2. Results and Discussion
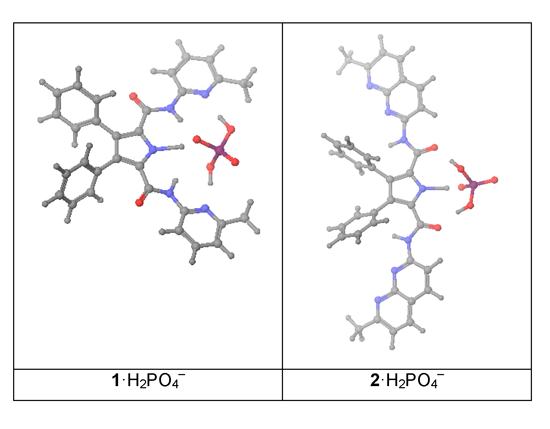
We report here the complexation studies of N2,N5-bis(6-methylpyridin-2-yl)-3,4-diphenyl-1H-pyrrole-2,5-dicarboxamide (1) and N2,N5-bis(7-methyl-1,8-naphthyridin-2-yl)-3,4-diphenyl-1H-pyrrole-2,5-dicarboxamide (2) with six monoanions of five different shapes: (i) spherical like Cl−; (ii, iii) trigonal planar or V-shaped like CH3CO2− and NO3−; (iv) tetrahedral like H2PO4− and BF4−; (v) octahedral like PF6−, all in the form of tetrabutylammonium salts.
The preparation of hosts
1 and
2, achieved by condensation of the 3,4-diphenyl-1
H-pyrrole-2,5-dicarbonyl dichloride with 2-amino-6-methylpyridine and 2-amino-7-methyl-1,8-naphthyridine, as well as their complete NMR characterization is reported elsewhere [
10]. The most significant proton signals used in the NMR titrations are the pyrrole NH singlet at 10.44 ppm, the amide NH singlet at 7.97 ppm and the H3' doublet at 7.99 ppm in host
1 and at 10.54 ppm, 8.37 ppm and 8.50 ppm in host
2.
Crystals of host
1 suitable for analysis by single crystal X-ray diffraction were obtained by recrystallization either from chloroform-hexane or from ethanol. It crystallizes in the monoclinic
P2
1/c space group.
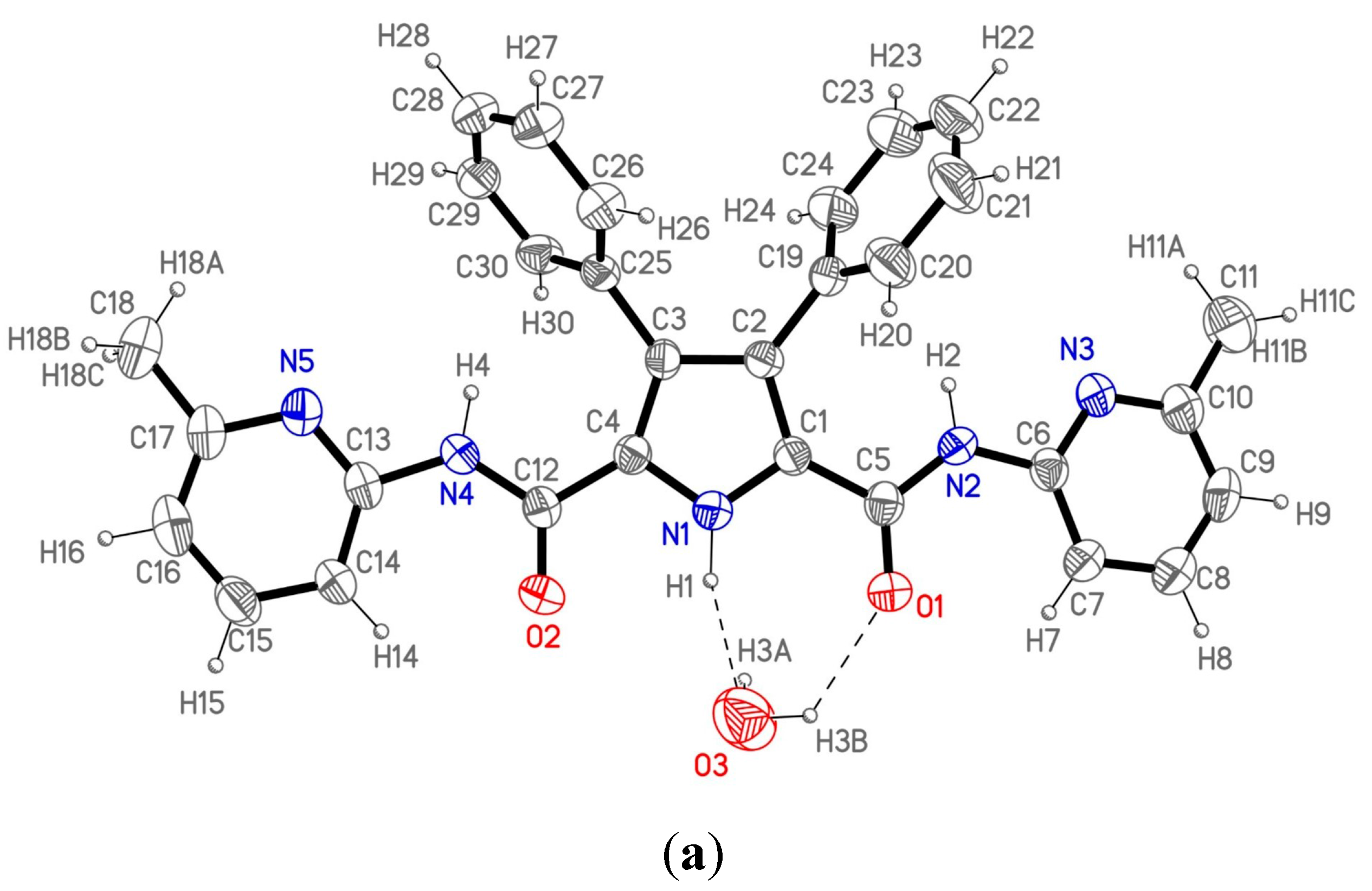
Figure 2a shows the labeling of the asymmetric unit and H-bonding data are collected in
Table 1.
Table 1.
Hydrogen bonds (Å and °) for compound 1.
Table 1.
Hydrogen bonds (Å and °) for compound 1.
| D-H···A | Symmetry Operation | d(D-H) | d(H···A) | d(D···A) | <(DHA) |
|---|
| N1-H1···O3 | | 1 | 2.18 | 3.151(5) | 162.5 |
| O3-H3B···O1 | | 1.07 | 2.09 | 2.855(4) | 126.2 |
| O3-H3A···N5(#1) | (#1) –x + 1, –y, –z | 1.05 | 2.07 | 3.097(5) | 165.6 |
