3.2. BeR2:C6X6 Binary Complexes
The binding energy and intermolecular distances of BeR
2:C
6X
6 complexes are listed in
Table 1. The binding energies of the complexes with benzene range between −26 kJ/mol and −47 kJ/mol; the BeCl
2 and BeH
2 complexes are the most stable and the least stable, respectively. The binding energies for the C
6F
6 range between −13 kJ/mol and −25 kJ/mol and are about half of the analogous ones with C
6H
6.
Table 1.
Binding energies (kJ/mol), intermolecular distances (Å) and R-Be-R bond angle (°) of the BeR2:C6X6 binary complexes.
Table 1.
Binding energies (kJ/mol), intermolecular distances (Å) and R-Be-R bond angle (°) of the BeR2:C6X6 binary complexes.
| System | Eb | Be···Z* | > R-Be-R | System | Eb | Be···Z* | > R-Be-R |
|---|
| BeH2:C6H6 | −25.7 | 2.575 | 157.5 | BeH2:C6F6 | −13.1 | 2.945 | 179.0 |
| BeF2:C6H6 | −41.4 | 2.214 | 146.4 | BeF2:C6F6 | −15.8 | 2.916 | 178.6 |
| BeCl2:C6H6 | −46.7 | 2.182 | 139.7 | BeCl2:C6F6 | −24.6 | 3.213 | 177.7 |
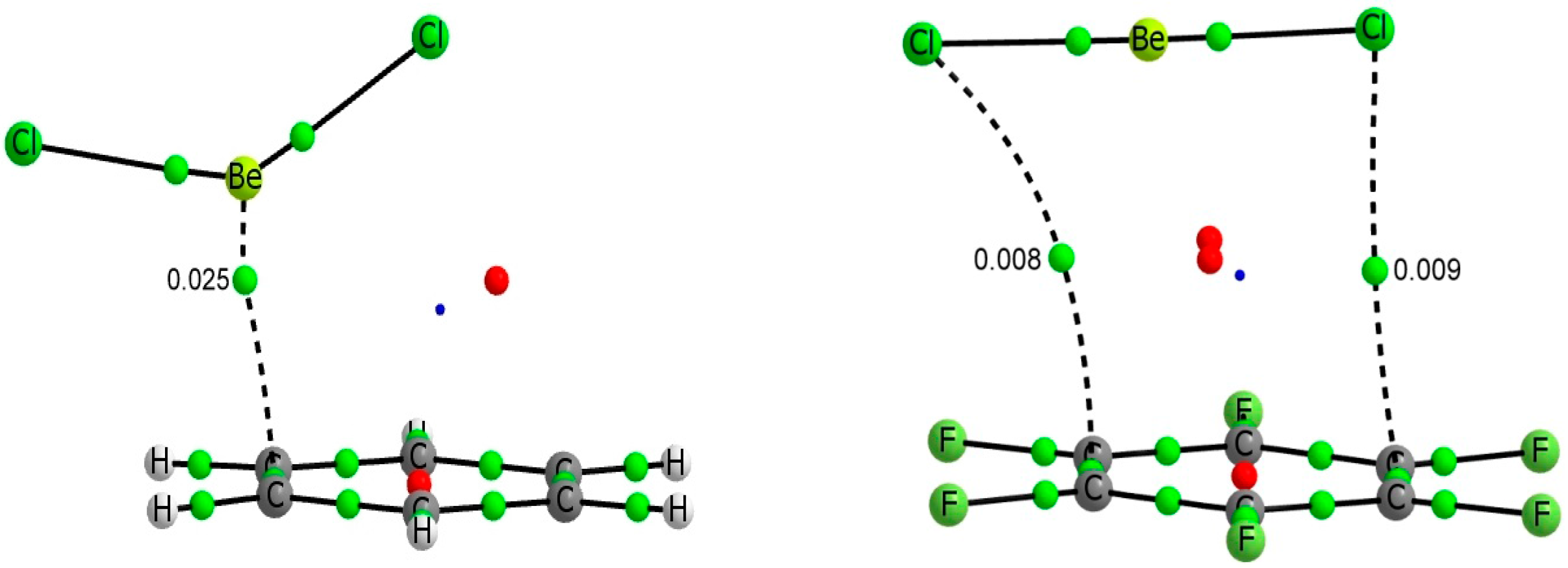
The molecular graph of the BeCl
2:C
6H
6 and BeCl
2:C
6F
6 complexes have been represented in
Figure 2, as a suitable case for BeR
2:C
6H
6 and BeR
2:C
6F
6 systems. Clear differences are observed between the two families of complexes. In complexes with C
6H
6, the beryllium atom of the BeR
2 derivatives is located above and close to one of the C-C bonds and slightly out of the aromatic ring while in the C
6F
6 family the Be is far from the C-C bond and placed close to the center of the aromatic ring.
Figure 2.
Molecular graph of BeR2:C6H6 (R = H, Cl) (left) and BeR2:C6F6 (R = H, Cl) (right) binary complexes. Green, red and blue dots denote BCPs, ring critical points and cage critical points respectively. The value of the electron density at the intermolecular BCP is indicated.
