
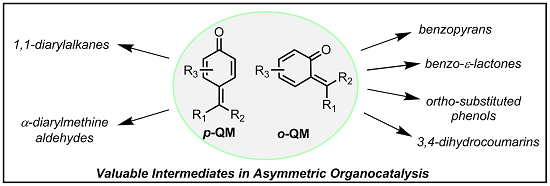
The Emergence of Quinone Methides in Asymmetric Organocatalysis



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
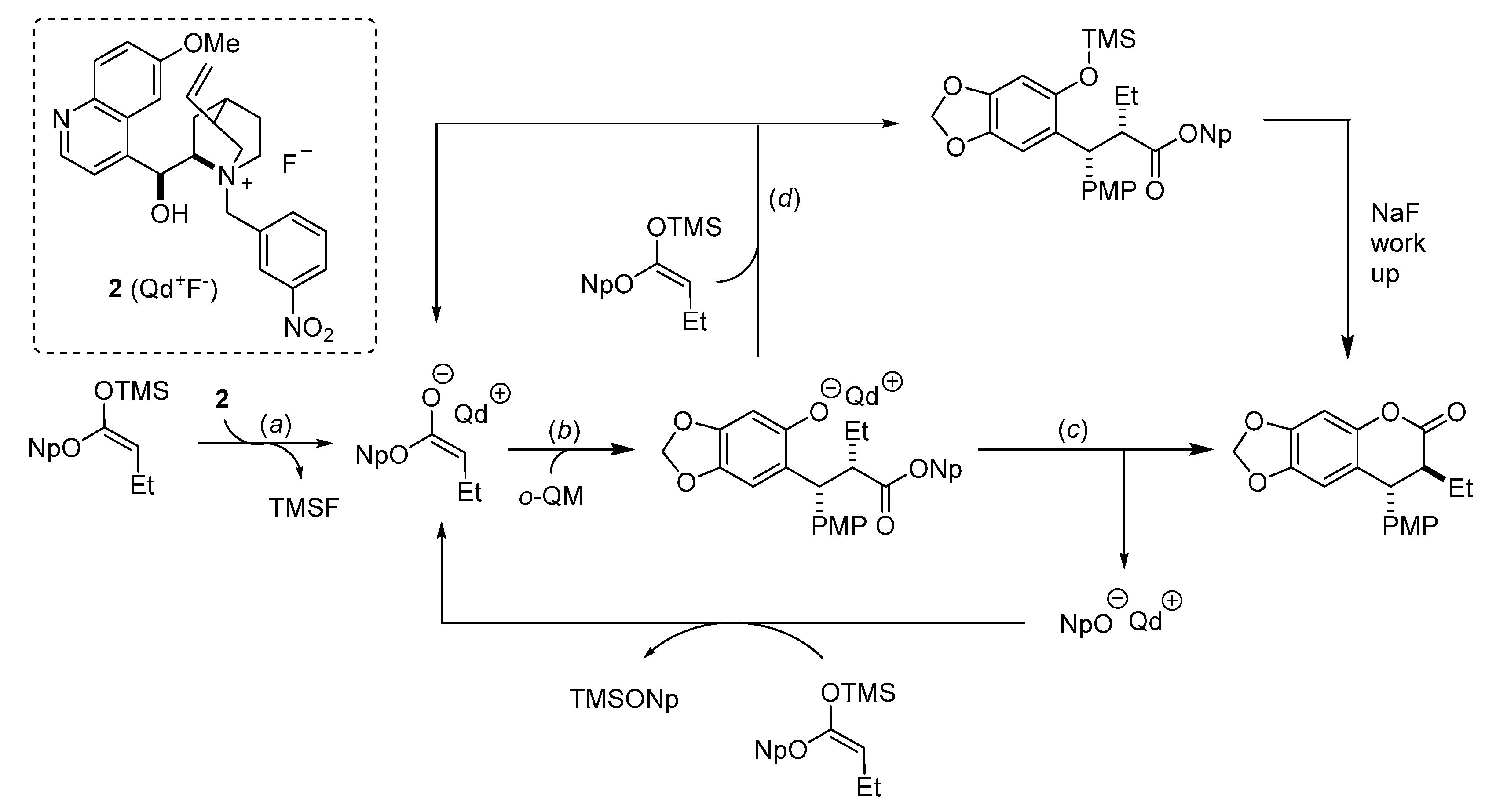
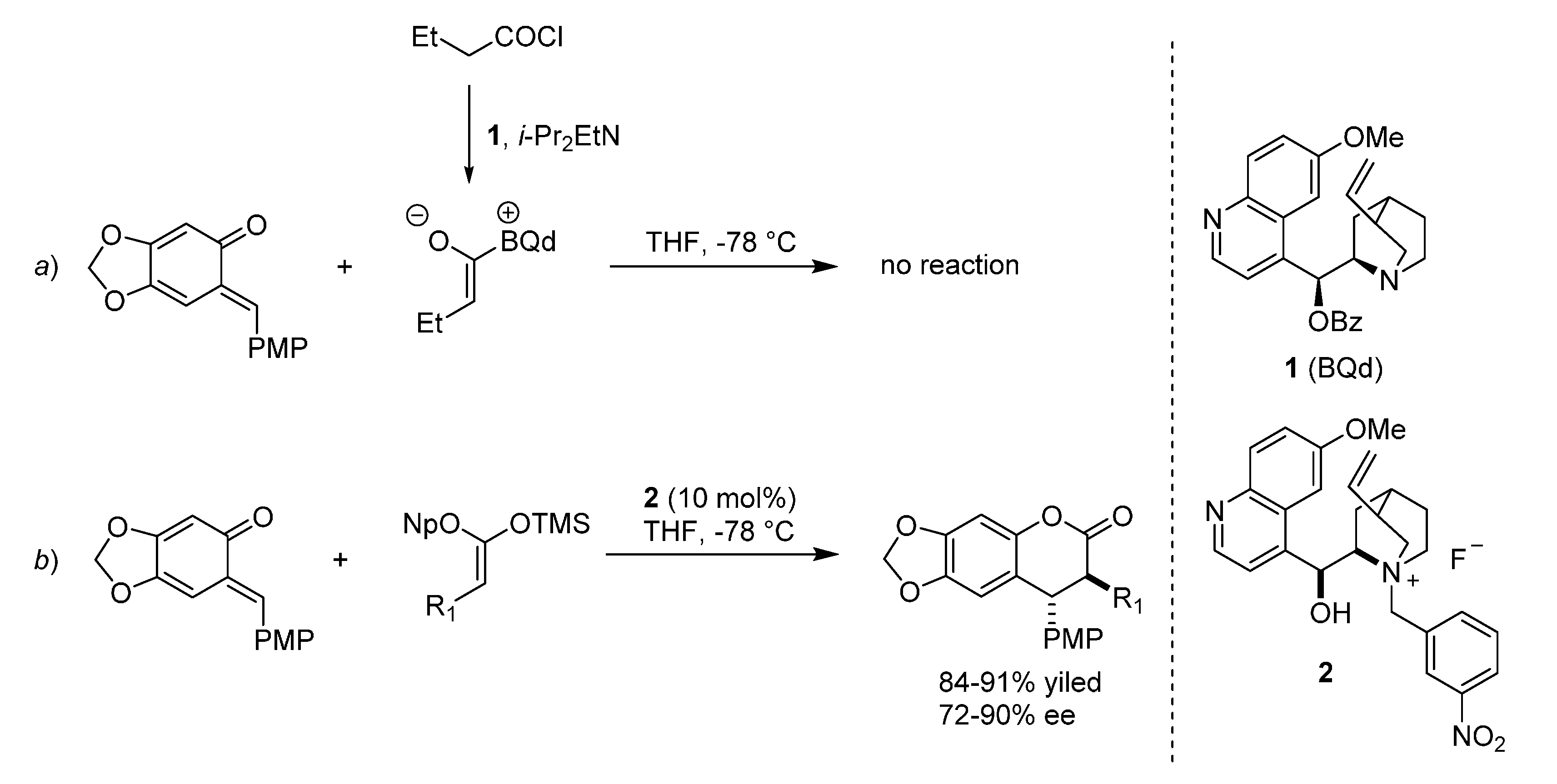{kind=link}
{kind=link}
