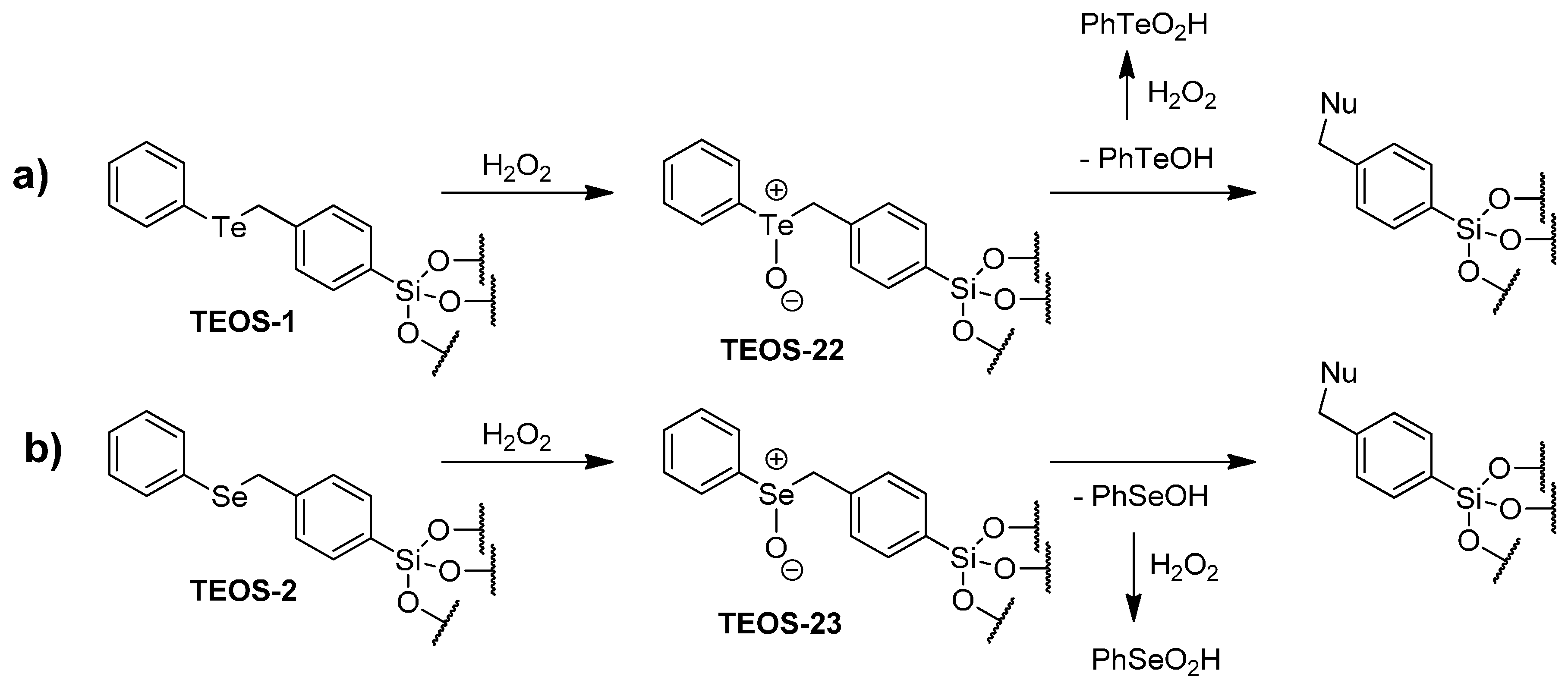
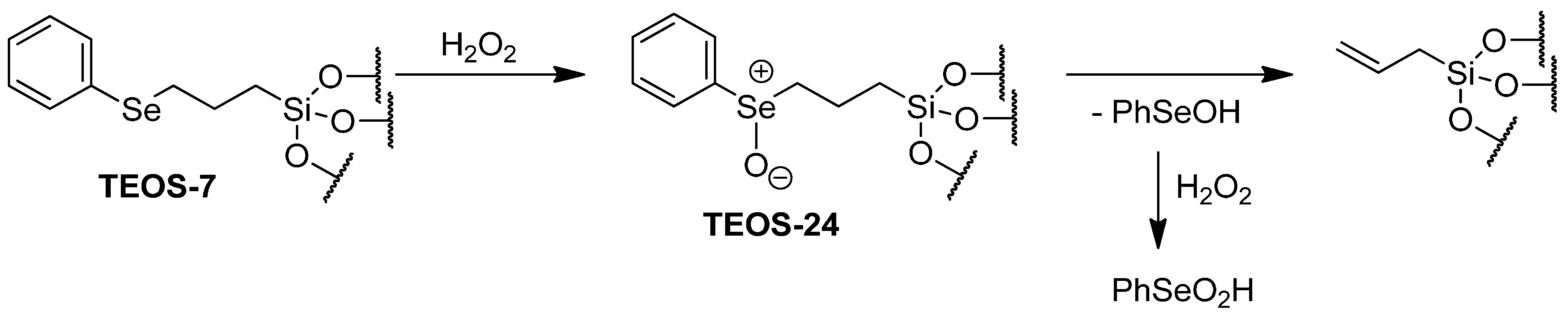
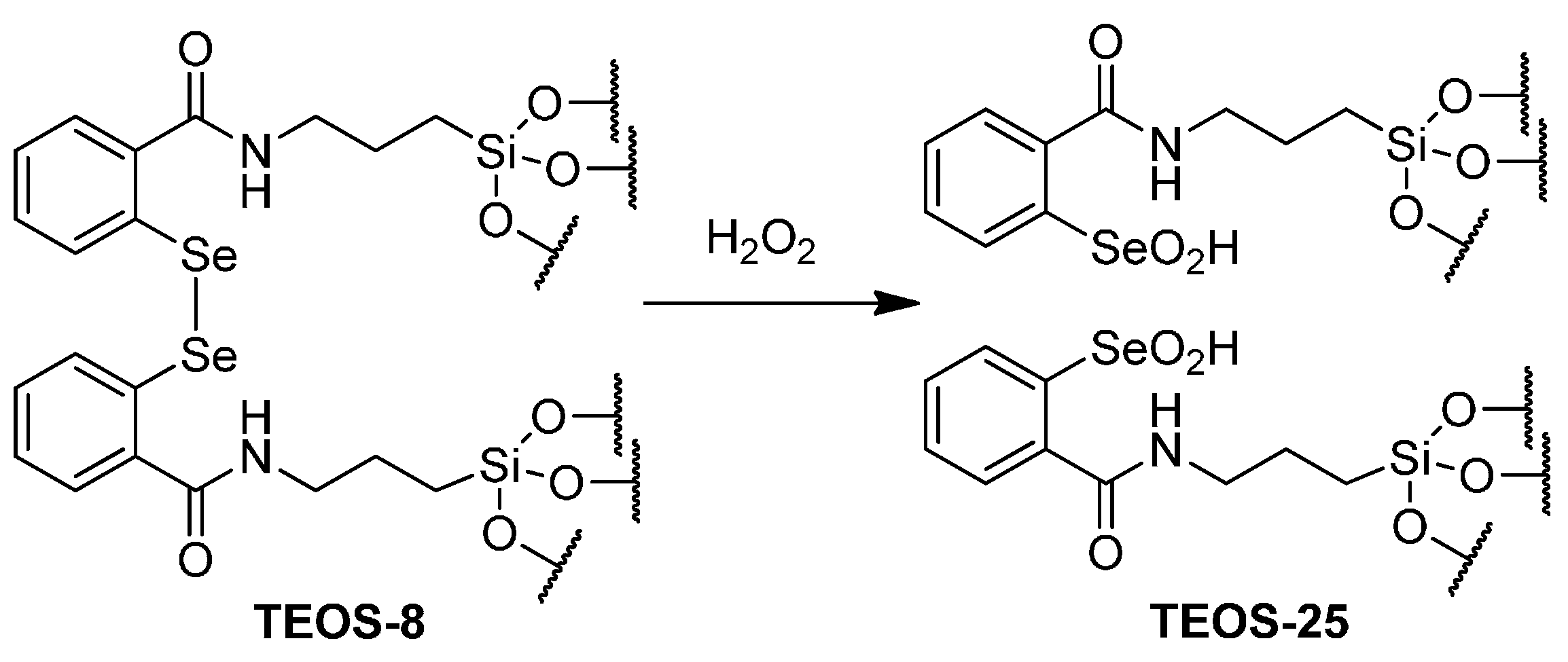
2.3. Catalytic Oxidation of Bromide with Hydrogen Peroxide


{kind=link}
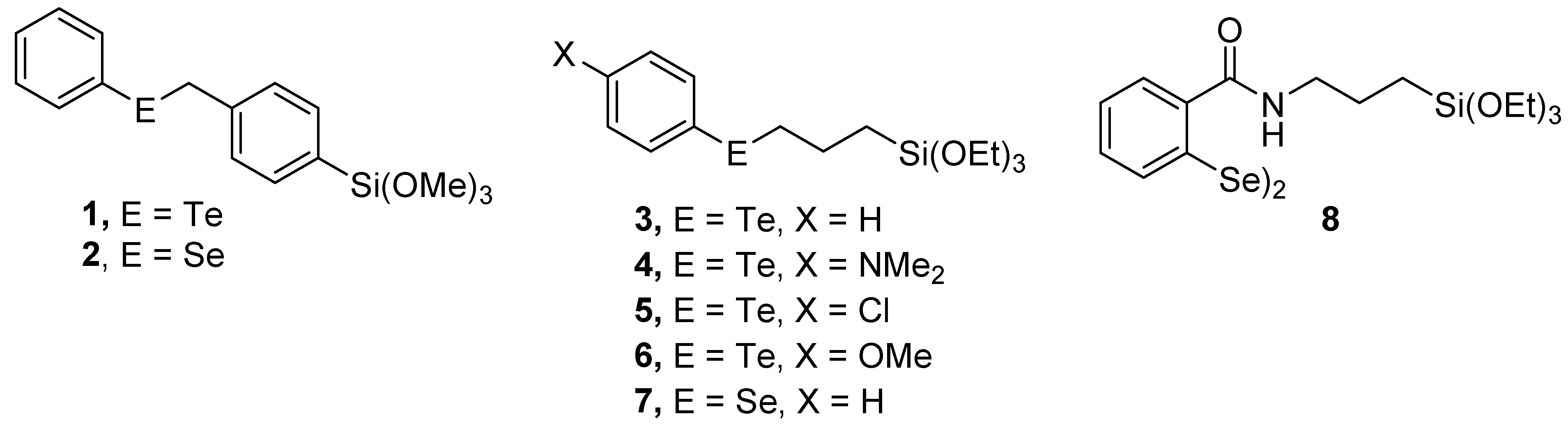{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | kobs (s−1) c | krel |
|---|---|---|
| blank | (8.35 ± 0.17) × 10−7 | 1.0 |
| 1 a | (5.71 ± 0.02) × 10−5 | 68 |
| 2 b | (1.27 ± 0.20) × 10−5 | 15 |
| 3 a | (1.91 ± 0.06) × 10−5 | 23 |
| 4 a | (3.64 ± 0.54) × 10−5 | 44 |
| 5 a | (3.61 ± 0.01) × 10−6 | 4.3 |
| 6 a | (6.93 ± 0.20) × 10−6 | 8.3 |
| 7 b | (8.03 ± 1.11) × 10−6 | 9.6 |
| 8 b | (1.37 ± 0.02) × 10−5 | 16 |
2.4. Catalytic Lifetimes and Recyclability
| Cycle | Catalyst | kobs (s−1) d | krel |
|---|---|---|---|
| Initial Reaction a | 1 | (5.71 ± 0.02) × 10−5 | 1.00 |
| First Recycle a | 1 | (1.16 ± 0.01) × 10−5 | 0.20 |
| Second Recycle a | 1 | (1.08 ± 0.20) × 10−5 | 0.19 |
| Third Recycle a | 1 | (5.69 ± 0.50) × 10−6 | 0.10 |
| Initial Reaction b | 2 | (1.27 ± 0.20) × 10−5 | 1.00 |
| First Recycle b | 2 | (3.44 ± 0.05) × 10−6 | 0.27 |
| Second Recycle b | 2 | (2.53 ± 0.01) × 10−6 | 0.20 |
| Third Recycle b | 2 | (1.96 ± 0.05) × 10−6 | 0.15 |
| Initial Reaction a | 3 | (1.91 ± 0.06) × 10−5 | 1.00 |
| First Recycle a | 3 | (2.48 ± 0.06) × 10−5 | 1.30 |
| Second Recycle a | 3 | (1.68 ± 0.09) × 10−5 | 0.88 |
| Third Recycle a | 3 | (2.00 ± 0.03) × 10−5 | 1.05 |
| Initial Reaction b | 7 | (8.03 ± 0.55) × 10−6 | 1.00 |
| First Recycle b | 7 | (4.45 ± 0.05) × 10−6 | 0.55 |
| Second Recycle b | 7 | (3.65 ± 0.44) × 10−6 | 0.45 |
| Third Recycle b | 7 | (3.65 ± 0.41) × 10−6 | 0.45 |
| Initial Reaction c | 8 | (1.24 ± 0.01) × 10−5 | 1.00 |
| First Recycle c | 8 | (1.34 ± 0.00) × 10−5 | 1.08 |
| Second Recycle c | 8 | (1.14 ± 0.00) × 10−5 | 0.92 |
| Third Recycle c | 8 | (1.46 ± 0.02) × 10−5 | 1.18 |



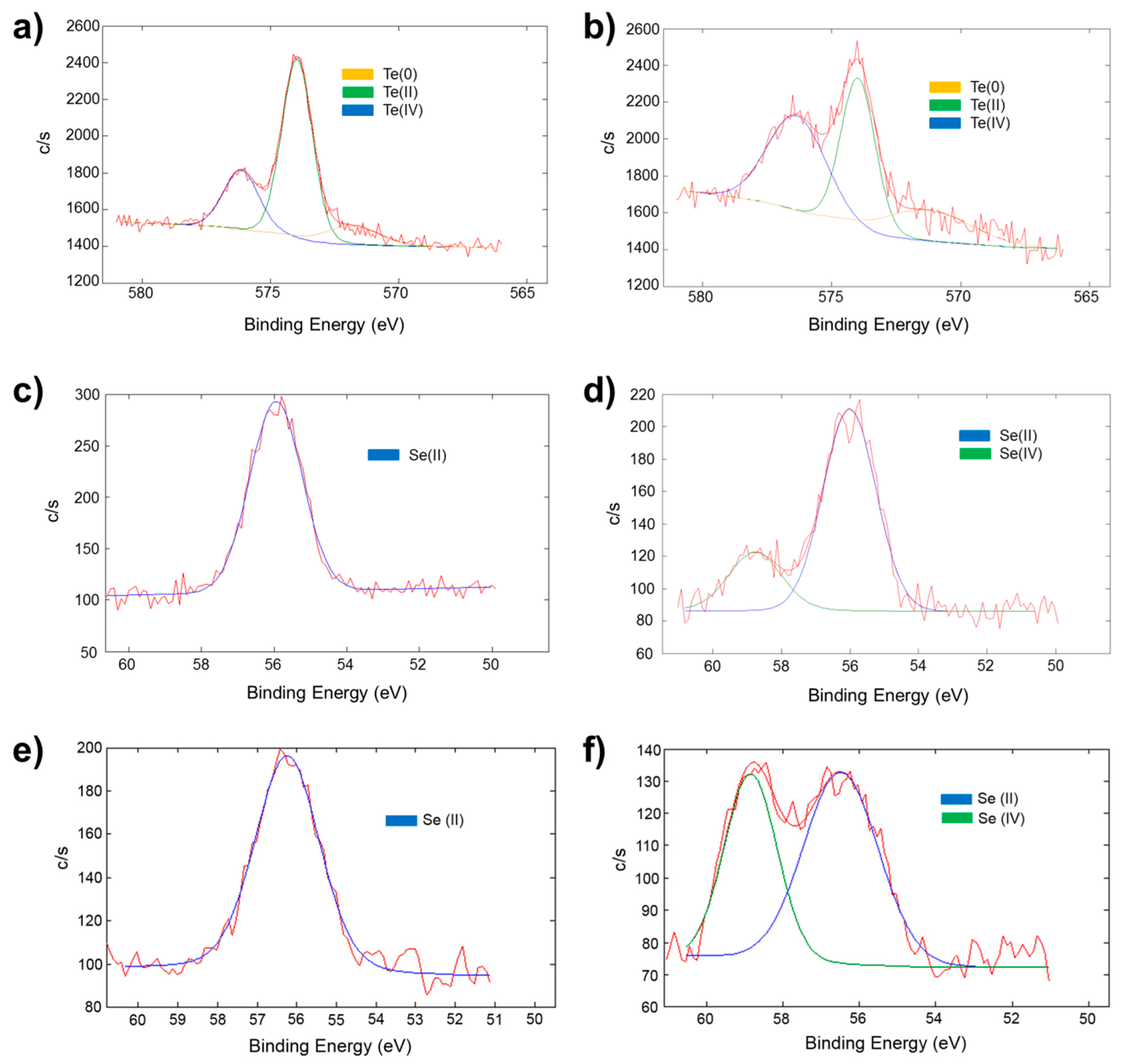
2.5. Oxidation-State Changes as Measured by X-ray Photoelectron Spectroscopy in the TEOS Xerogel/Chalcogenide Catalysts upon Exposure to Hydrogen Peroxide