Figure 2.
Molecular graph of BeR2:C6H6 (R = H, Cl) (left) and BeR2:C6F6 (R = H, Cl) (right) binary complexes. Green, red and blue dots denote BCPs, ring critical points and cage critical points respectively. The value of the electron density at the intermolecular BCP is indicated.
The NBO analysis offers some clue on the origin of the aforementioned differences between BeR
2:C
6H
6 and BeR
2:C
6F
6 complexes. In both cases the aromatic moiety behaves as a Lewis base with respect to the BeR
2 moiety, since a clear charge donation from the occupied π
cc orbitals of the aromatic into the empty p orbitals of Be and into the σ
BeR* antibonding orbital is detected from the calculated second order orbital perturbation energies. The former are responsible for the bending undergone by the BeR
2 moiety and the latter for the lengthening of the Be-R distances when BeR
2 forms part of the complex. The NBO analysis shows that for C
6H
6 complexes, the larger contribution comes from a couple of C=C bonds, reflecting that the orbital interaction energies strongly depend on the overlap of the interacting occupied and empty orbitals. Clearly, the specific interaction with two of the CC bonds is privileged with respect to an equal interaction with the six bonds because in the first situation the overlap is much more efficient. In the case of the C
6F
6, the aforementioned interactions are much weaker, since C
6F
6 is a much poorer electron donor than C
6H
6. Indeed, as indicated in
Table 2, the natural charges obtained within the NBO approach clearly show that the charge transfer from the aromatic systems towards the beryllium derivatives, is about three times larger when the aromatic is benzene than when it is C
6F
6.
Table 2.
NBO charges (e) of the aromatic system within the BeR2:C6X6 complexes.
Table 2.
NBO charges (e) of the aromatic system within the BeR2:C6X6 complexes.
| | NBO Charges (e) | | NBO Charges (e) |
|---|
| BeH2:C6H6 | 0.048 | BeH2:C6F6 | 0.017 |
| BeF2:C6H6 | 0.066 | BeF2:C6F6 | 0.005 |
| BeCl2:C6H6 | 0.116 | BeCl2:C6F6 | 0.012 |
However, also in C
6F
6 complexes there is a tendency to privilege the donation for only one couple of CC bonds. Actually, as shown in
Figure 2, the BeR
2 moiety does not sit strictly above the center of the ring, but it is also slightly displaced towards one of its CC bonds. However, since the interactions for C
6F
6 are much weaker than for benzene, the distance between both moieties is much longer, and the overlap does not privilege significantly the interaction with a specific pair of CC bonds, with respect to the others, leading to a more centered position of the BeR
2 subunit. The fact that C
6F
6 is a much poorer electron donor than C
6H
6 is also clearly mirrored on the fact that in the C
6H
6 complex, the disposition of the three atoms of the BeR
2 molecule is far from linearity, reaching R-Be-R angles of 140° in the strongest complex, while in the complexes with C
6F
6 the change of this angle is very small (less than 2.5°).
It is worth noting that the BeR2:C6F6 complexes with the beryllium atom along the C6 symmetry axes, which have a C2v symmetry, present one imaginary frequency and a very small relative energy (less than 2.0 kJ/mol) with respect to the equilibrium conformation, corresponding to a transition state between two identical structures.
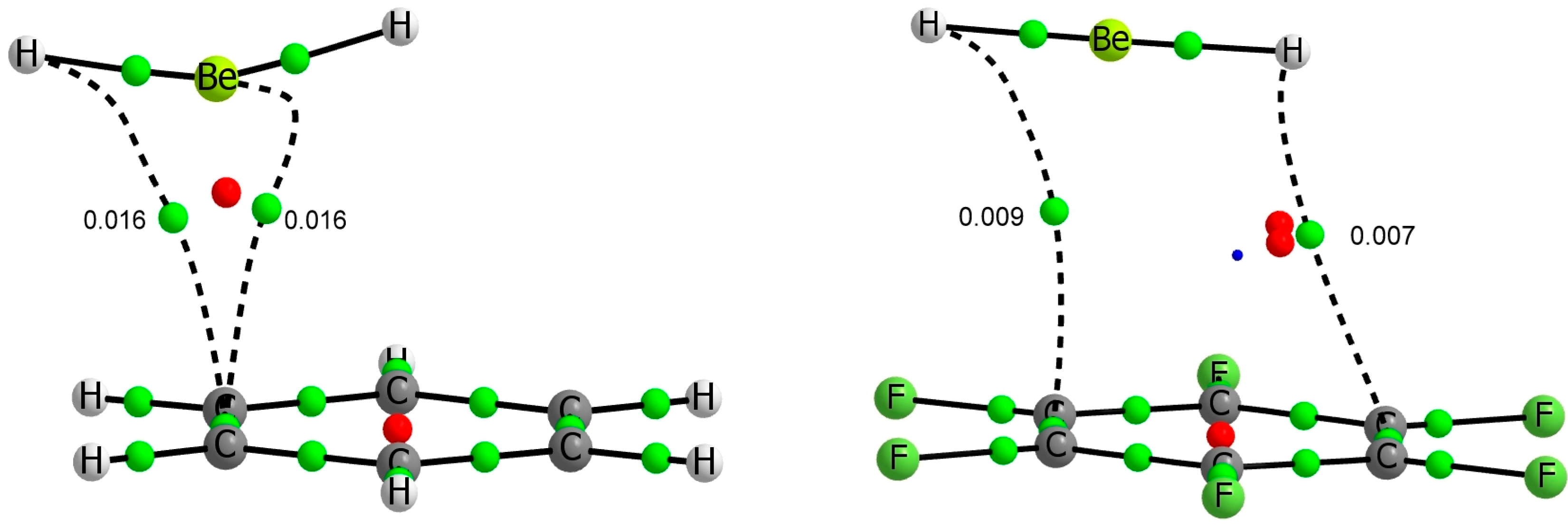
In line with the NBO analysis discussed above, the AIM approach shows the existence of just one intermolecular BCP between the beryllium atom and the centre of a C-C bond for complexes involving benzene (
Figure 2). The values of the electron density at these BCPs range between 0.016 (BeH
2) and 0.025 au (BeCl
2). Positive values of the Laplacian and negative total energy density (between −0.003 and −0.006 au) are found in the BCPs (see
Table 3), confirming that these interactions have a certain covalent character [
56].