The asymmetric unit consists of a single molecule of 1 and one crystallization water molecule. The molecule is not planar due to the twisted 3,4-disubstituted phenyl rings with respect to the pyrrole moiety; the dihedral angle between the latter and each of the two mentioned rings is 60.2(3)° and 66.9(3)°, respectively. The rest of the molecule could be considered nearly planar as the atoms in the pyrrole ring, the amido groups and the pyridine rings show an extended electronic conjugation in accordance with the bond distances found in this part of the molecule.
These molecules are not isolated as each one connects with the centrosymmetric one through two different water molecules appropriately situated to form hydrogen bonds (
Figure 2b) giving rise to a dimeric unit. Therefore, each dimeric entity consists of two host molecules linked by two bridging water molecules. Each water molecule forms three different hydrogen bonds with the two hosts in the dimeric unit. So, every water molecule interacts with the first one through a double hydrogen bond O3···N1–H1 and O3–H3B···O1 (the pyrrolic and the amido group respectively), and the third hydrogen bond is formed with the second molecule through O3-H3A and the N5’ atom, completing the dimeric moiety. These dimers are isolated, as no significant additional interactions between them have been found in the crystal structure.
Figure 2.
(
a) ORTEP plot (40% ellipsoid probability) of 1 showing the labeling of the asymmetric unit; (
b) View of the 1 dimer (1
2); (
c) The structure of 2 with a water and a DMSO molecule from reference [
10].
Figure 2.
(
a) ORTEP plot (40% ellipsoid probability) of 1 showing the labeling of the asymmetric unit; (
b) View of the 1 dimer (1
2); (
c) The structure of 2 with a water and a DMSO molecule from reference [
10].
When comparing the molecular structure of
1 (
Figure 2a) with that of host
2 already described by us in reference 10 (
Figure 2c) immediately the role of DMSO in changing the conformation of one of the lateral heterocycles (in this case, a 1,8-naphthyridine) is apparent. In its absence, one must expect a conformation similar to that of
1.