| “as Prepared” | 24 h Soaked in 1.0 × 10−4 M H2O2 | ||||||
|---|---|---|---|---|---|---|---|
| Te(0) Binding Energy (eV) | Te(II) Binding Energy (eV) | Te(IV) Binding Energy (eV) | Te(IV)/Te(II) Ratio | Te(0) Binding Energy (eV) | Te(II) Binding Energy (eV) | Te(IV) Binding Energy (eV) | Te(IV)/Te(II) Ratio |
| 571.7 ± 0.8 | 573.8 ± 0.5 | 575.9 ± 0.5 | 0.5 ± 0.1 | 571.3 ± 0.1 | 573.9 ± 0.1 | 576.3 ± 0.1 | 0.9 ± 0.2 |
| Xerogel | “as Prepared” | 24 h Soaked in 1.0 × 10−4 or 5.0 × 10−5 M H2O2 | ||||
|---|---|---|---|---|---|---|
| Se(II) Binding Energy (eV) | Se(IV) Binding Energy (eV) | Se(II)/Se(IV) Ratio | Se(II) Binding Energy (eV) | Se(IV) Binding Energy (eV) | Se(IV)/Se(II) Ratio | |
| TEOS-7 | 55.1 ± 0.2 | -- | -- | 55.4 ± 0.5 | 58.1 ± 0.5 | 0.2 ± 0.1 |
| TEOS-8 | 56.1 ± 0.1 | -- | -- | 56.4 ± 0.2 | 58.8 ± 0.2 | 0.4 ± 0.2 |

2.6. The Effect of Xerogel Composition on Rates of Reaction

| Xerogel | kobs (s−1) b | krel |
|---|---|---|
| Catalyst-free TEOS | (9.22 ± 0.04) × 10−7 | 0.07 |
| TEOS | (1.24 ± 0.01) × 10−5 | 1.00 |
| 10:90 DMAP/TEOS | (1.61 ± 0.02) × 10−5 | 1.30 |
| 10:90 MAP/TEOS | (1.51 ± 0.03) × 10−5 | 1.22 |
| 10:90 AP/TEOS | (2.05 ± 0.06) × 10−5 | 1.65 |
| 10:90 TMAP/TEOS | (3.87 ± 0.01) × 10−5 | 3.12 |
| 10:90 COE/TEOS | (1.53 ± 0.04) × 10−5 | 1.23 |
3. Experimental Section
3.1. General Information
3.2. General Procedure for Kinetic Experiments
3.3. General Procedure for Recycling Catalysts
3.4. Preparation of Catalysts
3.4.1. Preparation of Trimethoxy(4-(phenyltelluranyl)phenyl)silane (1)
3.4.2. Preparation of Trimethoxy(4-(phenylselanyl)phenyl)silane (2)
3.4.3. Preparation of (3-(Phenyltelluranyl)propyl)silane (3)
3.4.4. Preparation of 4-((3-(Triethoxysilyl)propyl)telluranyl)aniline (4)
3.4.5. Preparation of (3-((4-Chlorophenyl)telluranyl)propyl)triethoxysilane (5)
3.4.6. Preparation of Triethoxy(3-((4-methoxyphenyl)telluranyl)propyl)silane (6)
3.4.7. Preparation of Triethoxy(3-(phenylselanyl)propyl)silane (7)
3.4.8. Synthesis of 2,2′-Diselanediyldibenzoic Acid (16)
3.4.9. Synthesis of 2,2′-Diselanedibenzoyl Chloride [41] (17)