In the BeR
2:C
6F
6 complexes, mentioned above, the interaction is much weaker and more delocalized, the intermolecular BCPs link the R atoms with the aromatic ring through two opposite C-C bonds. The electron density at the BCPs is rather small (between 0.009 and 0.007 au) and the Laplacian and total energy density are positive or nearly zero (
Table 3).
Table 3.
AIM parameters (in au) for the BCPs corresponding to the intermolecular interactions in the BeR2:C6X6 binary systems, the electron density, ρBCP, its Laplacian, ∇2ρBCP, and the total electron energy density, HBCP.
Table 3.
AIM parameters (in au) for the BCPs corresponding to the intermolecular interactions in the BeR2:C6X6 binary systems, the electron density, ρBCP, its Laplacian, ∇2ρBCP, and the total electron energy density, HBCP.
| System | ρBCP | ∇2ρBCP | HBCP | Interaction |
|---|
| BeH2:C6H6 | 0.0157 | 0.0184 | -0.0028 | Be···π |
| BeF2:C6H6 | 0.0218 | 0.0409 | -0.0052 | Be···π |
| BeCl2:C6H6 | 0.0247 | 0.0577 | -0.0059 | Be···π |
| BeH2:C6F6 | 0.0085 | 0.0153 | -0.0001 | H···π |
| 0.0067 | 0.0192 | 0.0008 | H···π |
| BeF2:C6F6 | 0.0091 | 0.0263 | 0.0008 | F···π |
| 0.0084 | 0.0320 | 0.0012 | F···π |
| BeCl2:C6F6 | 0.0079 | 0.0180 | 0.0004 | Cl···π |
| 0.0085 | 0.0240 | 0.0008 | Cl···π |
The calculated HOMA aromaticity indexes for these complexes (See
Table S3 of the
Supplementary Materials) are very similar to the corresponding isolated aromatic molecules, being the largest differences 0.01 units.
The application of the MBIE partition method shows that for both families of compounds the distortion energy of the aromatic ring is very small, as it is also for the BeR
2 systems in the complexes with C
6F
6 (See
Table 4). In contrast, the distortion energies of the BeR
2 molecules in the complexes with C
6H
6 present values between 11 and 39 kJ/mol in agreement with the geometrical perturbation already discussed. Consequently, the interaction energy (Δ
2E) of these complexes reaches values up to −87 kJ/mol in the C
6H
6:BeCl
2 case while in the ones with C
6F
6 the values of Δ
2E are about four times smaller and very similar to those of the binding energies.
Table 4.
Many body Interaction energy (MBIE) partition terms (kJ/mol) in the BeR2:C6X6 binary systems.
Table 4.
Many body Interaction energy (MBIE) partition terms (kJ/mol) in the BeR2:C6X6 binary systems.
| System | Er(Ar) | Er(BeR2) | Δ2E(BeR2:C6H6) | System | Er(Ar) | Er(BeR2) | Δ2E(BeR2:C6F6) |
|---|
| BeH2:C6H6 | 0.2 | 10.7 | −36.6 | BeH2:C6F6 | 0.16 | 0.03 | −13.3 |
| BeF2:C6H6 | 0.5 | 26.3 | −68.2 | BeF2:C6F6 | 0.3 | 0.1 | −16.2 |
| BeCl2:C6H6 | 0.9 | 39.0 | −86.6 | BeCl2:C6F6 | 0.3 | 0.05 | −24.9 |
3.3. C6X6:Y− Binary Complexes
As expected from the characteristics of the molecular electrostatic potential discussed above, the equilibrium structure for C
6H
6:Y
− complexes is totally different from that of C
6F
6, in agreement with previous reports [
3,
57,
58,
59]. In the C
6H
6 complexes, the anion is located in the molecular plane, interacting simultaneously with two hydrogen atoms, whereas in the C
6F
6:Y
− complexes the anion sits on the
C6 symmetry axis and above the plane of the molecule.