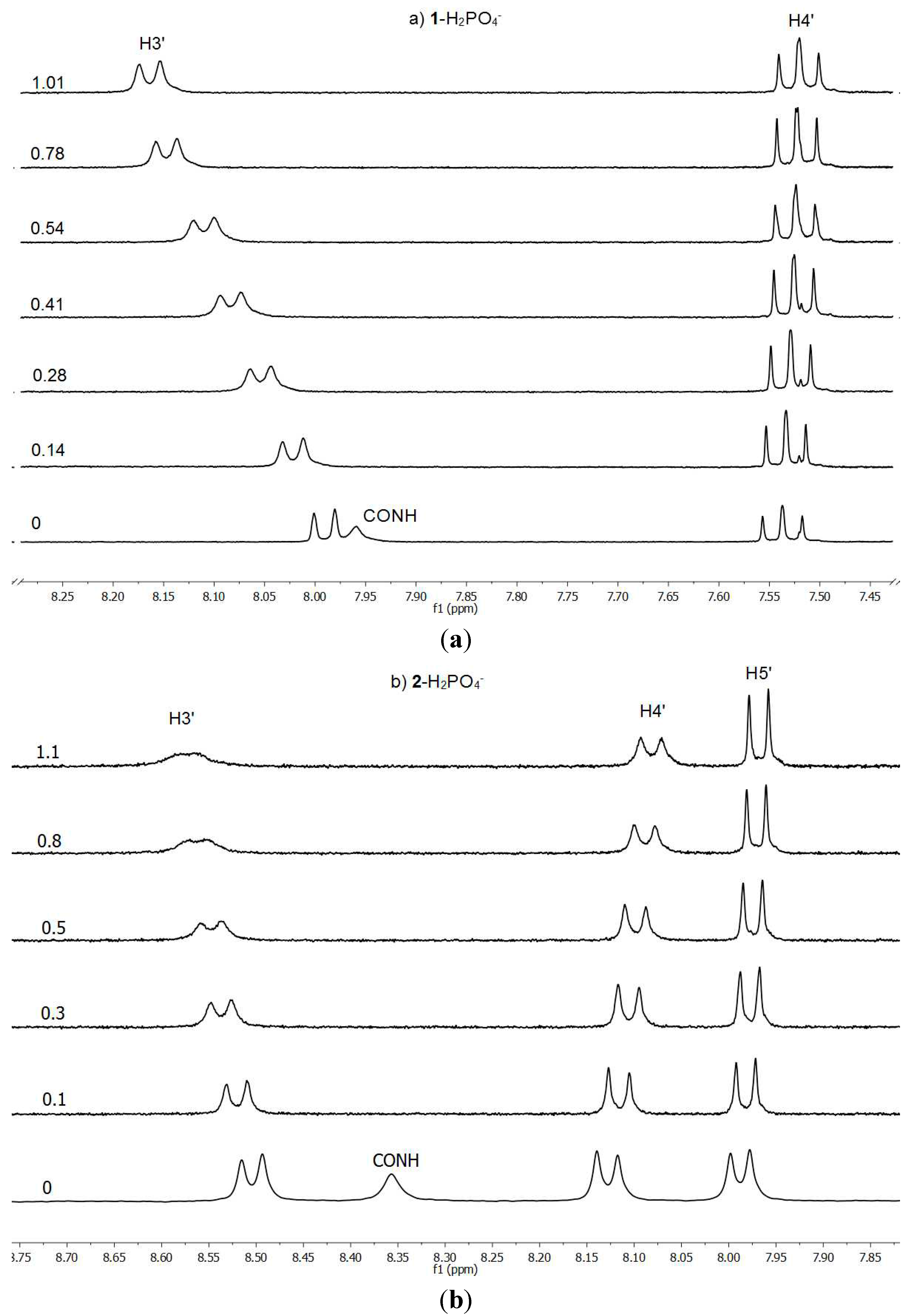
The binding properties have been determined by
1H-NMR titrations in deuterochloroform following the complexation effects on the chemical shifts (CIS) of the amide NH (and the pyrrole NH for BF
4−) and H3
' signals of the hosts
1 and
2, by addition of the anions as their tetrabutylammonium salts, and the association constants
Ka are reported in
Table 2. It is important to note that the acidic protons bonded to the N atoms disappear when the anion is added even, in some cases, at the first 10 μL addition of the solution containing the anion. The pyrrolic N–H is the first to disappear (it is only observed for the
1/BF
4− combination), then both amidic N–Hs that could be used for the titration only with the acetate anion. This is an indication that, in some cases, there are equilibria involving host anions and neutral guests.