3.4.10. Synthesis of 2,2′-Diselanediylbis(N-(3-(triethoxysilyl)propyl)benzamide 8
3.5. Preparation of Monoliths
3.5.1. Preparation of 2.5 mol % 1 in TEOS
3.5.2. Preparation of 0.5 mol % 2 in TEOS
3.5.3. Preparation of 2.5 mol % 3 in TEOS
3.5.4. Preparation of 2.5 mol % 4 in TEOS
3.5.5. Preparation of 2.5 mol % 5 in TEOS
3.5.6. Preparation of 2.5 mol % 6 in TEOS
3.5.7. Preparation of 0.5 mol % 7 in TEOS
3.5.8. Preparation of 0.5 mol % 8 in TEOS
3.5.9. Preparation of 0.5 mol % 8 in 10:90 DMAP/TEOS
3.5.10. Preparation of 0.5 mol % 8 in 10:90 MAP/TEOS
3.5.11. Preparation of 0.5 mol % 8 in 10:90 AP/TEOS
3.5.12. Preparation of 0.5 mol % 8 in 10:90 COE/TEOS
3.5.13. Synthesis of 3-Trimethoxysilyl-Propyltrimethylammonium Iodide [[19]] (TMAP)
3.5.14. Preparation of 0.5 mol % 8 in 10:90 TMAP/TEOS
3.6. Formation of Thin Films
3.6.1. Cleaning Microscope Slides
3.6.2. Spin Coating
3.6.3. Preparation of 5 mol % 3 in TEOS
3.6.4. Preparation of 20 mol % 7 in TEOS
3.6.5. Preparation of 10 mol % (8) in TEOS
3.7. X-ray Photoelectron Spectroscopy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Strukul, G. Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Spring Science & Business Media: Dordrecht, Germany, 1992. [Google Scholar]
- Ten Brink, G.J.; Vis, J.M.; Arends, I.W.C.E.; Sheldon, R.A. Selenium-Catalyzed Oxidations with Aqueous Hydrogen Peroxide. 2. Baeyer-Villiger Reactions in Homogeneous Solution. J. Org. Chem. 2001, 66, 2429–2433. [Google Scholar] [CrossRef] [PubMed]
- Ten Brink, G.J.; Fernandes, B.C.M.; van Vliet, M.C.A.; Arends, I.W.C.E.; Sheldon, R.A. Selenium catalyzed oxidations with aqueous hydrogen peroxide. Part I. Epoxidation reactions in homogenous solution. J. Chem. Soc. Perkin Trans. 1 2001, 3, 224–228. [Google Scholar]
- Drabowicz, J.; Mikolajczyk, M. A Facile and Selective Oxidation of Organic Sulphides and Sulphoxides with Hydrogen Peroxide/Selenium Dioxide System. Synthesis 1978, 10, 758–759. [Google Scholar] [CrossRef]