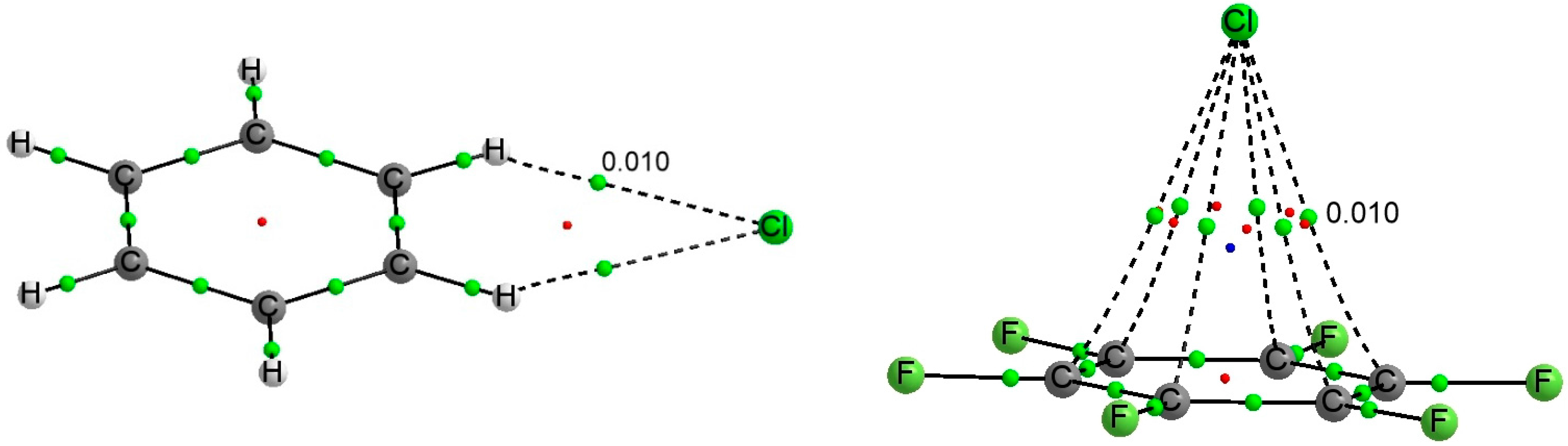
Figure 3 shows the molecular graph of two representative C
6X
6:Y
− complexes.
Figure 3.
Molecular graph of the C6H6:Cl− (left) and C6F6:Cl− (right) complexes. Green, red and blue dots denote BCPs, ring and cage critical points respectively. The value of the electron density at the intermolecular BCP is indicated.
Figure 3.
Molecular graph of the C6H6:Cl− (left) and C6F6:Cl− (right) complexes. Green, red and blue dots denote BCPs, ring and cage critical points respectively. The value of the electron density at the intermolecular BCP is indicated.
The binding energies of these complexes (
Table 5) show that the C
6F
6:Y
− complexes are almost twice more stable than the C
6H
6:Y
− ones, in contrast with the results obtained for the BeR
2:C
6X
6 complexes, simply because in the complexes with BeR
2 the aromatic ring behaves as a Lewis base
versus a rather strong Lewis acid, whereas in the complexes with Y
− they behave as a Lewis acid, which can only accept electrons in the π* antibonding orbitals. The nature of the halide has a small effect on the binding energy, the complexes with chloride being slightly more stable than with bromide. The MBIE partition (
Table 4) shows very small distortion energies for the aromatic systems and consequently, the interaction energies (Δ
2E) are very similar to the binding ones.
The molecular graph of these complexes (see
Figure 3 for two examples) shows two degenerate Y···H BCPs in the C
6H
6:Y
− complexes, corresponding to the two hydrogen bonds between the halogen anion and the CH groups of benzene, and six Y···C BCP in C
6F
6:Y
−. Those BCPs show similar values of the electron density, 0.010 au for the chloride complexes and 0.009 au for the bromide ones. In all cases, the BCPs show positive values of the Laplacian and total energy density.
Table 5.
Binding energy (kJ/mol), intermolecular distance (Å), distortion energy and Δ2E (kJ/mol) in the C6X6:Y− binary systems within the MBIE partition method.
Table 5.