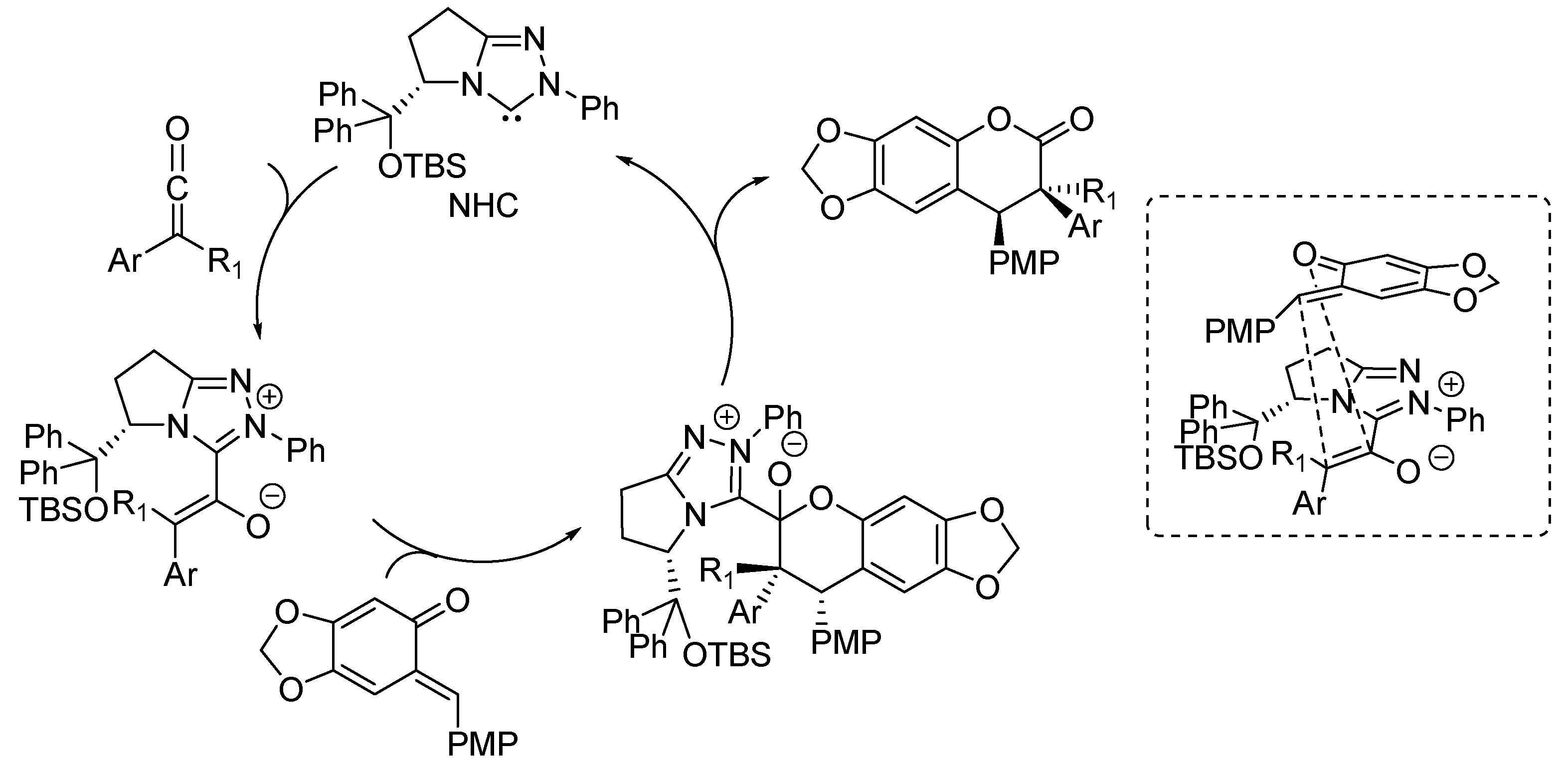
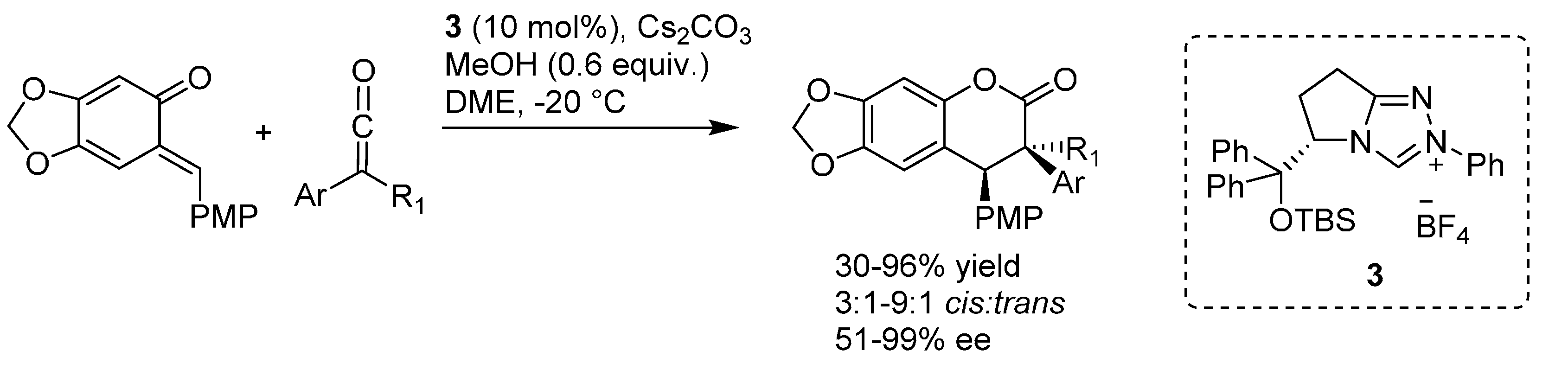{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
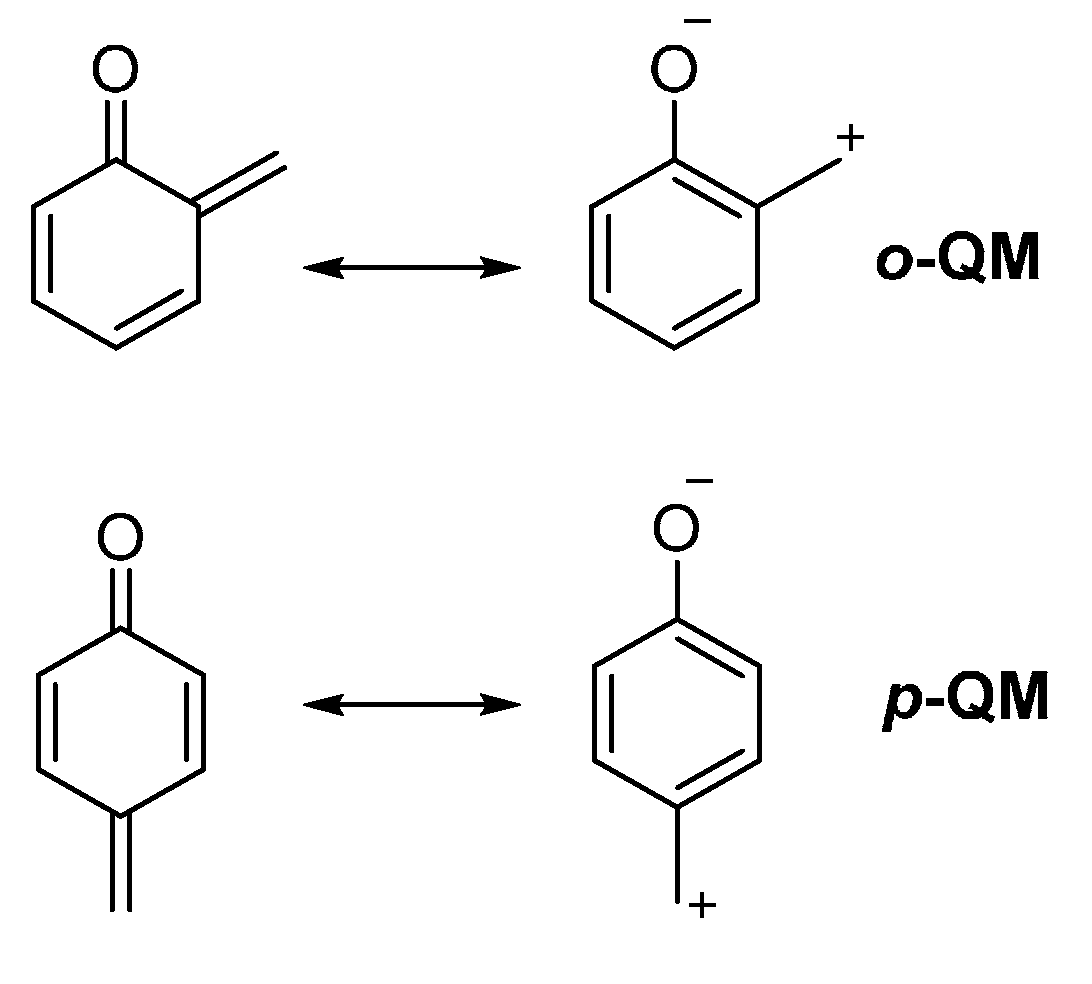
1. Introduction


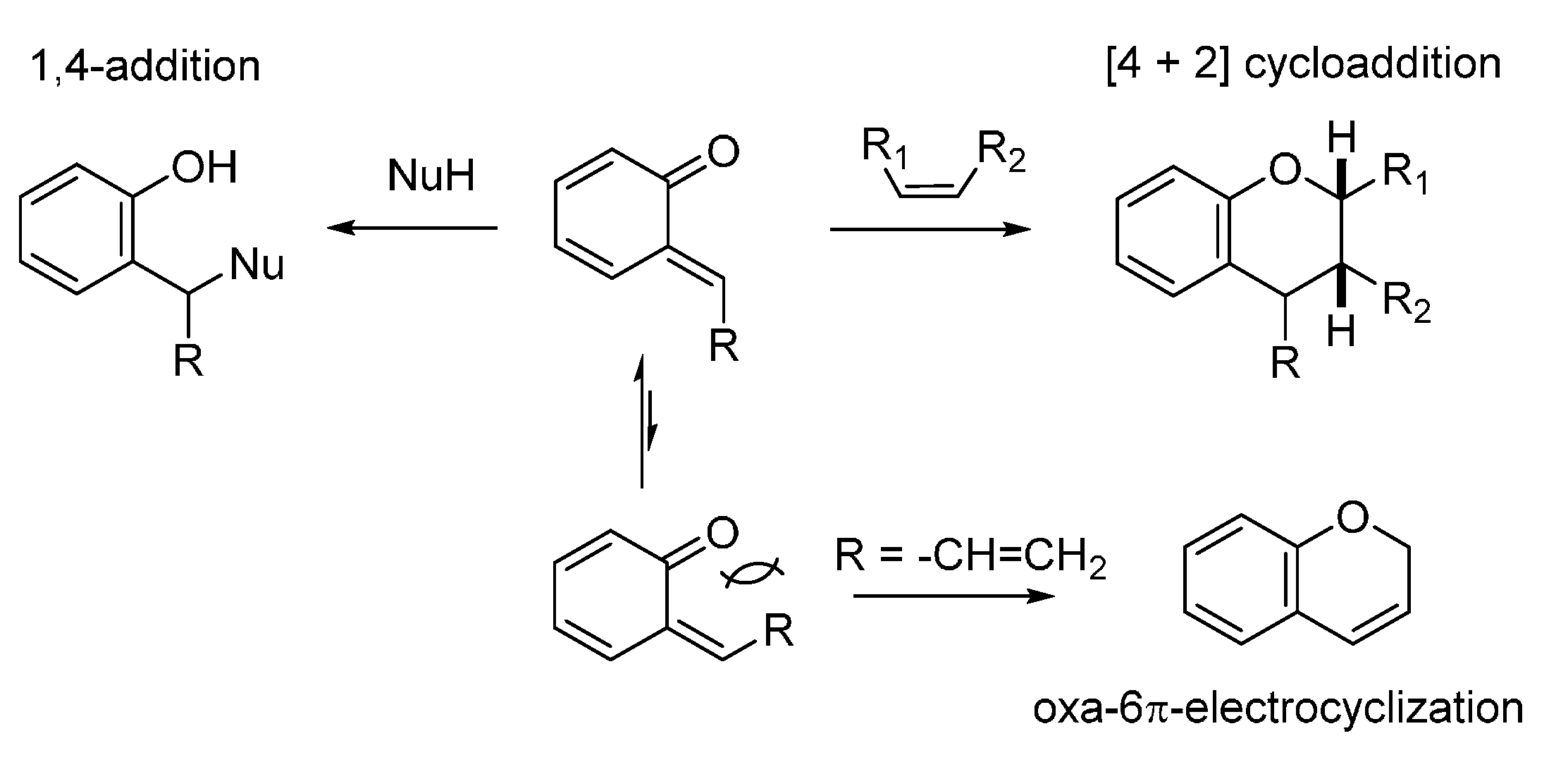
2. Catalytic Asymmetric Reactions with Stabilized o-QMs