Table 2.
Experimental determination of the association constants Ka (M−1).
Table 2.
Experimental determination of the association constants Ka (M−1).
| Host | Guest | H3' | Amide NH | Mean |
|---|
| 1 | BF4− | 702 ± 22 | 595 a ± 123 | 648.5 |
| 1 | CH3CO2− | 37 ± 8 | 24 ± 5 | 30.5 |
| 1 | NO3− | 0 | --- | |
| 1 | H2PO4− | 10005 ± 65 | --- | |
| 1 | PF6− | 0 | --- | |
| 1 | Cl− | 0 | --- | |
| 2 | BF4− | 0 | --- | |
| 2 | CH3CO2− | 312 ± 48 | 294 ± 30 | 303 |
| 2 | NO3− | 0 | --- | |
| 2 | H2PO4− | 440 ± 47 | --- | |
| 2 | PF6− | 0 | --- | |
| 2 | Cl− | 3748 ± 106 | --- | |
As we hypothesized in the introduction, the presence of the 2-methylpyridyl substituent makes receptor 1 highly selective for H2PO4− since it does not recognize NO3−, PF6− and Cl− and only binds moderately to BF4− and very weakly with CH3CO2−. However, the behavior of receptor 2 is different and this needs an explanation that we will provide later on.
The experimental values of
Ka have been transformed into Δ
G values (Δ
G = −RT Ln
Ka, R = 8.314 J·K
−1·mol
−1, T = 298.15 K). Anions for what
Ka values could be determined, which excludes the nitrate and hexafluorophosphate anions, are reported in
Table 3.
The lowest Ka that has been determined corresponds to 1·CH3CO2− (30.5 M−1); in the cases of 1·Cl− and 2·BF4−Ka is not equal to zero but a lower value, that we have assumed Ka = 10 corresponding to ΔG = −5.7; but this value is to be considered with caution.
We have calculated the total Gibbs free energies (
G in Hartree) of the three species, the host H, the guest G and the complex C, and from these values we have calculated Δ
G (C-H-G):
GComplex −
GHost −
GGuest (
Table 3), for the six most interesting cases.
Table 3.
The experimental and calculated host-guest interaction energies (ΔG) (underlined, assumed values). All Δ values in kJ·mol−1.
Table 3.
The experimental and calculated host-guest interaction energies (ΔG) (underlined, assumed values). All Δ values in kJ·mol−1.
| Host | Guest | Exp.
Ka | Exp. ΔG | Calc. ΔG |
|---|
| 1 | BF4− | 648.5 | −16.1 | −57.2 |
| 1 | CH3CO2− | 30.5 | −8.5 | −131.8 |
| 1 | H2PO4− | 10005 | −23.0 | −104.6 |
| 2 | BF4− | 10 | −5.7 | −69.3 |
| 2 | CH3CO2− | 303 | −14.3 | −140.2 |
| 2 | H2PO4− | 440 | −15.2 | −75.9 |
The calculations were carried out at the B3LYP/6-31G(d,p) level (see
Section 3.3. Computational Details). The host, when isolated, was supposed having a
C2 symmetry with a geometry preorganized for the complexation. In some cases, to favor the complexation with some guests, like the acetate anion,
Cs symmetry of the host was considered, however at the end of the optimization process, the
C2 geometry was again obtained.