Binding energy (kJ/mol), intermolecular distance (Å), distortion energy and Δ2E (kJ/mol) in the C6X6:Y− binary systems within the MBIE partition method.
| System | Eb | Y···HC | Er(C6H6) | Δ2E(C6H6:Y) | System | Eb | Y···Z* | Er(C6F6) | Δ2E(C6F6:Y) |
|---|
| C6H6:Br− | −34.4 | 2.902 | 1.2 | −35.6 | C6F6:Br− | −65.8 | 3.433 | 0.7 | −66.6 |
| C6H6:Cl− | −35.9 | 2.743 | 1.6 | −37.5 | C6F6:Cl− | −67.1 | 3.290 | 0.9 | −67.9 |
The NBO analysis indicates a larger charge transfer for the C6H6:Y− complexes (−0.026 and −0.027 e, for Y = Br and Cl, respectively) than for the C6F6:Y− ones (−0.013 and −0.012 e), as a consequence of the rather different nature of both kinds of interactions, since, as indicated above the former are stabilized through intermolecular C-H···Y− hydrogen bonds and the latter through Y−-π interactions. Coherently, the second order perturbation analysis indicates a charge transfer in the C6H6:Y− complexes from the lone pairs of the anions towards the σCH* antibonding orbitals with interaction energies up to 7.4 kJ/mol, while in the C6F6:Y− ones, the expected charge transfer between the lone pair of the anions and the πCC* antibonding orbitals of the aromatic systems is very small (<0.7 kJ/mol).
3.4. BeR2:C6X6:Y− Ternary Complexes
The binding energy and intermolecular distances of the BeR
2:C
6X
6:Y
− (R = H, F, Cl; X = H, F; Y = Cl, Br) ternary complexes have been listed in
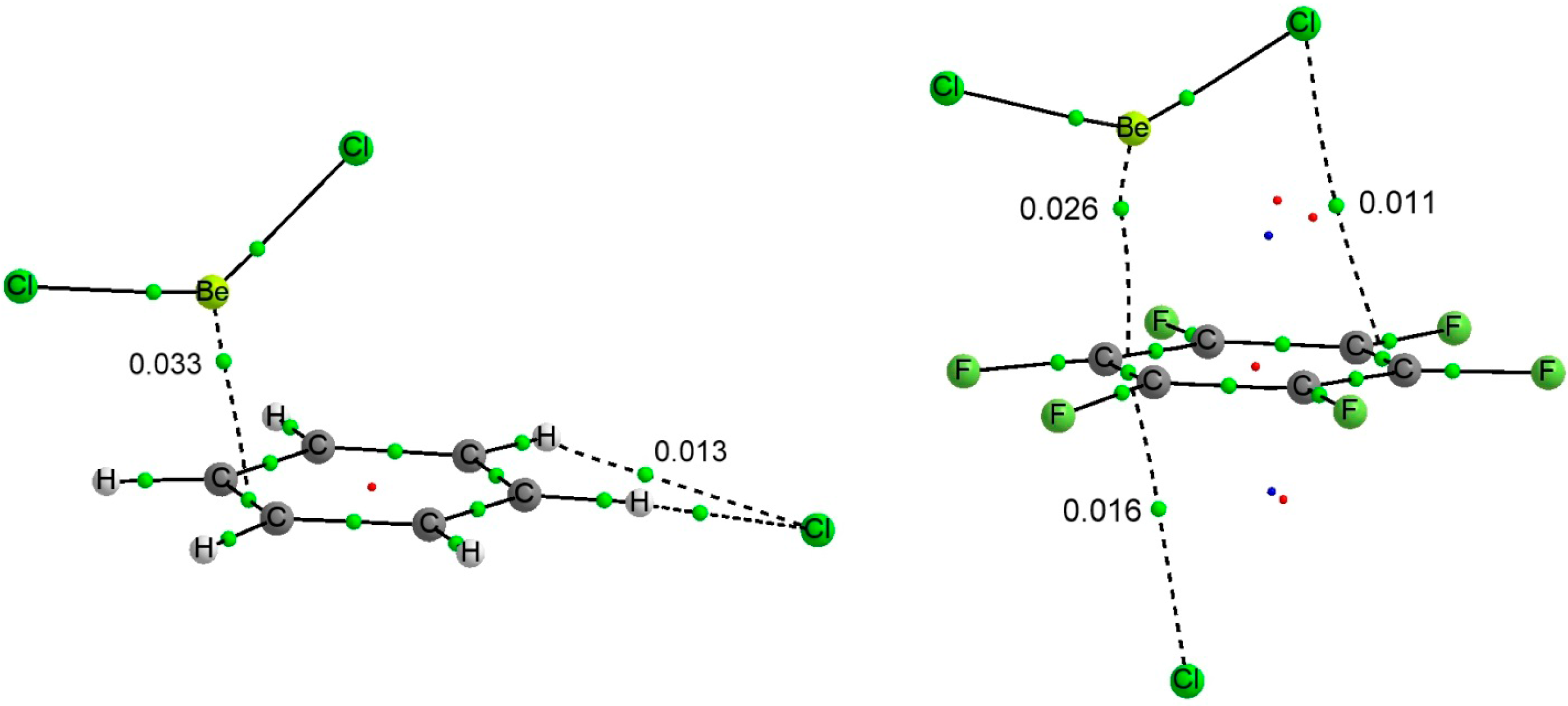
Table 6. The molecular graphs of two representative ternary complexes have been represented in
Figure 4.
Table 6.
Binding energy (kJ/mol), intermolecular distances (Å) and R-Be-R bond angle (°) of the ternary complexes. The variations with respect to the corresponding binary complexes are also added.
Table 6.