- Reich, H.J.; Chow, F.; Peake, S.L. Seleninic Acids as Catalysts for Oxidation of Olefins and Sulfides Using Hydrogen Peroxide. Synthesis 1978, 4, 299–300. [Google Scholar] [CrossRef]
- Ten Brink, G.J.; Vis, J.M.; Arends, I.W.C.E.; Sheldon, R.A. Selenium catalysed oxidations with aqueous hydrogen peroxide. Part 3. Oxidation of carbonyl compounds under mono/bi/triphasic conditions. Tetrahedron 2002, 58, 3977–3983. [Google Scholar] [CrossRef]
- Murahashi, S.; Tatsuki, S. Selenium Dioxide Catalyzed Oxidation of Secondary Amines with Hydrogen Peroxide. Simple Synthesis of Nitrones from Secondary Amines. Tetrahedron Lett. 1987, 28, 2383–2386. [Google Scholar] [CrossRef]
- Back, T.G.; Moussa, Z. Remarkable activity of a novel phenylseleninate ester as a glutathione peroxidase mimetic and its facile in situ generation from allyl 3-hydroxypropyl selenide. J. Am. Chem. Soc. 2002, 124, 12103–12105. [Google Scholar] [CrossRef]
- Press, D.J.; McNeil, N.M.R.; Hambrook, M.; Back, T.G. Effects of methoxy substituents on glutathione peroxidase-like activity of cyclic seleninate esters. J. Org. Chem. 2014, 79, 9394–9401. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G.; Singh, H.B. Synthetic organoselenium compounds as antioxidants: Glutathione peroxidaze activity. Chem. Soc. Rev. 2000, 29, 347–357. [Google Scholar] [CrossRef]
- Goodman, M.A.; Detty, M.R. Selenoxides as Catalysts for the Activation of Hydrogen Peroxide. Bromination of Organic Substrates with Sodium Bromide and Hydrogen Peroxide. Organometallics 2004, 23, 3016–3020. [Google Scholar] [CrossRef]
- Francavilla, C.; Drake, M.D.; Bright, F.V.; Detty, M.R. Dendrimeric Organochalcogen Catalysts for the Activation of Hydrogen Peroxide: Improved Catalyst Activity through Statistical Effects and Cooperativity in Successive Generations. J. Am. Chem. Soc. 2001, 123, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.D.; Bright, F.V.; Detty, M.R. Dendrimeric Organochalcogen Catalyst for the Activation of Hydrogen Peroxide: Origins of the “Dendrimer Effect” with Catalyst Terminating in Phenylseleno Groups. J. Am. Chem. Soc. 2003, 125, 12558–12566. [Google Scholar] [CrossRef] [PubMed]