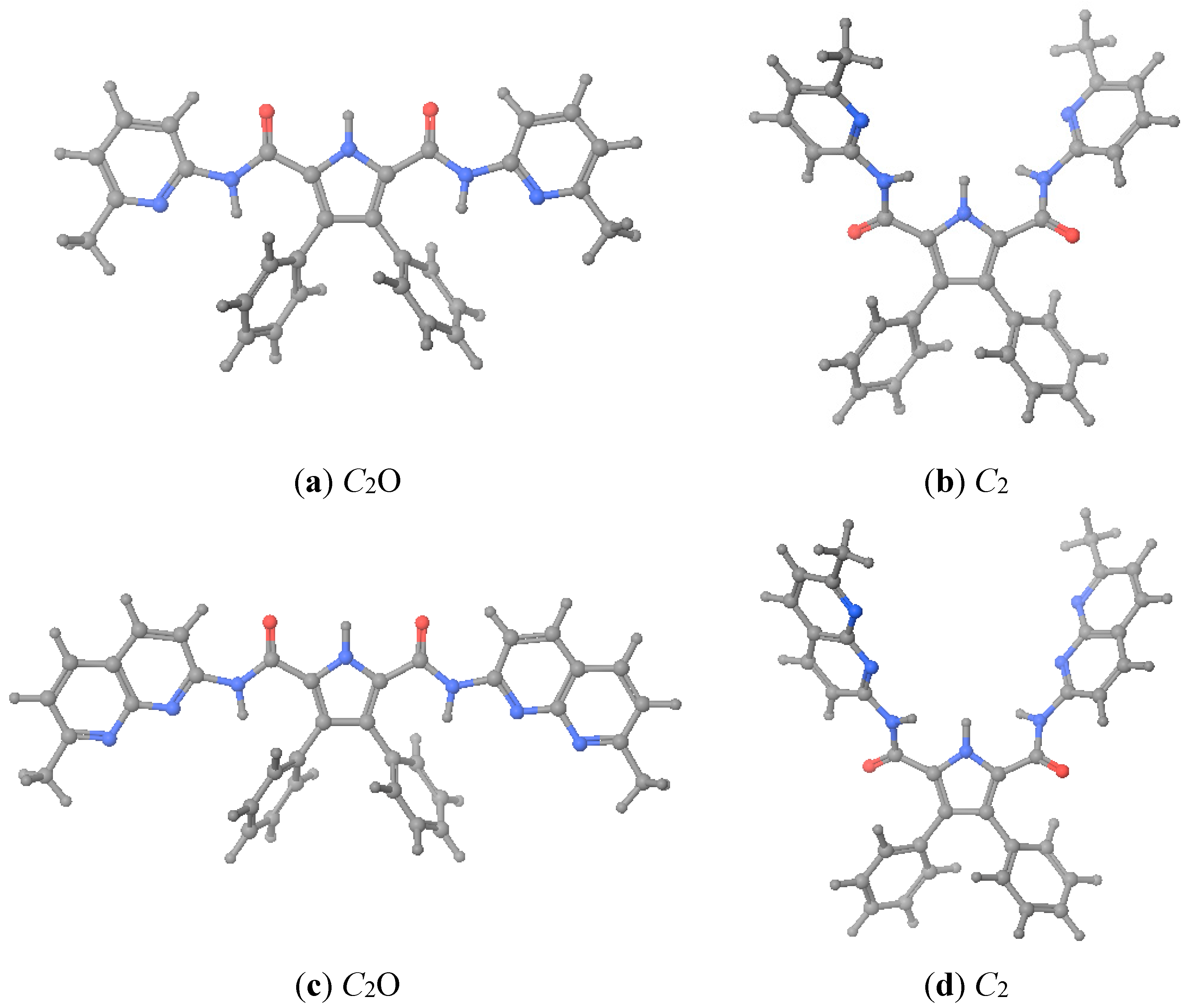
As described in the foregoing section, hosts
1 and
2 crystallized with geometries different from that was assumed as preorganized. When we calculated the energies corresponding to the X-ray geometries (close to
C2O, see
Figure 3), we found that, after optimization, they were 54–58 kJ·mol
−1 more stable (see
Table 4). Therefore, when we calculated the complexation energies we have used the energies corresponding to the more stable X-ray geometries. Note that the
Grel values are lower than the
Erel ones.
Figure 3.
Optimized geometries of hosts 1 and 2. (a) 1·X-ray; (b) 1·preorganized; (c) 2·X-ray; (d) 2·preorganized. C2O is the geometry where the C=O of the urea groups pointed towards the pyrrole NH.
Figure 3.
Optimized geometries of hosts 1 and 2. (a) 1·X-ray; (b) 1·preorganized; (c) 2·X-ray; (d) 2·preorganized. C2O is the geometry where the C=O of the urea groups pointed towards the pyrrole NH.
Table 4.
Energies (E) and free energies (G) in hartrees as well as corresponding energy differences in kJ·mol−1 of hosts 1 and 2 using as starting points the X-ray and preorganized geometries.
Table 4.