Binding energy (kJ/mol), intermolecular distances (Å) and R-Be-R bond angle (°) of the ternary complexes. The variations with respect to the corresponding binary complexes are also added.
| System | Eb | Be···Z* | ∆Be···Z* | Y···Z* | ∆Y···Z* | ∠ R-Be-R | ∆∠ R-Be-R |
|---|
| BeH2:C6H6: Br− | −80.3 | 2.185 | −0.390 | 2.820 | −0.082 | 145.7 | −11.8 |
| BeH2:C6H6:Cl− | −83.2 | 2.177 | −0.398 | 2.658 | −0.085 | 145.2 | −12.3 |
| BeF2:C6H6:Br− | −104.6 | 2.089 | −0.125 | 2.802 | −0.100 | 138.1 | −8.3 |
| BeF2:C6H6:Cl− | −107.9 | 2.084 | −0.130 | 2.639 | −0.104 | 137.7 | −8.7 |
| BeCl2:C6H6 :Br− | −118.7 | 2.042 | −0.140 | 2.778 | −0.124 | 132.0 | −7.7 |
| BeCl2:C6H6:Cl− | −122.4 | 2.034 | −0.148 | 2.616 | −0.127 | 131.7 | −8.0 |
| BeH2:C6F6:Br− | −96.7 | 2.413 | −0.532 | 3.204 | −0.229 | 155.8 | −23.2 |
| BeH2:C6F6:Cl− | −99.0 | 2.396 | −0.549 | 3.031 | −0.259 | 154.8 | −24.2 |
| BeF2:C6F6:Br− | −112.8 | 2.270 | −0.646 | 3.156 | −0.277 | 144.6 | −34.0 |
| BeF2:C6F6:Cl− | −115.6 | 2.261 | −0.655 | 2.990 | −0.300 | 143.9 | −34.7 |
| BeCl2:C6F6:Br− | −122.5 | 2.253 | −0.960 | 3.126 | −0.307 | 137.9 | −39.8 |
| BeCl2:C6F6:Cl− | −125.8 | 2.242 | −0.971 | 2.957 | −0.333 | 137.2 | −40.5 |
The binding energies in the ternary complexes range between −80 and −126 kJ/mol. The C6F6 complexes are always more stable than the analogous with C6H6. As in the case of the binary complexes, the ranking based on the beryllium derivative is BeH2 > BeF2 > BeCl2 and the difference between the binding energy in the chloride and bromide complexes is small, the chloride complexes always being more stable than the bromide ones. An excellent linear correlation is obtained between the binding energies in the C6F6 vs. the C6H6 series (R2 = 0.999).
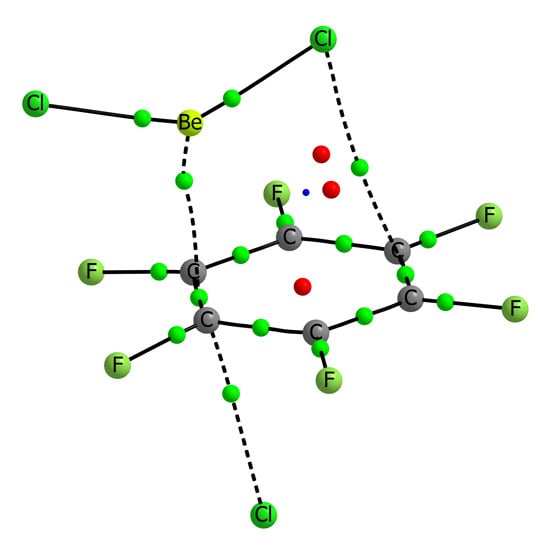
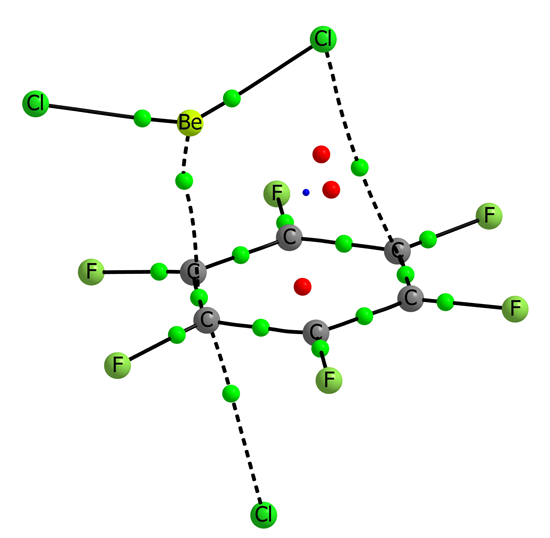
Figure 4.
Molecular graph of BeCl2:C6H6:Cl− (left) and BeCl2:C6F6:Cl− (right). The value of the electron density at the intermolecular BCPs is indicated.
Figure 4.
Molecular graph of BeCl2:C6H6:Cl− (left) and BeCl2:C6F6:Cl− (right). The value of the electron density at the intermolecular BCPs is indicated.