3. Catalytic Asymmetric Reactions with o-QMs Generated in Situ
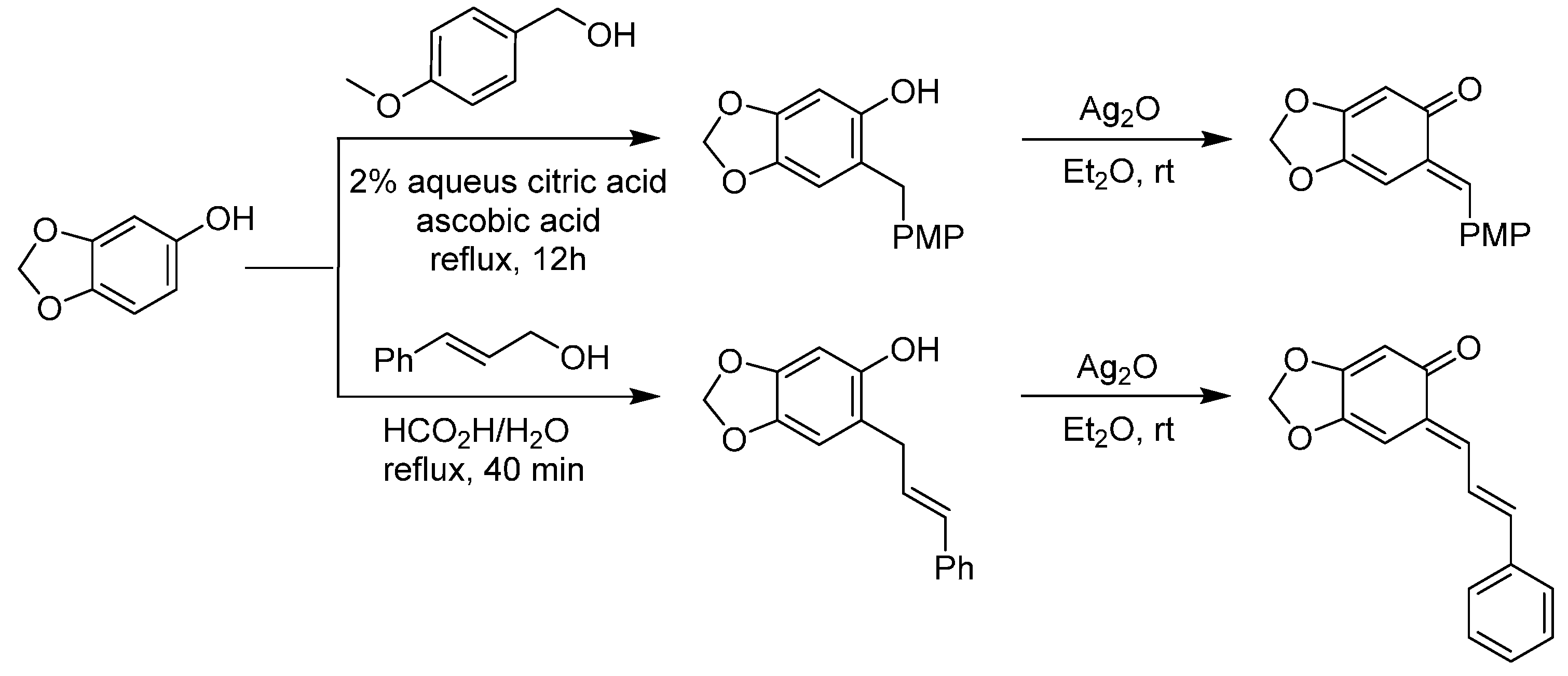
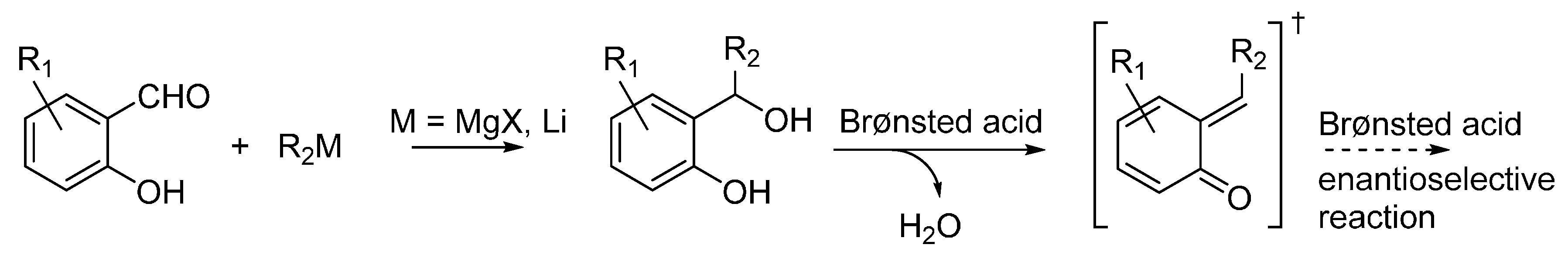
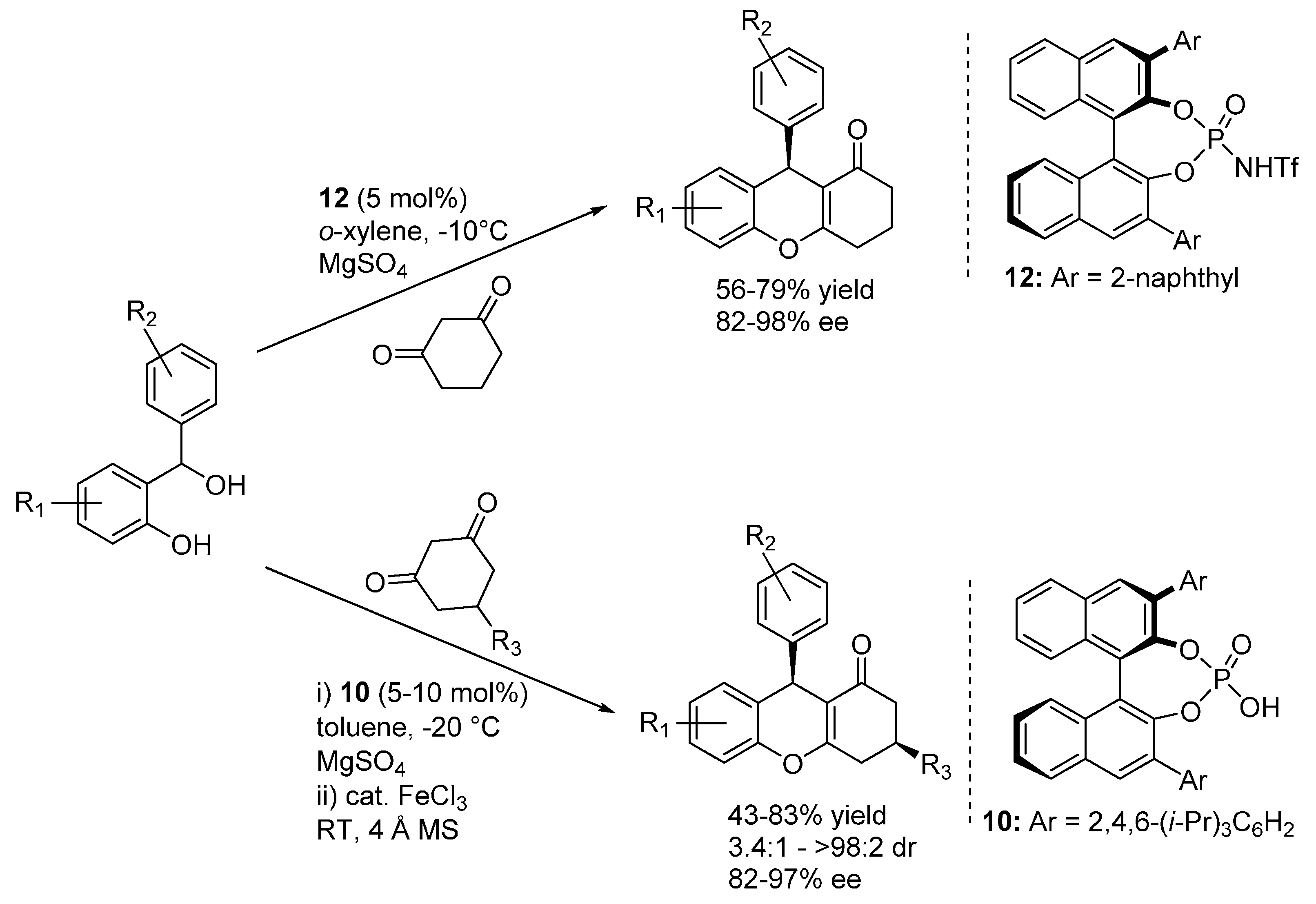
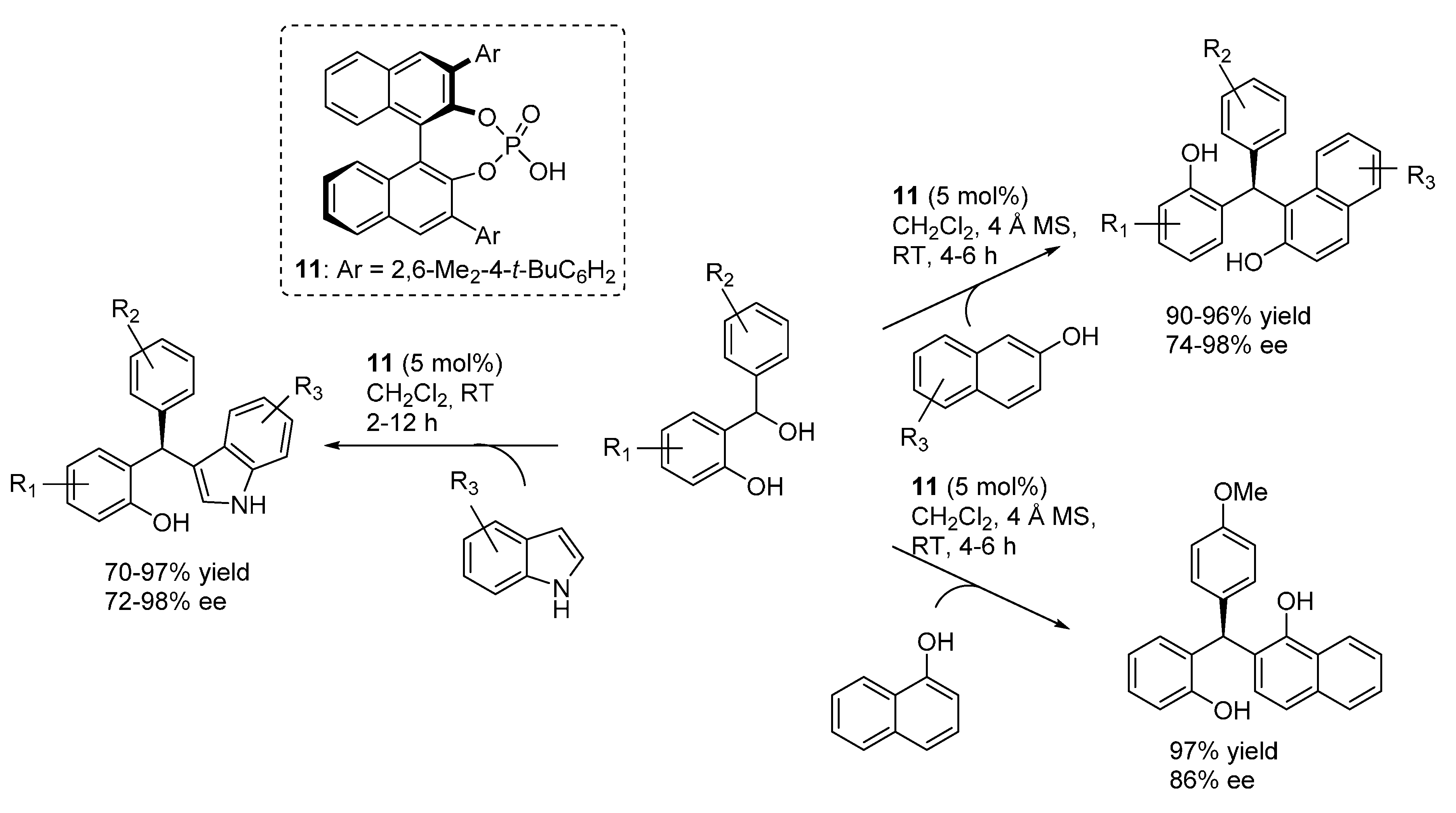
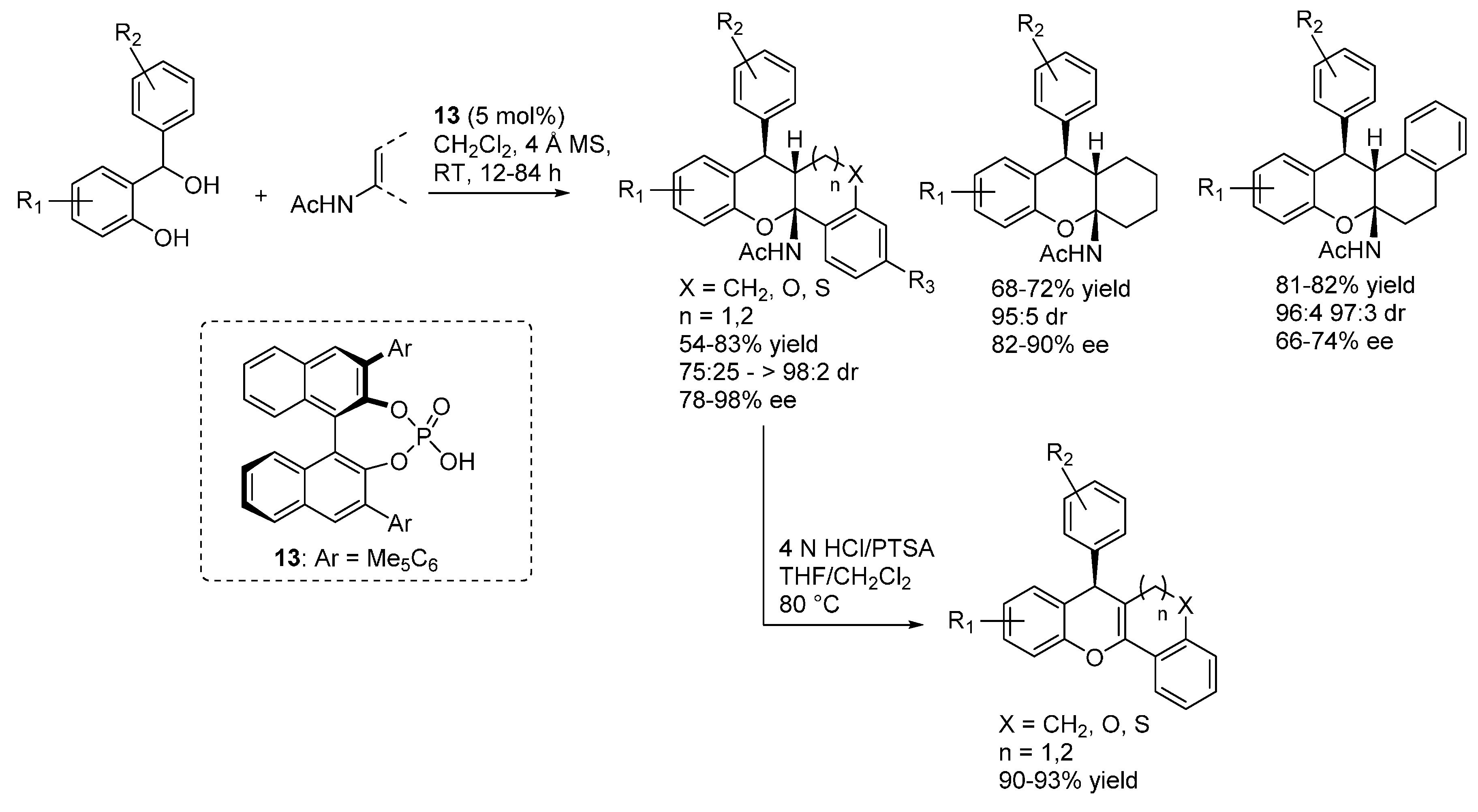