- Alberto, E.E.; Braga, A.L.; Detty, M.R. Imidazolium-containing diselenides for catalytic oxidations with hydrogen peroxide and sodium bromide in aqueous solutions. Tetrahedron 2012, 68, 10476–10481. [Google Scholar] [CrossRef]
- Drake, M.D.; Bateman, M.A.; Detty, M.R. Substituent Effects in Arylseleninic Acid-Catalyzed Bromination of Organic Substrates with Sodium Bromide and Hydrogen Peroxide. Organometallics 2003, 22, 4158–4162. [Google Scholar] [CrossRef]
- Detty, M.R.; Zhou, F.; Friedman, A.E. Positive Halogens from Halides and Hydrogen Peroxide with Organotellurium Catalysts. J. Am. Chem. Soc. 1996, 118, 313–318. [Google Scholar] [CrossRef]
- Higgs, D.E.; Nelen, M.I.; Detty, M.R. Iodination of Organic Substrates with Halide Salts and H2O2 Using an Organotelluride Catalyst. Org. Lett. 2001, 3, 349–352. [Google Scholar] [PubMed]
- Alberto, E.E.; Muller, L.M.; Detty, M.R. Rate Accelerations of Bromination Reactions with NaBr and H2O2 via the Addition of Catalytic Quantities of Diaryl Ditellurides. Organometallics 2014, 33, 5571–5581. [Google Scholar] [CrossRef]
- Bennett, S.M.; Ying, T.; McMaster, D.; Bright, F.V.; Detty, M.R. A Xerogel-Sequestered Selenoxide Catalyst for Bromination with Hydrogen Peroxide and Sodium Bromide in an Aqueous Environment. J. Org. Chem. 2008, 73, 6849–6852. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Finlay, J.A.; Kowalke, G.L.; Meyer, A.E.; Bright, F.V.; Callow, M.E.; Callow, J.A.; Wendt, D.E.; Detty, M.R. Hybrid xerogel films as novel coatings for antifouling and fouling release. Biofouling 2005, 21, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.M.; Finlay, J.A.; Gunari, N.; Wells, D.D.; Meyer, A.E.; Walker, G.C.; Callow, M.E.; Callow, J.A.; Bright, F.V.; Detty, M.R. The role of surface energy and water wettability in aminoalkyl/fluorocarbon/hydrocarbon-modified xerogel surfaces in the control of marine biofouling. Biofouling 2010, 26, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Evariste, E.; Gatley, C.M.; Detty, M.R.; Callow, M.E.; Callow, J.A. The performance