The geometrical parameters listed in
Table 6 already provide some clues about the cooperativity in the ternary complexes. The intermolecular distances between the aromatic systems and the beryllium derivatives are reduced up to 0.40 Å in the C
6H
6 series and up to 0.97 Å in the C
6F
6 ones when comparing to the corresponding binary complexes. In C
6H
6 complexes, the larger effects are observed for complexes with BeH
2 and for the BeCl
2 for C
6F
6 complexes. Similar shortening is observed for the intermolecular distances between the anions and the aromatic rings. The larger effect observed in both series corresponds to the complexes with BeCl
2 being the calculated shortening 0.13 and 0.30 Å in the C
6H
6 and C
6F
6 series, respectively.
Another geometrical parameter that changes from the binary to the ternary complexes is the R-Be-R bond angle which is always smaller in the latter ones. The largest effect is observed in the BeCl2:C6F6:Y− complexes, where the variation of the R-Be-R bond angle on going from the binary to the ternary complexes is 40°.
As in the case of the binary complexes, the calculated HOMA aromaticity indexes for the ternary complexes (See
Table S3) are almost identical to those of the corresponding isolated aromatic molecules, being the largest differences 0.02 units.
The MBIE partition terms of the ternary complexes have been gathered in
Table 7. The distortion energy in the aromatic molecules is small in all cases (between +1.7 and +4.3 kJ/mol), but larger than in binary complexes, while those of the beryllium derivatives complexed with C
6H
6 range between +26 and +59 kJ/mol and in the complexes with C
6F
6 between +12 and +44 kJ/mol, are also larger than in the binary complexes. The three Δ
2E terms and the Δ
3E one for all the compounds are negative. The largest stabilization energy is the Δ
2E(BeR
2:Ar) for the C
6H
6 complexes and Δ
2E(Ar:Y) for the C
6F
6 ones. For the C
6H
6 complexes the second most important term is the Δ
2E(Ar:Y) followed by the Δ
3E(BeR
2:Ar:Y) one, the least important one being the Δ
2E(BeR
2:Y). In the C
6F
6 complexes, Δ
2E(BeR
2:Y) is of similar magnitude to that of Δ
2E(BeR
2:Ar) in the BeR
2:C
6F
6:Y for R = H and F while for R = Cl, Δ
2E(BeR
2:Ar) is more important than Δ
2E(BeR
2:Y). The negative value of ∆
3E, which indicates strong cooperativity, ranges between −21 and −35 kJ/mol in the C
6H
6 complexes and between −13 and −24 kJ/mol in the C
6F
6 ones. The Δ
2E(BeR
2:Ar) term is always larger in absolute value in the ternary complexes than in the binary ones while the Δ
2E(Ar:Y) one is slightly smaller in absolute value in the ternary than in the corresponding binary complexes.
Table 7.
Many body Interaction energy (MBIE) partition term (kJ/mol) in the ternary systems *.
Table 7.
Many body Interaction energy (MBIE) partition term (kJ/mol) in the ternary systems *.
| System | Er(Ar) | Er(BeR2) | Δ2E(BeR2:Ar) | Δ2E(Ar:Y) | Δ2E(BeR2:Y) | Δ3E(BeR2:Ar:Y) |
|---|
| BeH2:C6H6: Br− | 2.1 | 25.8 | −48.2 | −34.5 | −4.3 | −21.1 |
| BeH2:C6H6:Cl− | 2.6 | 26.5 | −48.7 | −36.5 | −4.5 | −22.6 |
| BeF2:C6H6:Br− | 2.5 | 43.3 | −81.7 | −33.8 | −11.0 | −24.0 |
| BeF2:C6H6:Cl− | 3.0 | 44.3 | −82.3 | −35.8 | −11.5 | −25.6 |
| BeCl2:C6H6 :Br− | 3.4 | 58.4 | −103.0 | −33.1 | −11.6 | −32.8 |
| BeCl2:C6H6:Cl− | 4.0 | 59.3 | −103.6 | −35.1 | −12.1 | −34.8 |
| BeH2:C6F6:Br− | 1.7 | 12.4 | −16.1 | −65.2 | −16.7 | −12.9 |
| BeH2:C6F6:Cl− | 1.8 | 13.5 | −16.3 | −66.3 | −18.3 | −13.5 |
| BeF2:C6F6:Br− | 2.4 | 29.2 | −32.7 | −64.2 | −32.3 | −15.1 |
| BeF2:C6F6:Cl− | 2.4 | 30.4 | −33.1 | −65.2 | −34.7 | −15.4 |
| BeCl2:C6F6:Br− | 4.3 | 42.2 | −49.5 | −63.5 | −33.0 | −22.9 |
| BeCl2:C6F6:Cl− | 4.3 | 43.8 | −50.2 | −64.5 | −35.5 | −23.8 |
The topology of the molecular graph of the BeR
2:C
6H
6:Y
− complexes is similar to the sum of those of the corresponding dimers. However the electron density values in the intermolecular BCPs (
Table 8) are larger in the ternary complexes than in the corresponding binary ones [0.033 vs. 0.0025 au in the Be-π BCP and 0.013 vs. 0.010 in the Cl···HC interaction in the BeCl
2:C
6H
6:Cl
− complex and its corresponding binary complexes,
Figure 2,
Figure 3 and
Figure 4] in agreement with the shorter intermolecular distances found in the former complexes and the relationship between the electron density at the BCP and the interatomic distance [
60,
61,
62,
63,
64,
65,
66], and with the negative values of the ∆
3E terms. As a consequence of the substantial reinforcement of both the beryllium bonds and the interaction between the aromatic and the anion Y
− on going from the binary complexes to the triads, the molecular graph of the triads BeR
2:C
6F
6:Y
−, presents a single intermolecular BCP between the anion and the aromatic ring (
Figure 4) in contrast to the six BCPs found in the binary complexes (
Figure 3) and a BCP connecting the beryllium atom with the aromatic ring while in the binary complexes the two BCPs were between the R groups and the aromatic ring. Consistently, for the BeR
2:C
6H
6:Y
−, both the electron density at the BCP connecting the beryllium atom with the aromatic ring and at the CH···Y
− hydrogen bonds are much larger in the triad than in the corresponding binary complexes.