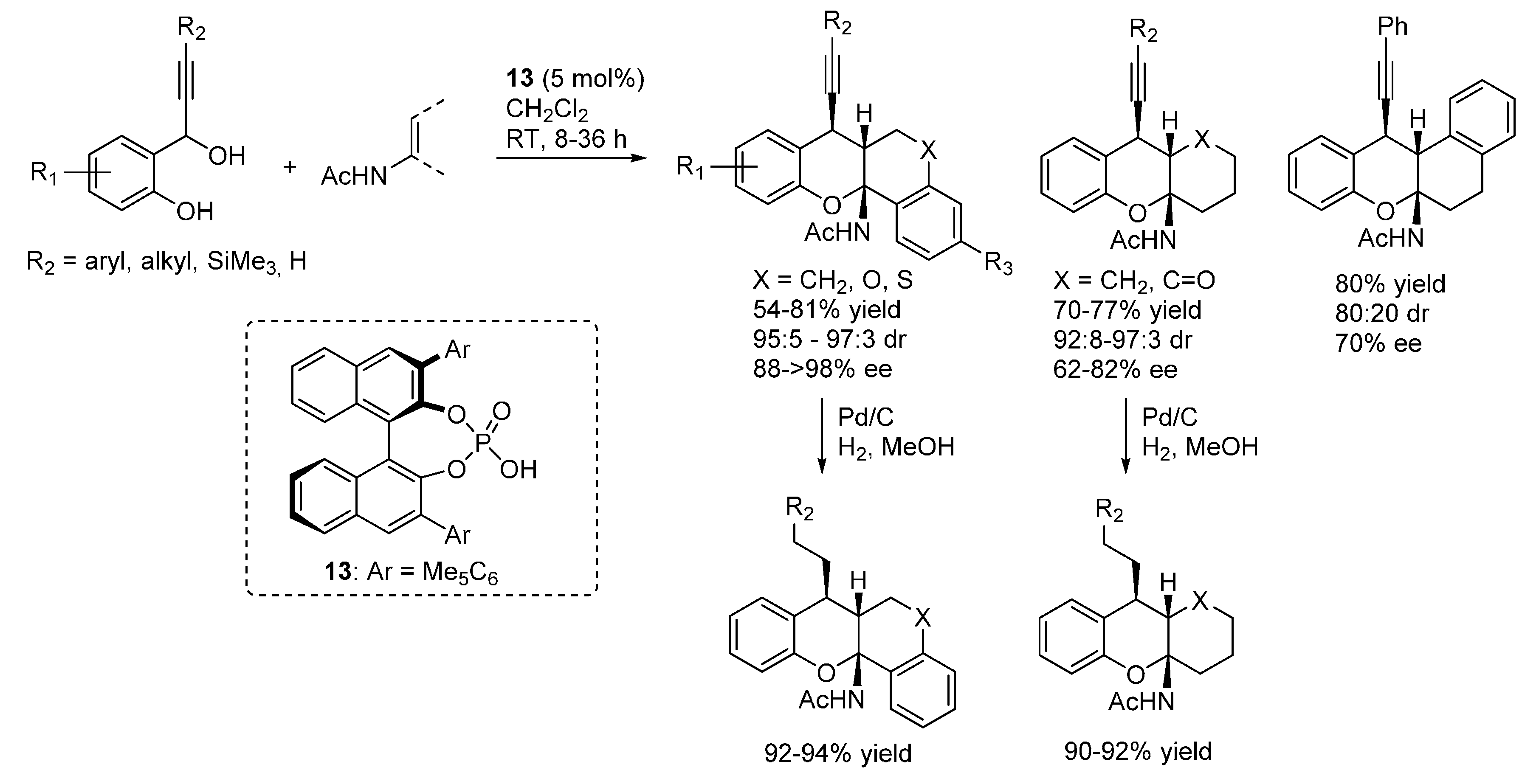
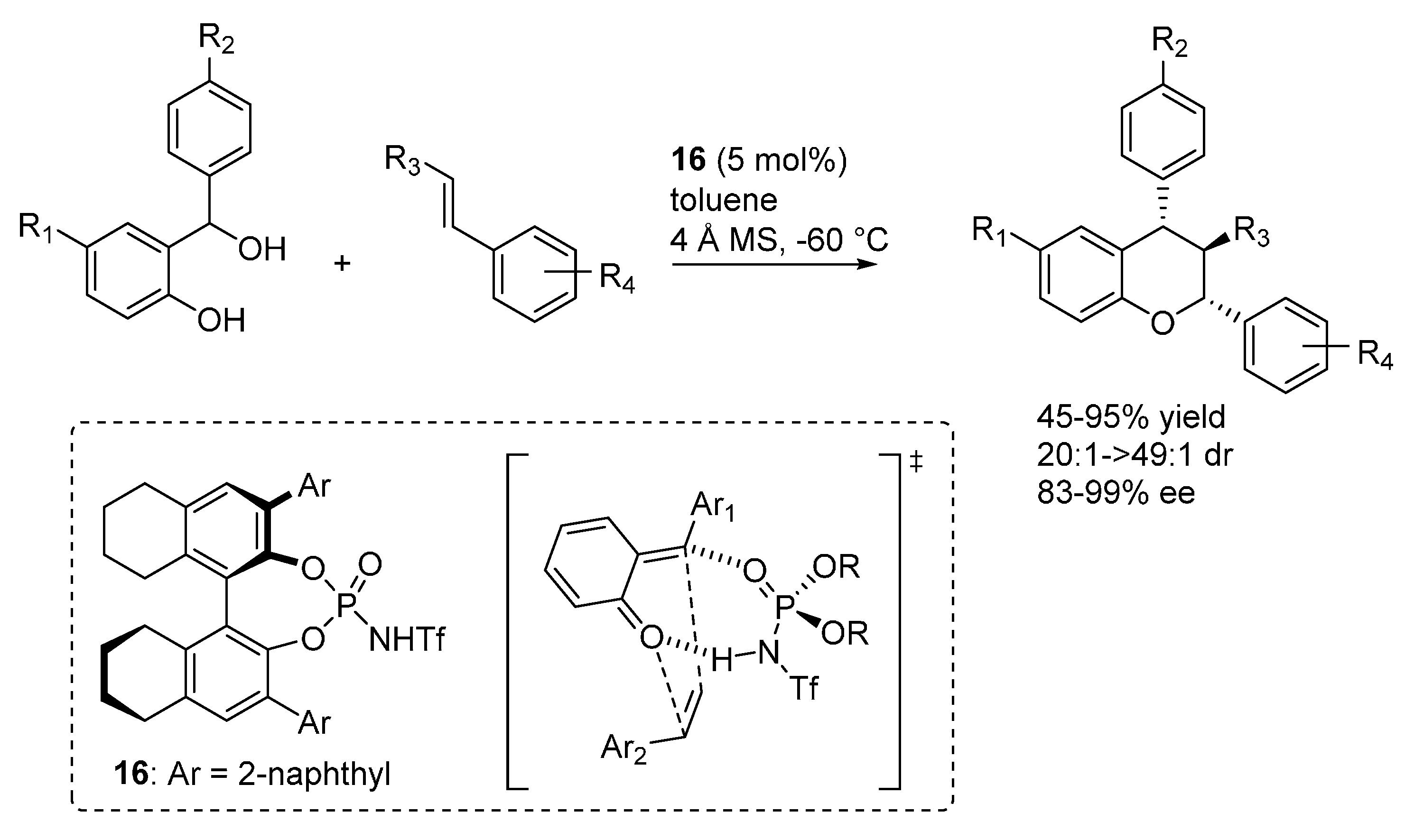
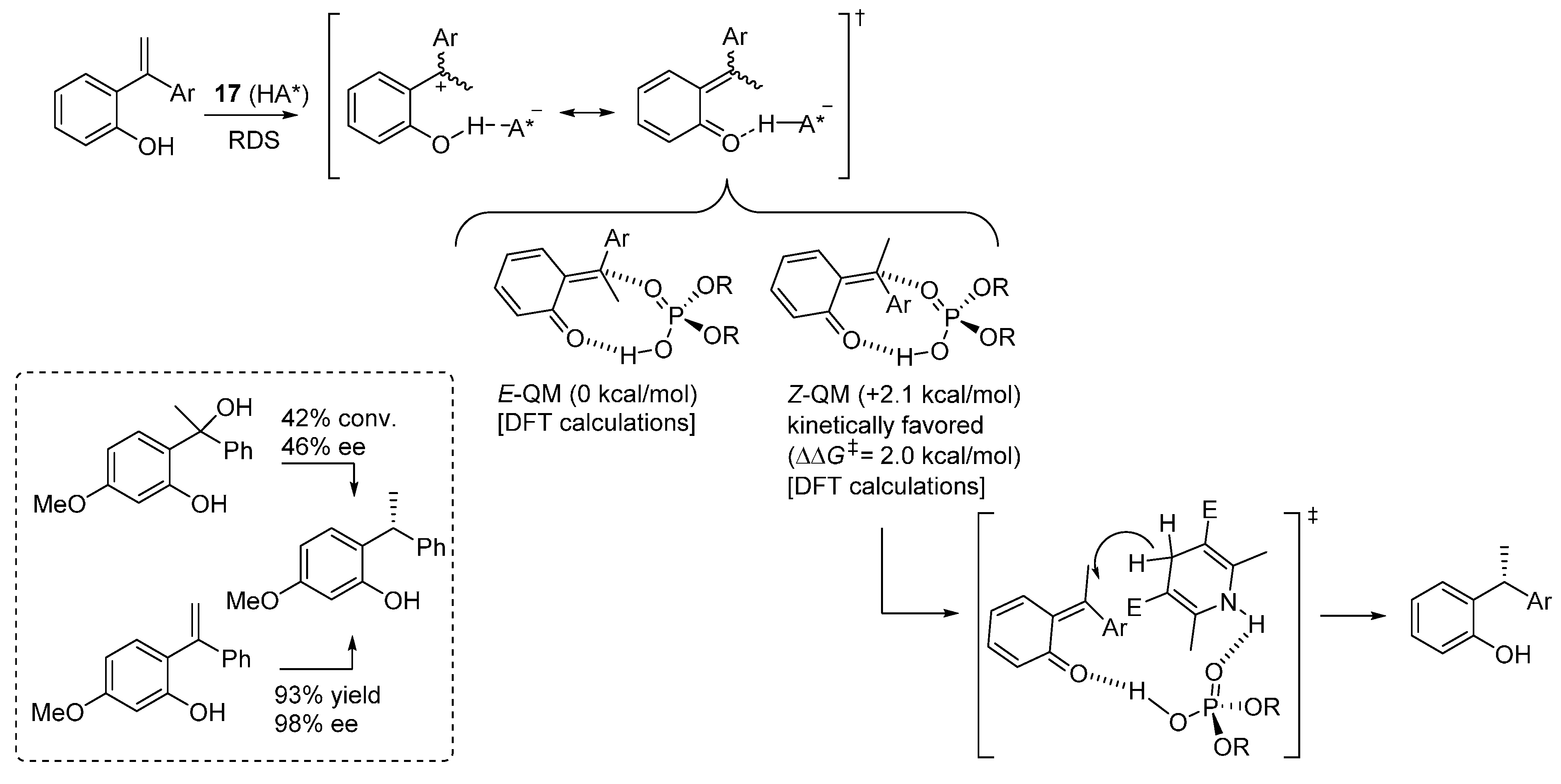
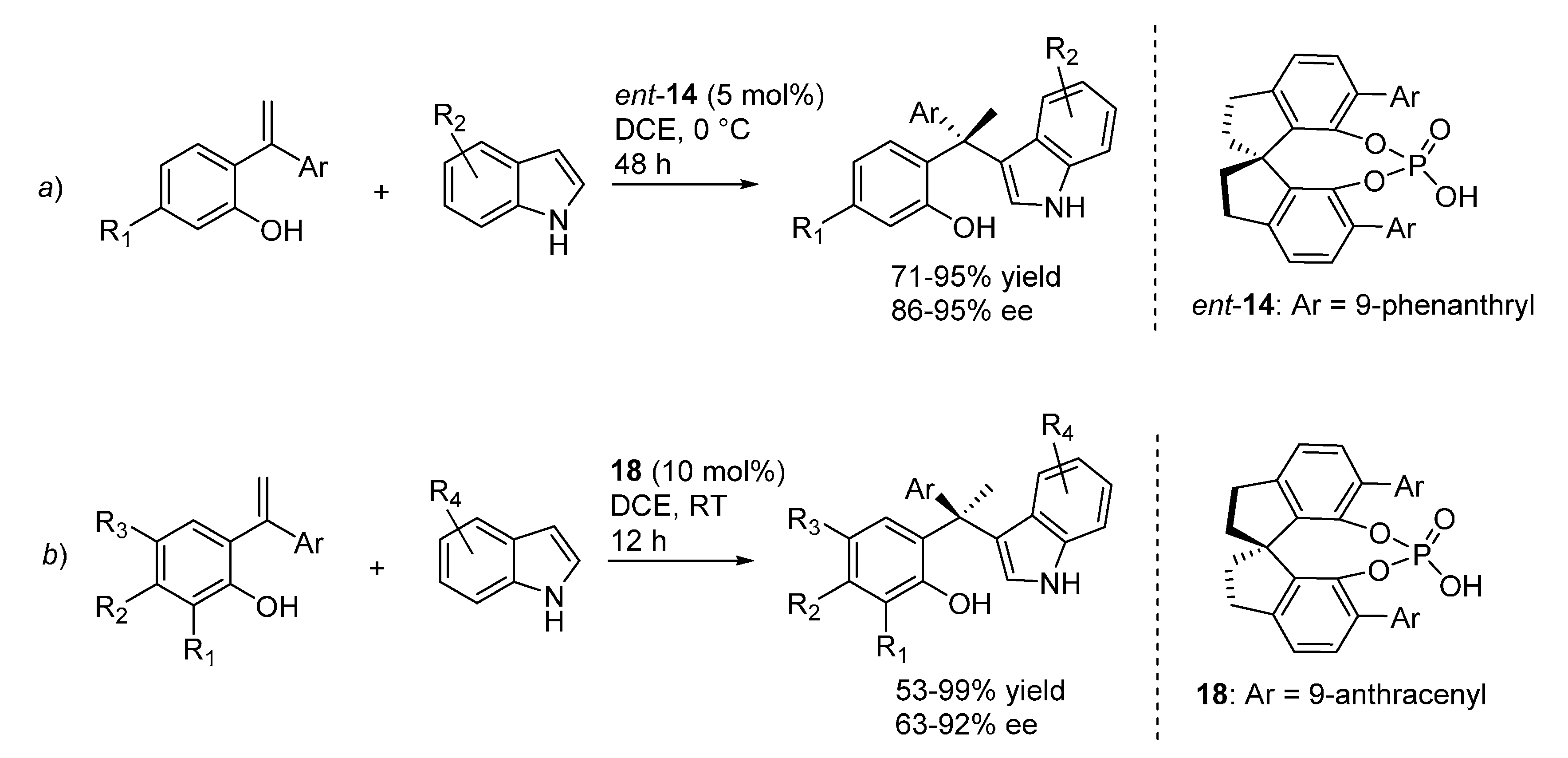
3.1. o-QMs Generated in Situ by Dehydration of Ortho-Hydroxybenzylic Alcohols under Brønsted Acid Conditions