of aminoalkyl/fluorocarbon/hydrocarbon-modified xerogel coatings against the marine alga Ectocarpus crouaniorum: Relative roles of surface energy and charge. Biofouling 2013, 29, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Finlay, J.A.; Bennett, S.M.; Brewer, L.H.; Sokolova, A.; Clay, G.; Gunari, N.; Meyer, A.E.; Walker, G.C.; Wendt, D.E.; Callow, M.E.; et al. Barnacle settlement and the adhesion of protein and diatom microfouling to xerogel films with varying surface energy and water wettability. Biofouling 2010, 26, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Bailey, J.J.; Waltz, G.T.; Brewer, L.H.; Finlay, J.A.; Fornalik, J.; Wendt, D.E.; Callow, M.E.; Callow, J.A.; Bright, F.V.; et al. Spontaneous multiscale phase separation within flurinated xerogel coatings for fouling-release surfaces. Biofouling 2012, 28, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Cilz, N.; Daniels, J.; Stafslien, S.J.; Brewer, L.H.; Wendt, D.E.; Bright, F.V.; Detty, M.R. A comparison of the antifouling/foul-release characteristics of non-biocidal xerogel and commerical coatings towards micro- and macrofouling organsims. Biofouling 2012, 28, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Gunari, N.; Brewer, L.H.; Bennett, S.M.; Sokolova, A.; Kraut, N.D.; Finlay, J.A.; Meyer, A.E.; Walker, G.C.; Wendt, D.E.; Callow, M.E.; et al. The control of marine biofouling on xerogel surfaces with nanometer-scale topography. Biofouling 2011, 27, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Selvaggio, P.; Tusa, S.; Detty, M.R.; Bright, F.V.; Ciriminna, R.; Pagliaro, M. Ecofriendly protection from biofouling of the monitoring system as Pantelleria’s Cala Gadir underwater archaeological site, Sicily. Int. J. Naut. Arch. 2009, 38, 417–421. [Google Scholar] [CrossRef]