Energies (E) and free energies (G) in hartrees as well as corresponding energy differences in kJ·mol−1 of hosts 1 and 2 using as starting points the X-ray and preorganized geometries.
| Host | Structure | E (Hartrees) | Erel (kJ·mol−1) | G (Hartrees) | Grel (kJ mol−1) |
|---|
| 1 | X-ray | −1582.58265 | 0.0 | −1582.15847 | 0.0 |
| preorganized | −1582.56048 | 58.2 | −1582.13729 | 55.6 |
| 2 | X-ray | −1921.95091 | 0.0 | −1921.45682 | 0.0 |
| preorganized | −1921.93051 | 53.6 | −1921.44290 | 36.5 |
Due to the difficulty of obtaining a geometry of the complex with the lowest possible energy, consequence of the high number of degrees of freedom of these systems, the different complexes were built up maximizing the number of stabilizing interactions between host and guest, especially, the maximum number of hydrogen bonds. The calculated free energy results are shown in
Table 5.
Table 5.
Results of the B3LYP/6-31G(d,p) calculations. The column reporting the symmetry of the complex indicates the geometry of the closest one, e.g., C2 means that the geometry is close to C2.
Table 5.
Results of the B3LYP/6-31G(d,p) calculations. The column reporting the symmetry of the complex indicates the geometry of the closest one, e.g., C2 means that the geometry is close to C2.
| Complex | G (Host) (Hartrees) | G (Guest) (Hartrees) | G (Complex) (Hartrees) | Symmetry | ΔG (C-H-G) (Hartrees) | ΔG (kJ·mol−1) |
|---|
| 1·BF4− | −1582.15847 | −424.51203 | −2006.69234 | C2 | −0.0218 | −57.2 |
| 1·CH3CO2− | −1582.15847 | −228.48139 | −1810.69007 | C2 | −0.0502 | −131.8 |
| 1·H2PO4− | −1582.15847 | −643.59035 | −2225.78867 | Csh | −0.0398 | −104.6 |
| 2·BF4− | −1921.45682 | −424.51203 | −2345.99517 | C2 | −0.0264 | −69.3 |
| 2·CH3CO2− | −1921.45682 | −228.48139 | −2149.99162 | C2 | −0.0534 | −140.2 |
| 2·H2PO4− | −1921.45682 | −643.59035 | −2565.07605 | CsO | −0.0289 | −75.9 |
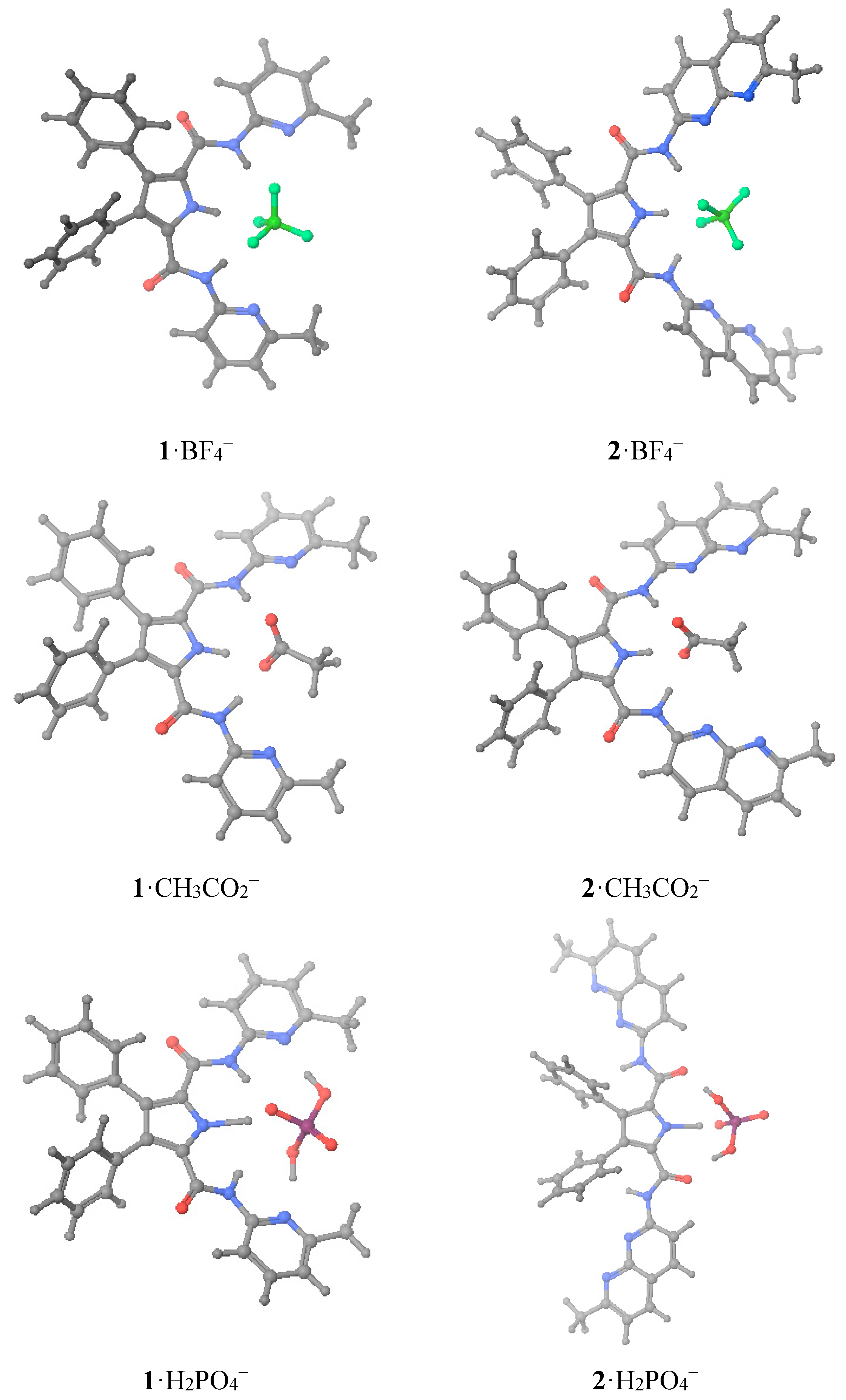
That is, we have theoretically calculated both hosts
1 and
2 (
Table 4) plus the complexes they formed with three monoanions, BF