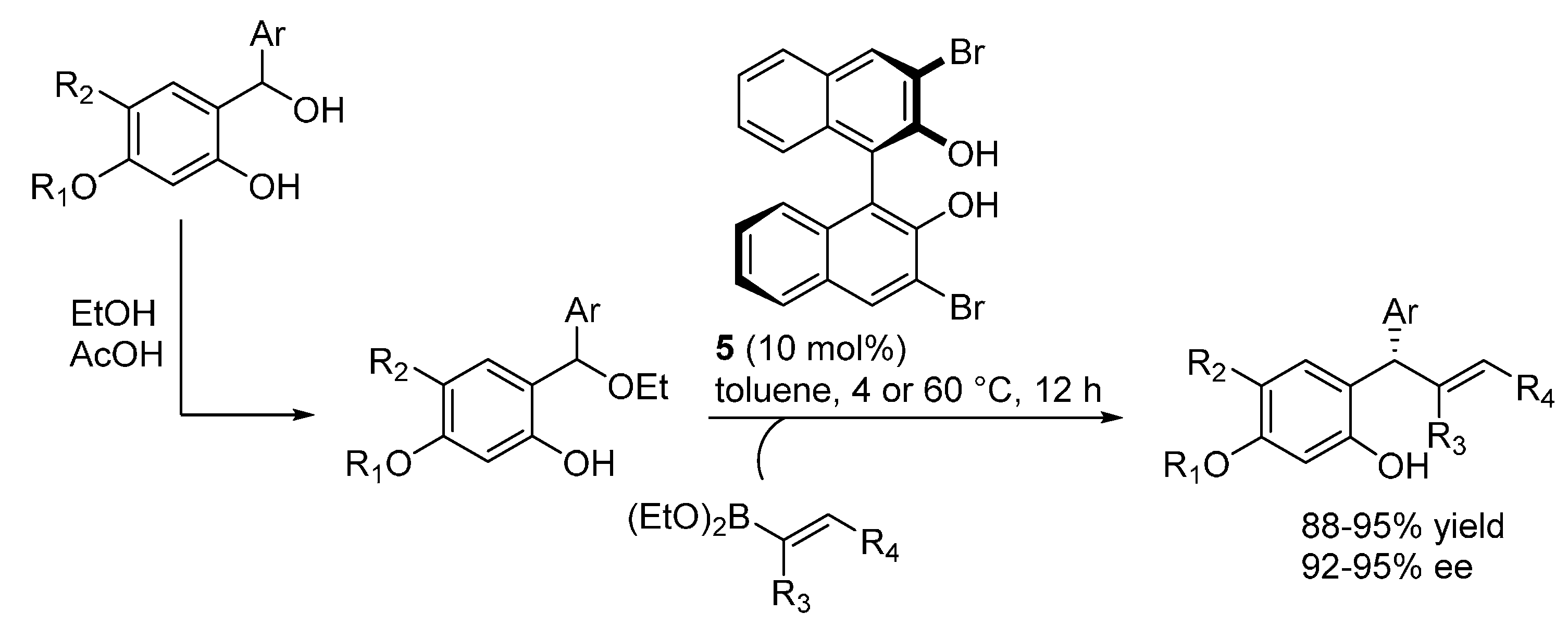
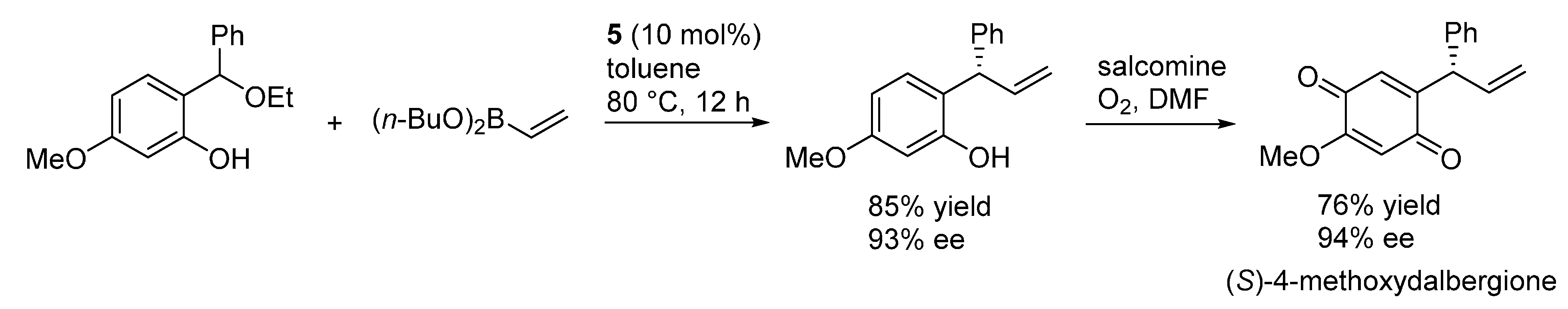
3.2. o-QMs Generated in Situ by Alcohol Elimination from Ortho-Hydroxybenzylic Ethers under Lewis Acid Conditions


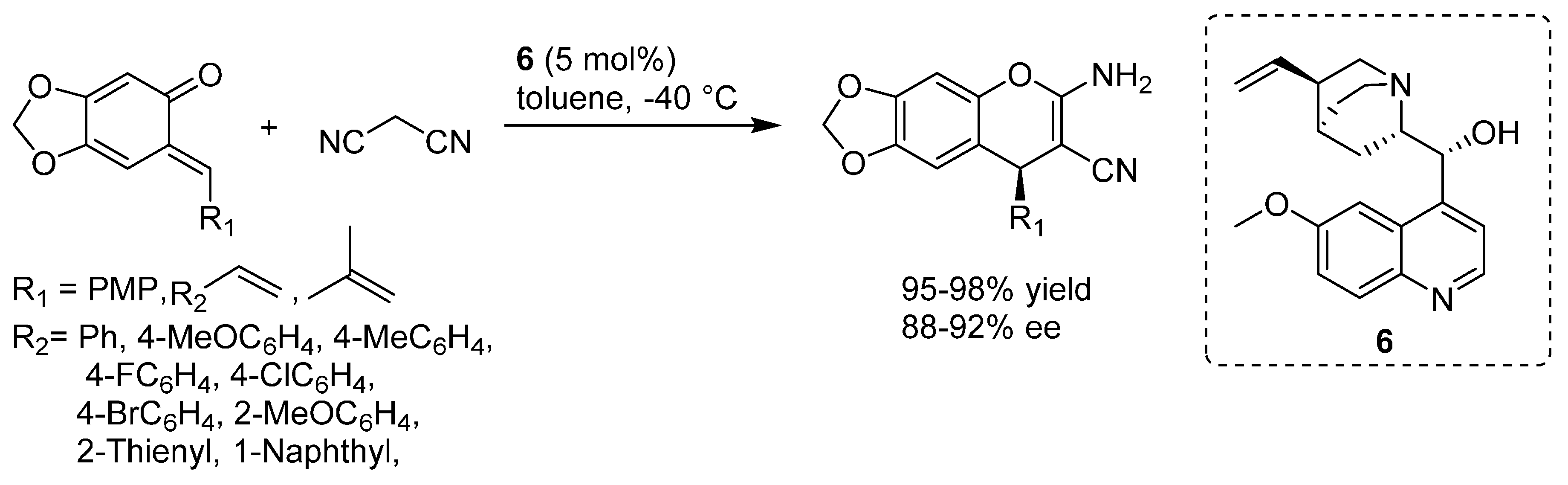