Table 8.
AIM parameters (in au) for the BCPs corresponding to the Be···π and π···Y interactions in the ternary systems, the electron density, ρBCP, its Laplacian, ∇2ρBCP, and the total electron energy density, HBCP.
Table 8.
AIM parameters (in au) for the BCPs corresponding to the Be···π and π···Y interactions in the ternary systems, the electron density, ρBCP, its Laplacian, ∇2ρBCP, and the total electron energy density, HBCP.
| System | Be···π | π···Y− |
|---|
| ρBCP | ∇2ρBCP | HBCP | ρBCP | ∇2ρBCP | HBCP |
|---|
| BeH2:C6H6: Br− | 0.0224 | 0.0278 | −0.0059 | 0.0111 | 0.0288 | 0.0005 |
| BeH2:C6H6:Cl− | 0.0227 | 0.0295 | −0.0060 | 0.0123 | 0.0347 | 0.0007 |
| BeF2:C6H6:Br− | 0.0280 | 0.0860 | −0.0048 | 0.0115 | 0.0299 | 0.0005 |
| BeF2:C6H6:Cl− | 0.0283 | 0.0872 | −0.0048 | 0.0128 | 0.0362 | 0.0007 |
| BeCl2:C6H6 :Br− | 0.0326 | 0.0974 | −0.0065 | 0.0120 | 0.0314 | 0.0005 |
| BeCl2:C6H6:Cl− | 0.0330 | 0.0990 | −0.0070 | 0.0130 | 0.0380 | 0.001 |
| BeH2:C6F6:Br− | 0.0164 | 0.0182 | −0.0041 | 0.0131 | 0.0364 | 0.0011 |
| BeH2:C6F6:Cl− | 0.0169 | 0.0190 | −0.0043 | 0.0146 | 0.0455 | 0.0016 |
| BeF2:C6F6:Br− | 0.0222 | 0.0373 | −0.0060 | 0.0140 | 0.0399 | 0.0012 |
| BeF2:C6F6:Cl− | 0.0226 | 0.0409 | −0.0060 | 0.0155 | 0.0494 | 0.0017 |
| BeCl2:C6F6:Br− | 0.0252 | 0.0336 | −0.0082 | 0.0147 | 0.0421 | 0.0012 |
| BeCl2:C6F6:Cl− | 0.0257 | 0.0377 | −0.0082 | 0.0163 | 0.0524 | 0.0018 |
Similar reinforcements of both non-covalent interactions become evident when the NBO analysis is employed, reflected in much larger charge transfer towards the beryllium derivative from both the anion and the aromatic systems (
Table 9). At the same time, the second order perturbation analysis shows an increment of the charge transferred from the C-C bonds of the aromatic systems towards the empty ones of the beryllium that corresponds to E(2) stabilization values of 98 and 21 kJ/mol in the BeH
2:C
6X
6:Cl
−, with X = H and F, respectively.
Table 9.
Charge (e) of the monomers in the ternary complex.
Table 9.
Charge (e) of the monomers in the ternary complex.
| System | Aromatic | BeR2 | Y− |
|---|
| BeH2:C6H6: Br− | 0.087 | −0.124 | −0.963 |
| BeH2:C6H6:Cl− | 0.088 | −0.126 | −0.962 |
| BeF2:C6H6:Br− | 0.058 | −0.010 | −0.959 |
| BeF2:C6H6:Cl− | 0.059 | −0.101 | −0.958 |
| BeCl2:C6H6 :Br− | 0.115 | −0.161 | −0.953 |
| BeCl2:C6H6:Cl− | 0.117 | −0.163 | −0.953 |
| BeH2:C6F6:Br− | 0.067 | −0.104 | −0.963 |
| BeH2: C6F6:Cl− | 0.070 | −0.107 | −0.962 |
| BeF2: C6F6:Br− | 0.116 | −0.167 | −0.949 |
| BeF2: C6F6:Cl− | 0.120 | −0.172 | −0.948 |
| BeCl2: C6F6:Br− | 0.060 | −0.102 | −0.958 |
| BeCl2: C6F6:Cl− | 0.063 | −0.107 | −0.957 |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