- McMaster, D.M.; Bennett, S.M.; Tang, Y.; Finlay, J.A.; Kowalke, G.L.; Nedved, B.; Bright, F.V.; Callow, M.E.; Callow, J.A.; Wendt, D.E.; et al. Antifouling character of “active” hybrid xerogel coatings with sequestered catalysts for the activation of hydrogen peroxide. Biofouling 2009, 25, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.J.; Hoeger, C.A.; Willis, W.W., Jr. Organoselenium Chemistry. Characterization of Reactive Intermediates in the Selenoxide Syn Elimination: Selenenic Acids and Selenolseleninate Esters. J. Am. Chem. Soc. 1982, 104, 2937–2940. [Google Scholar] [CrossRef]
- Reich, H.J.; Reich, I.L.; Renga, J.M. Organoselenium Chemstiry. α-Phenylseleno Carbonyl Compounds as Precursors for α,β-Unsaturated Ketones and Esters. J. Am. Chem. Soc. 1973, 95, 5813–5815. [Google Scholar] [CrossRef]
- Reich, H.J.; Renga, J.M.; Reich, I.L. Organoselenium Chemistry. Converstion of Ketones to Enones by Selenoxide Syn Elimination. J. Am. Chem. Soc. 1975, 97, 5434–5447. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Young, M.W.; Lauer, R.F. Reactions of Selenoxides: Thermal Syn-elimination and H218O Exchange. Tetrahedron Lett. 1973, 22, 1979–1982. [Google Scholar] [CrossRef]
- Vickerman, J.C.; Gilmore, I.S. Surface Analysis-The Principal Techniques, 2nd ed.; John Wiley & Sons Ltd: London, UK, 2009. [Google Scholar]
- Detty, M.R.; Lenhart, W.C.; Gassman, P.G.; Callstrom, M.R. XPS and 125Te NMR of Organotellurium Compounds. 2. Oxatellurolyium Halides and Dioxatellurapentalenes and Their Products of Oxidative Halogen Addition. Organometallics 1989, 8, 886–870. [Google Scholar] [CrossRef]
- Detty, M.R.; Lenhart, W.C.; Gassman, P.G.; Callstrom, M.R. XPS and 125Te NMR studies of organotellurium compounds. I. Tellurapyrans, tellurapyranones, tellurapyrylium salts, and their benzo analogues in both the tellurium(II) and tellurium(IV) oxidation states. Organometallics 1989, 8, 861–865. [Google Scholar] [CrossRef]
- Shenasa, M.; Sainkar, S.; Lichtman, D. XPS study of some selected selenium compounds. J. Electron. Spectrosc. Relat. Phenom. 1986, 40, 329–337. [Google Scholar] [CrossRef]
- Wang, L.; Cao, W.; Yi, Y.; Xu, H. Dual redox responsive coassemblies of diselenide-containing block copolymers and polymer lipids. Langmuir 2014, 30, 5628–5636. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Cornelius, M.; Morell, J.; Froba, M. Silica-based mesoporous organic-inorganic hybrid materials. Angew. Chem. 2006, 45, 3216–3251. [Google Scholar] [CrossRef] [PubMed]