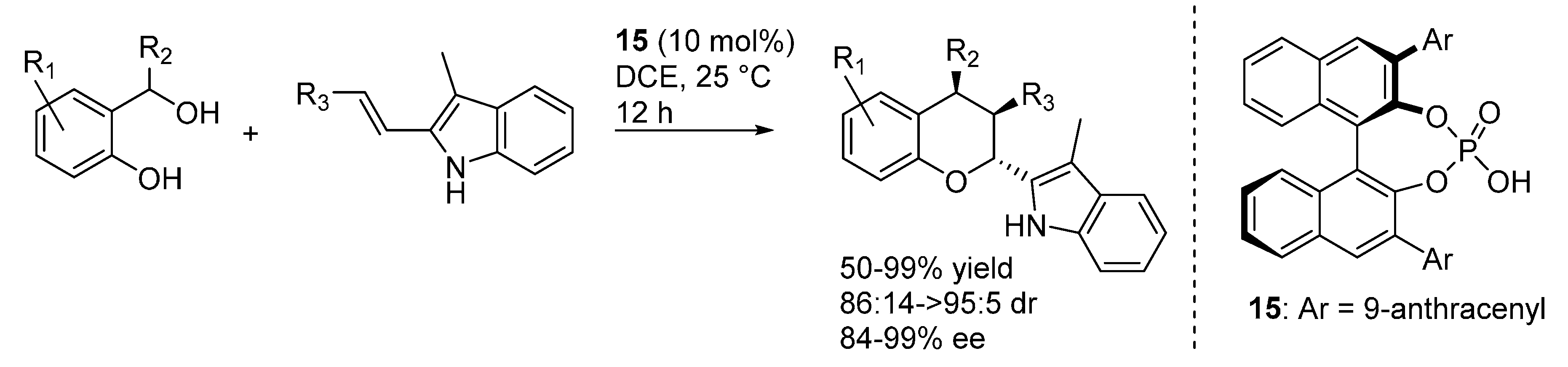
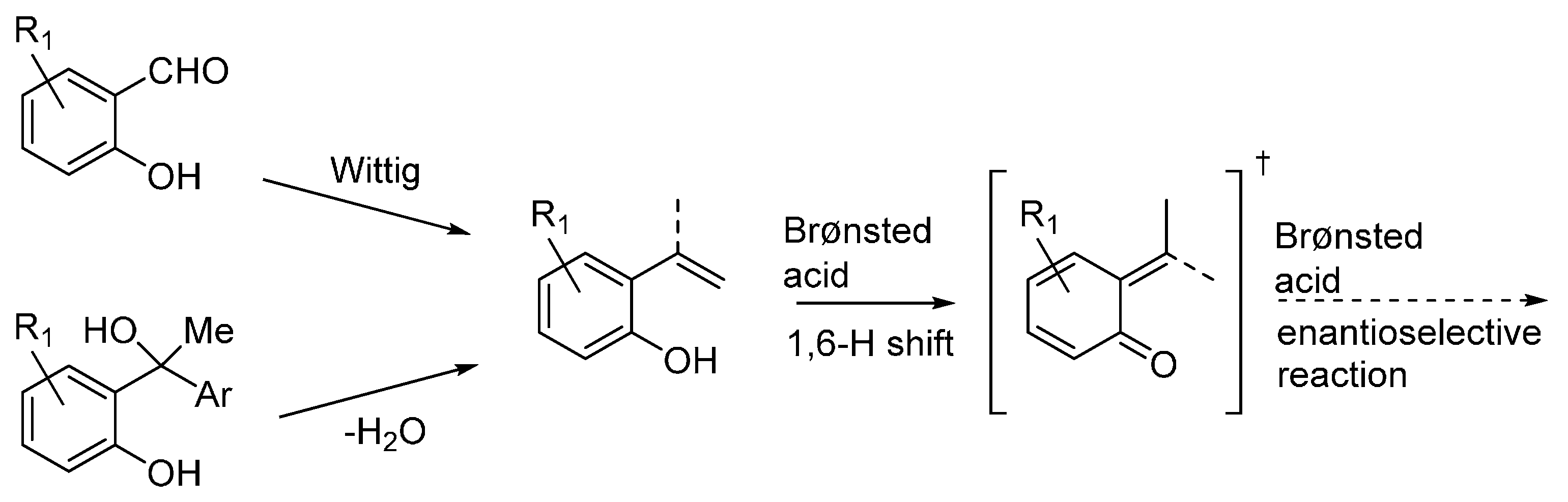
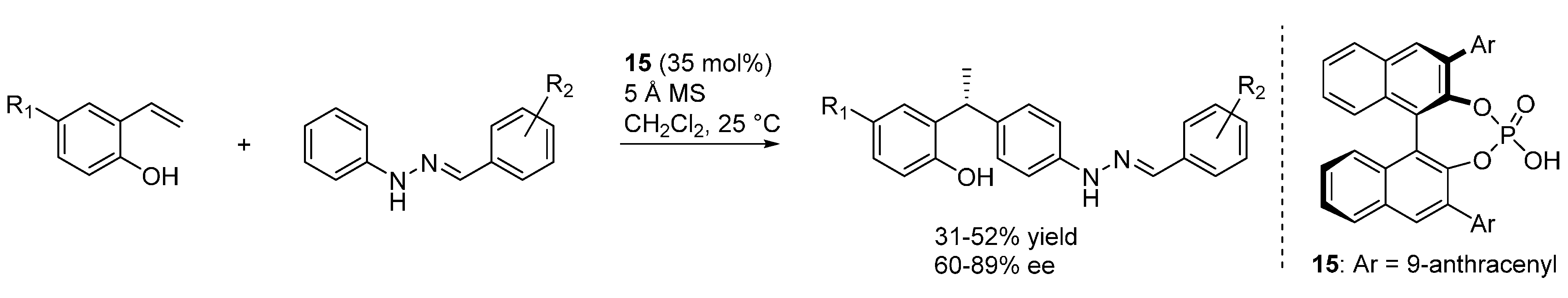
3.3. o-QMs Generated in Situ by 1,6-H Shift of Ortho-Hydroxystyrenes under Brønsted Acid Conditions





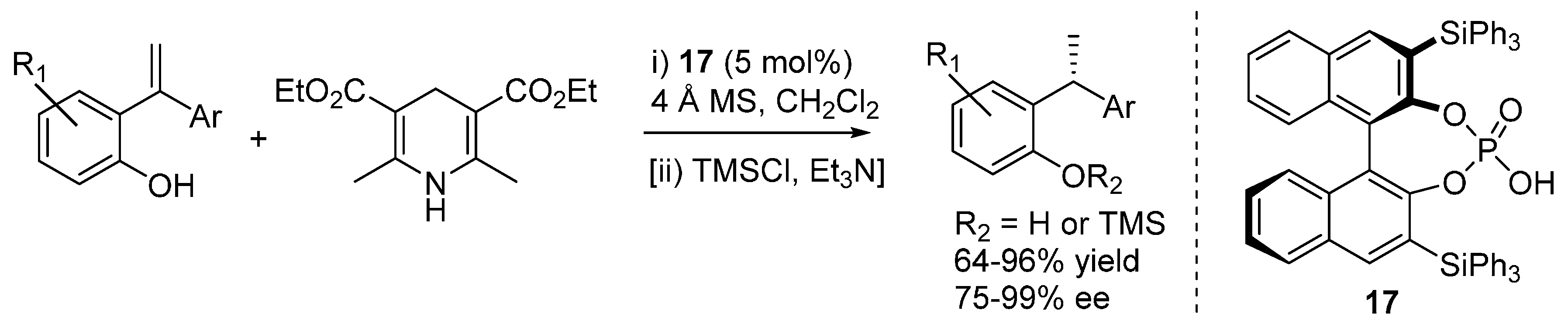