4−, CH
3CO
2− and H
2PO
4− (
Table 5) whose structures are represented in
Figure 4. Interestingly, a total of three energetic minima were obtained for the complex
1· H
2PO
4−, two with
C2 geometry and the other with a
CsO geometry (see
Figure 3). Both
Figure 4 and
Table 6 depict the calculated as the most stables geometries.
A very interesting result of
Figure 4 and
Table 6 concerns the structures
1·H
2PO
4− and
2·H
2PO
4−. For the pyridinic derivative (host
1), the phosphate not only binds to the pyrrolic N–H by the negatively charged O atom, but also forms two O–H···N HBs with the basic picoline N atoms. This results in a very strong interaction (Δ
Gexp = −23.0 kJ·mol
−1) with the proton of the pyrrole partly transferred to the anion (see
Table 6). The situation of the
2·H
2PO
4− is very different: here the methylnaphthyridine does not play any role and the interaction is sustained by three HBs: one N–H···O
(−) and two O–H···O, which is much less efficient (Δ
Gexp = −15.2 kJ·mol
−1) , although the pyrrolic N–H is similarly elongated. The different behavior of the two hosts is probably related to the fact that pyridine is a much stronger base than 1,8-naphthyridine, 5.20
vs. 3.39 p
Ka units [
14]. In the free host, the N–H bond lengths of the pyrrole and the amides are 1.009 and 1.012 Å for the C
2O and 1.012 and 1.014 Å for the C
2, respectively. The formation of the complexes results in a small elongation of the pyrrolic and amide NHs, the usual effect of the hydrogen bonds. In the complex
1.BF
4− the pyrrole N–H length is 1.026 Å, the shortest of all the complexes (the same happens for the amide N–H bond lengths); this could account for the observation of this proton during the titrations (
Table 2).
The only exception concerns the dihydrogen phosphate anion where a considerable lengthening is observed, indicative of a proton partly transferred from the pyrrole to the guest: dN(pyrrole)−···O4PH3.
Table 6.
Calculated geometries of the hydrogen bonds.
![Molecules 20 09862 i001]()
Table 6.