- Engman, L.; Stern, D.; Cotgreave, I.A.; Andersson, C.M. Thiol Peroxidase Activity of Diaryl Ditellurides as Determined by a 1H-NMR Method. J. Am. Chem. Soc. 1992, 114, 9737–9743. [Google Scholar] [CrossRef]
- Erben, F.; Kleeblatt, D.; Sonneck, M.; Hein, M.; Feist, H.; Fahrenwaldt, T.; Fischer, C.; Matin, A.; Iqbal, J.; Plotz, M.; et al. Synthesis and antiproliferative activity of selenoindirubins and selenoindirubin-N-glycosides. Org. Biomol. Chem. 2013, 11, 3963–3978. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Li, P.; Sun, X.; Yang, S.; Wang, B.; Han, K. A fluorescent probe for rapid detection of thiols and imaging of thiols reducing repair and H2O2 oxidative stress cycles in living cells. Chem. Commun. (Camb.) 2013, 49, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatley, C.M.; Muller, L.M.; Lang, M.A.; Alberto, E.E.; Detty, M.R. Xerogel-Sequestered Silanated Organochalcogenide Catalysts for Bromination with Hydrogen Peroxide and Sodium Bromide. Molecules 2015, 20, 9616-9639. https://doi.org/10.3390/molecules20069616
Gatley CM, Muller LM, Lang MA, Alberto EE, Detty MR. Xerogel-Sequestered Silanated Organochalcogenide Catalysts for Bromination with Hydrogen Peroxide and Sodium Bromide. Molecules. 2015; 20(6):9616-9639. https://doi.org/10.3390/molecules20069616
Chicago/Turabian StyleGatley, Caitlyn M., Lisa M. Muller, Meredith A. Lang, Eduardo E. Alberto, and Michael R. Detty. 2015. "Xerogel-Sequestered Silanated Organochalcogenide Catalysts for Bromination with Hydrogen Peroxide and Sodium Bromide" Molecules 20, no. 6: 9616-9639. https://doi.org/10.3390/molecules20069616
APA StyleGatley, C. M., Muller, L. M., Lang, M. A., Alberto, E. E., & Detty, M. R. (2015). Xerogel-Sequestered Silanated Organochalcogenide Catalysts for Bromination with Hydrogen Peroxide and Sodium Bromide. Molecules, 20(6), 9616-9639. https://doi.org/10.3390/molecules20069616