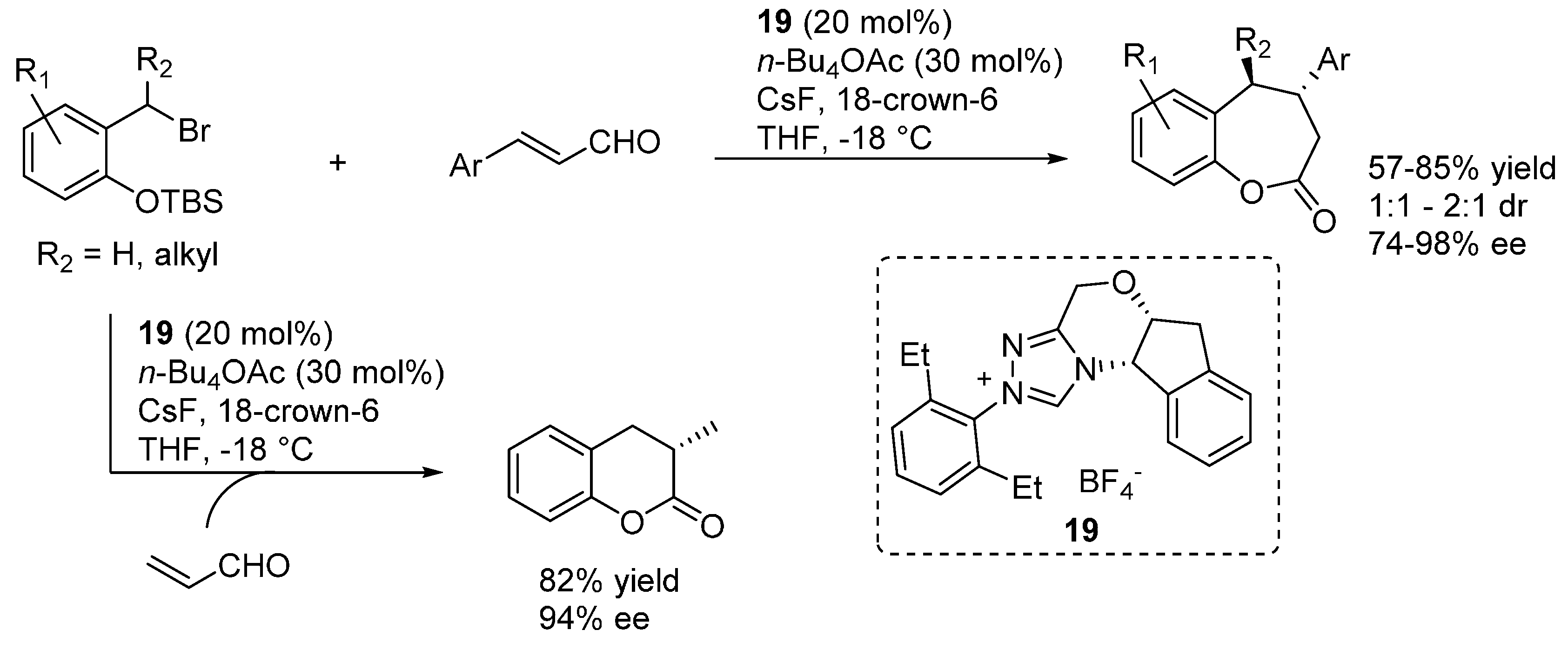
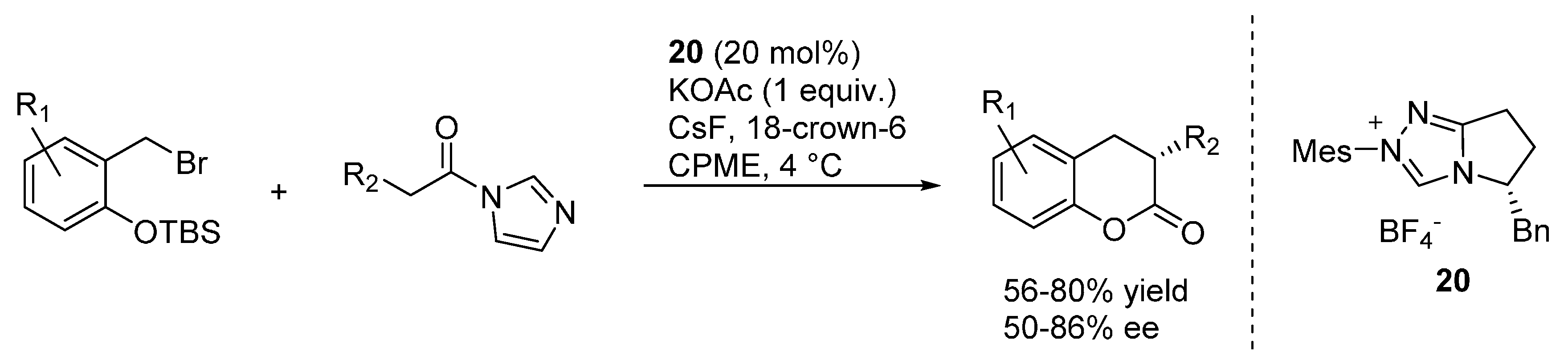
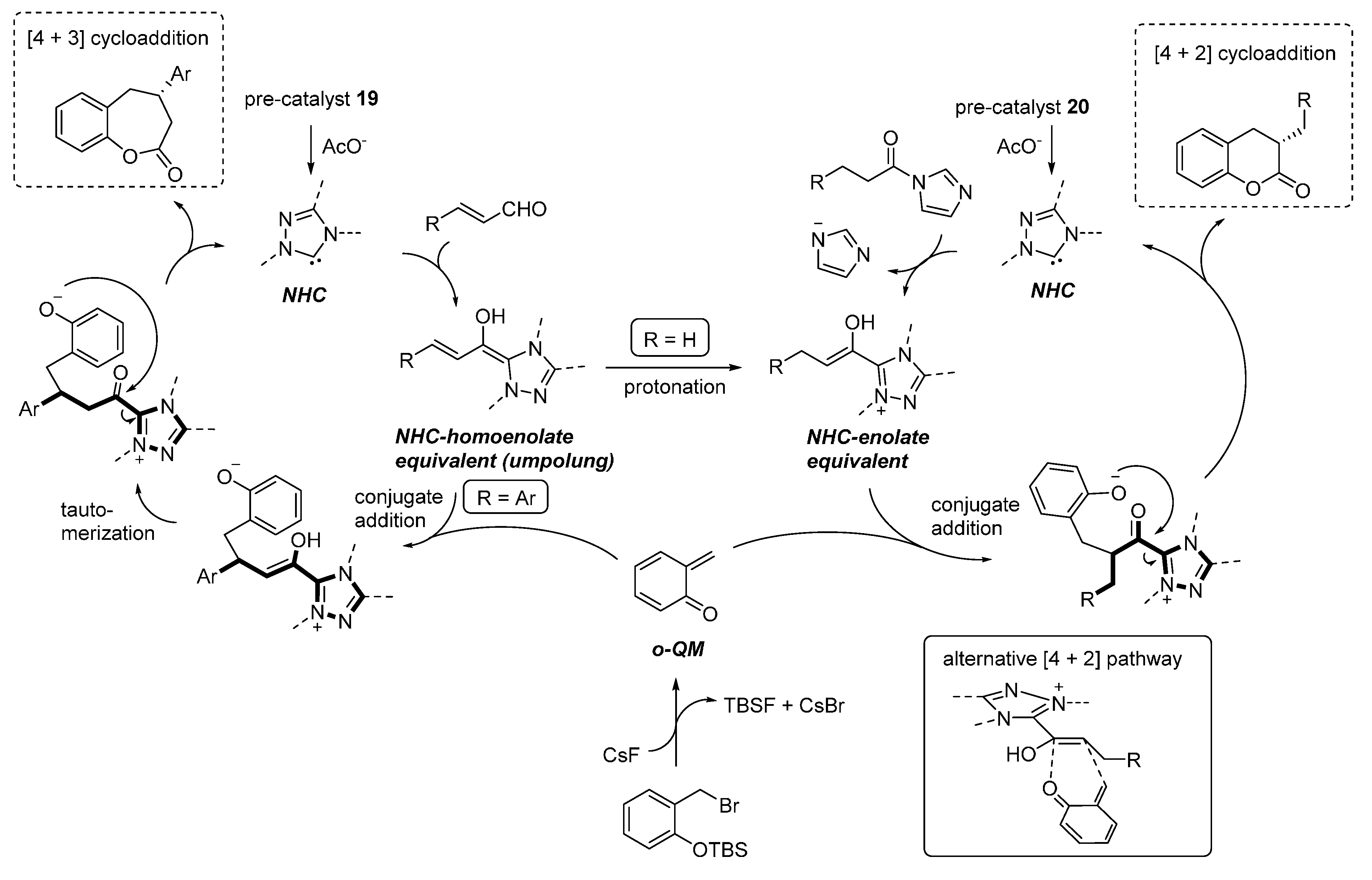
3.4. o-QMs Generated in Situ by Desilylation—Halide Elimination from Ortho-Silyloxy Benzylic Halides under Lewis Basic Conditions




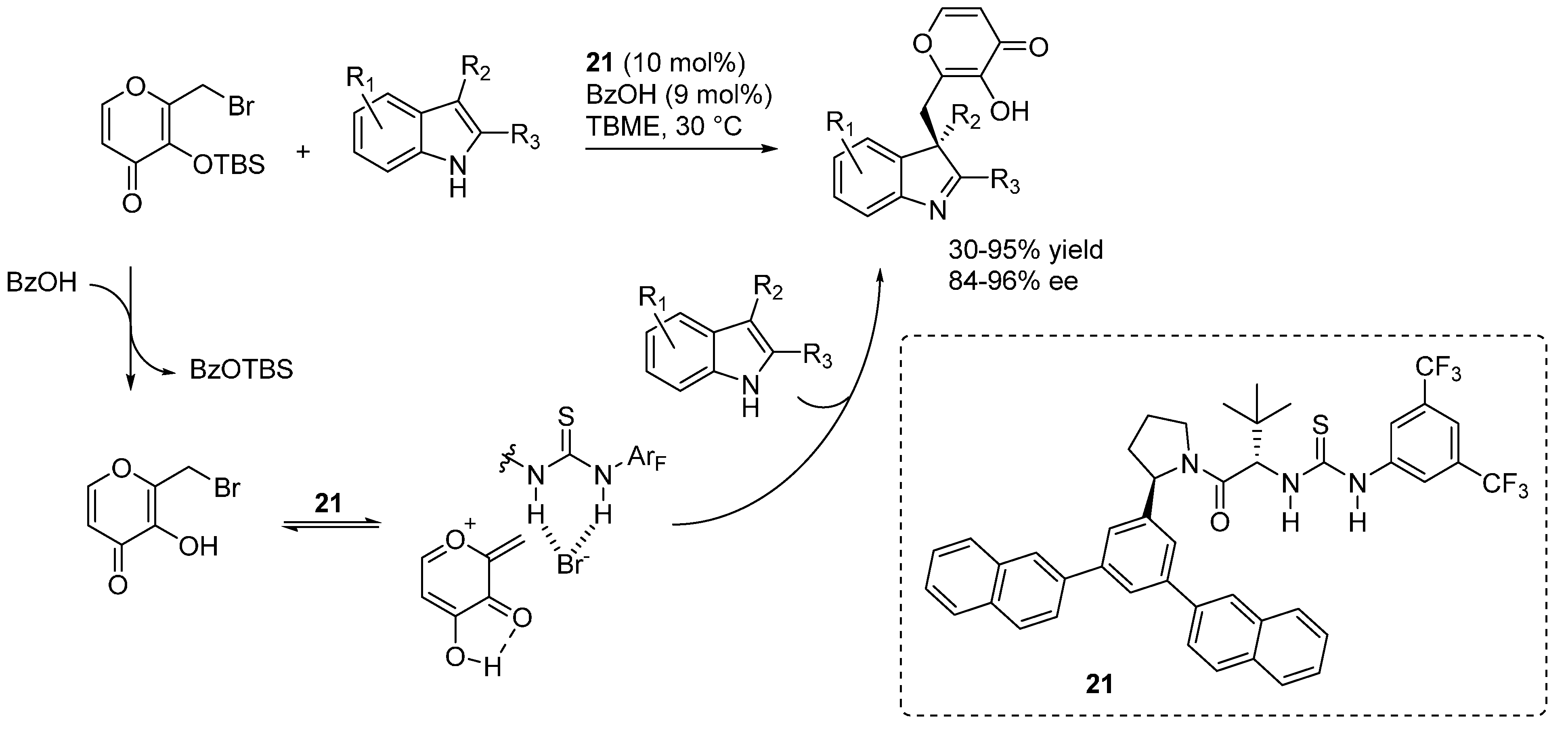