Calculated geometries of the hydrogen bonds. ![Molecules 20 09862 i001]()
| Host | Guest | Distances (Å) | Angles (°) |
|---|
| 1 | BF4− | dN(pyrrole)–H = 1.026 | (pyrrole)N–H···F1 = 158.4 |
| dN(amide)–H = 1.026 | (amide)N–H···F2 = 167.5 |
| dF1···H–N(pyrrole) = 1.752 | (amide)N–H···F3 = 160.4 |
| dF2···H–N(amide) = 1.774 | |
| dF3···H–N(amide) = 1.823 | |
| 2 | BF4− | dN(pyrrole)–H = 1.026 | (pyrrole)N–H···F1 = 124.7 |
| dN(amide)–H = 1.024 | (pyrrole)N–H···F2 = 164.2 |
| dF1···H–N(pyrrole) = 2.214 | (amide)N–H···F1 = 158.5 |
| dF2···H–N(pyrrole) = 1.703 | (amide)N–H···F2 = 163.4 |
| dF1···H–N(amide) = 1.812 | |
| dF2···H–N(amide) = 1.864 | |
| 1 | CH3CO2− | dN(pyrrole)–H = 1.071 | (pyrrole)N–H···O1 =167.1 |
| dN(amide)–H = 1.041 | (amide)N–H···O1 =171.8 |
| dO1···H–N(pyrrole) = 1.562 | (amide)N–H···O2 =175.0 |
| dO1···H–N(amide) = 1.785 | |
| dO2···H–N(amide) = 1.743 | |
| 2 | CH3CO2− | dN(pyrrole)–H = 1.086 | (pyrrole)N–H···O1 =168.6 |
| dN(amide)–H = 1.045 | (amide)N–H···O1 =172.6 |
| dO1···H–N(pyrrole) = 1.511 | (amide)N–H···O2 =173.5 |
| dO1···H–N(amide) = 1.747 | |
| dO2···H–N(amide) = 1.694 | |
| 1 | H2PO4− | dN(pyrrole)–H = 1.727 | (pyrrole)N–H···O1 = 161.1 |
| dN(amide)–H = 1.016 | (amide)N–H···O1 = 170.1 |
| dO1···H–N(pyrrole) = 1.014 | O2–H···N(pyridine) = 169.8 |
| dO1···H–N(amide) = 1.921 | O3–H···N(pyridine) = 166.6 |
| dO2–H···N(pyridine) = 1.753 | |
| dO3–H···N(pyridine) = 1.694 | |
| 2 | H2PO4− | dN(pyrrole)–H = 1.723 | (pyrrole)N–H···O1 = 173.5 |
| dO1···H–N(pyrrole) = 1.006 | O2–H···O=C(amide) =169.1 |
| dO2–H···O=C(amide) = 1.844 | O3–H···O=C(amide) =176.7 |
| dO2–H···O=C(amide) = 1.807 | |
Figure 4.
Optimized geometries of the six studied complexes.
Figure 4.
Optimized geometries of the six studied complexes.
The calculated Δ
G values of
Table 3 and
Table 5 show that there is a very good relationship between the values of both hosts for the same guests in the case of BF
4− and CH
3CO
2− anions (
2/
1 ratio ≈ 1.1) but not for the H
2PO
4− anion (
2, −75.9;
1, −104.6 kJ·mol
−1, ratio ≈ 0.7). This is a very significant result. In absolute values, calculated ∆
G varies in the order CH
3CO
2− >> H
2PO
4− > BF
4−. This is the order found in some experimental studies, for instance using thiosemicarbazide neutral sensors [
15]. The tetrafluoroborate anion is often used for cationic host recognition of anions because it has no affinity for the receptors [
16,
17].
The experimental ∆
G values ordering depends on the host:
1, H
2PO
4− > BF
4− >> CH
3CO
2;
2, H
2PO
4− > CH
3CO
2− >> BF
4−. Thus, they are different and none follow the calculated values. Why so? The experimental value of
1·BF
4− (−16.1 kJ·mol
−1) is probably overestimated and that of
2·BF
4−(−5.7 kJ·mol
−1) underestimated, thus the correct order for both hosts would be H
2PO
4− > CH
3CO
2− >> BF
4−. Thus compared with the calculated order, there is an inversion between acetate and dihydrogenphosphate. There are two possible explanations, one experimental and the other theoretical. Although we have used a deuterochloroform of the best available quality (stored over silver wire, see Experimental, to prevent the formation of DCl), traces of water cannot be avoided, and if present they could modify the
Ka values in a non-predictive way (specific solvation). The H
2PO
4− anion, according to the calculations, is able to deprotonate the receptor in the gas phase, leading to an anion host plus phosphoric acid (this a well-known phenomenon [
4,
15]), at least partially, and this is an acid-base equilibrium difficult to assess in solution. These explanations, alone or in combination, are the probable cause of the discrepancies between experiments and calculations. What it is the most important theoretical result is that both receptors are very similar when hosting CH
3CO
2− and BF
4− but very different towards H
2PO
4− explaining why
1 is highly selective for H
2PO
4−, contrarily to
2.



 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