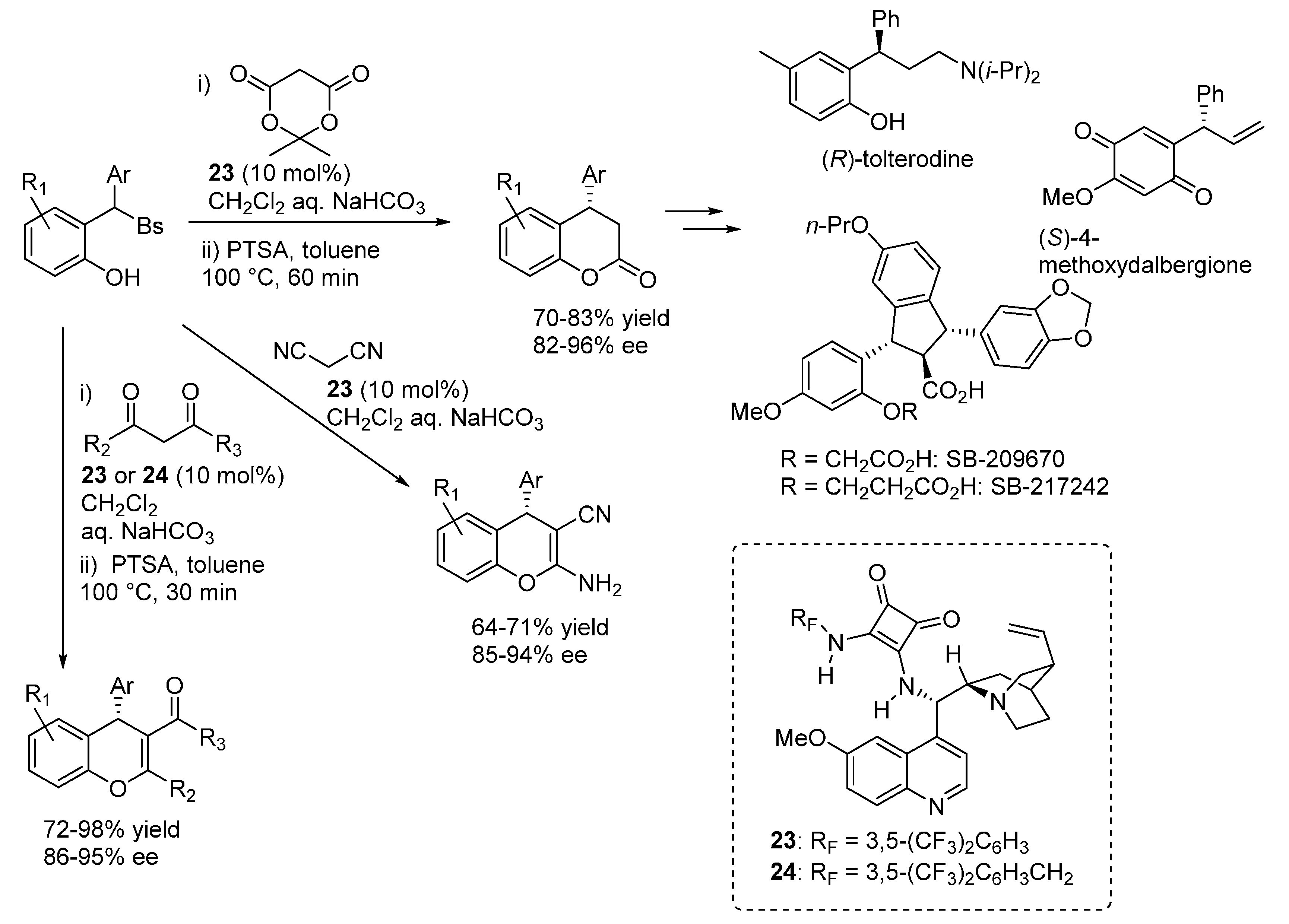
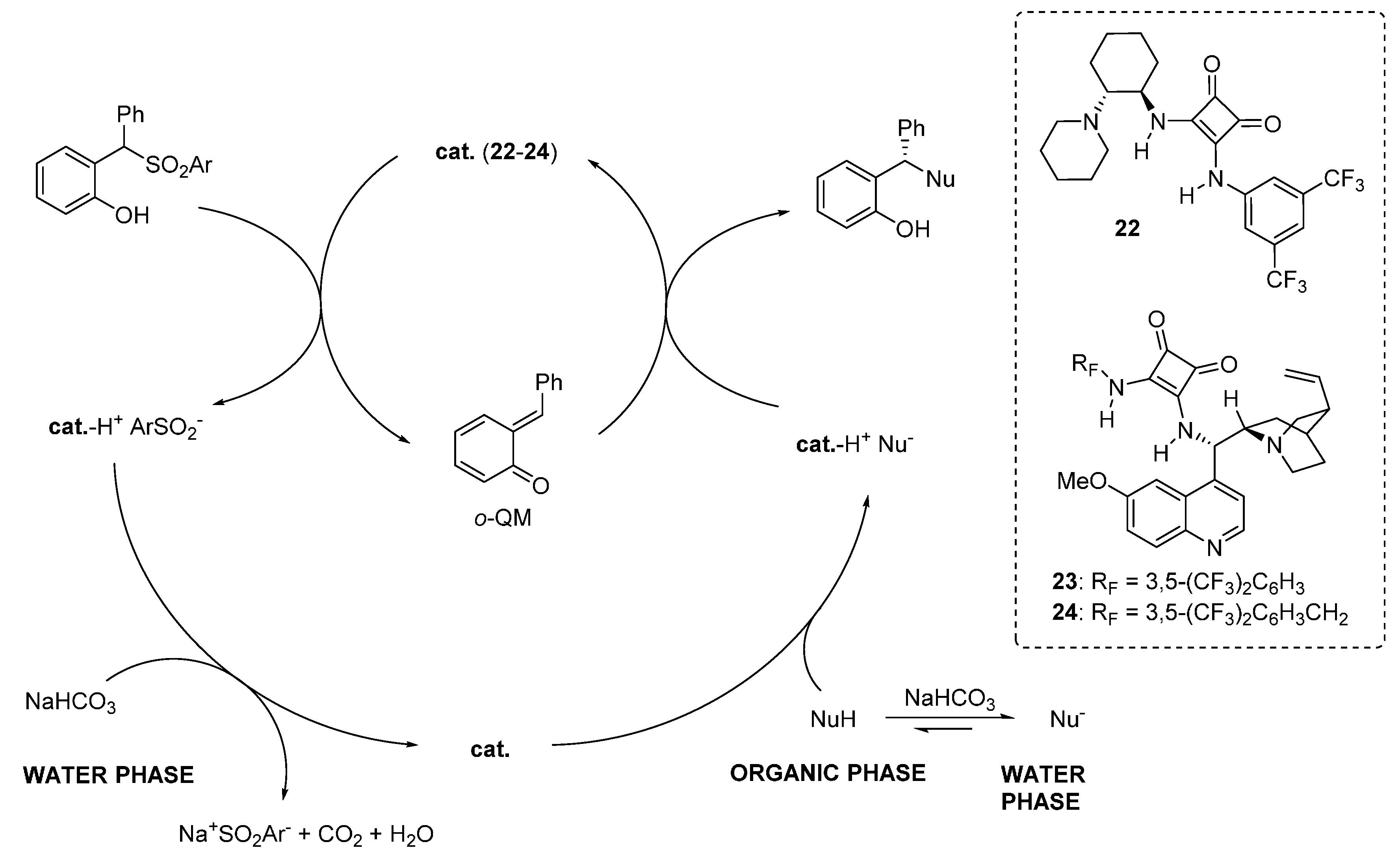
3.5. o-QMs Generated in Situ by Desilylation—Halide Elimination from Ortho-Silyloxy Benzylic Halides under Brønsted Acid Conditions

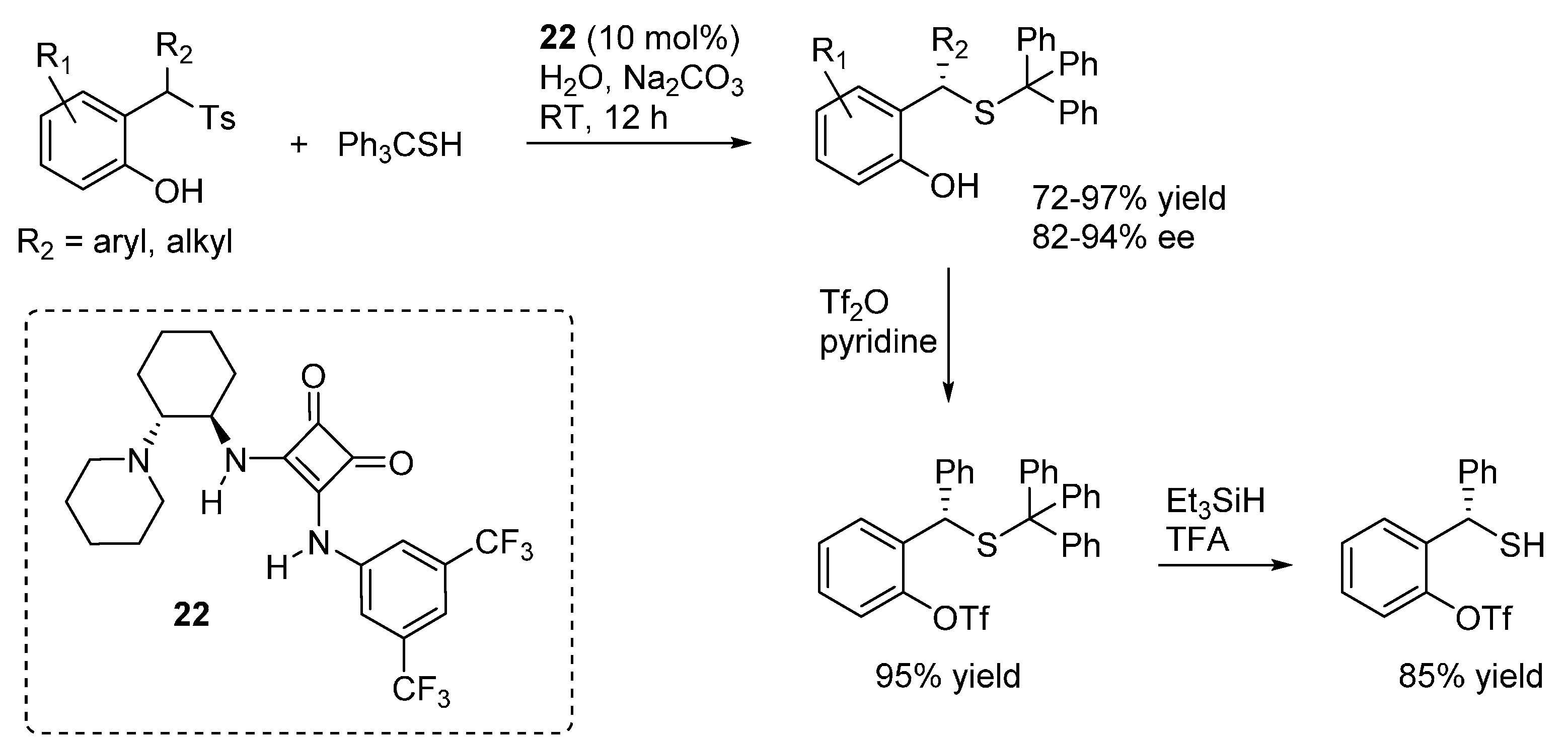
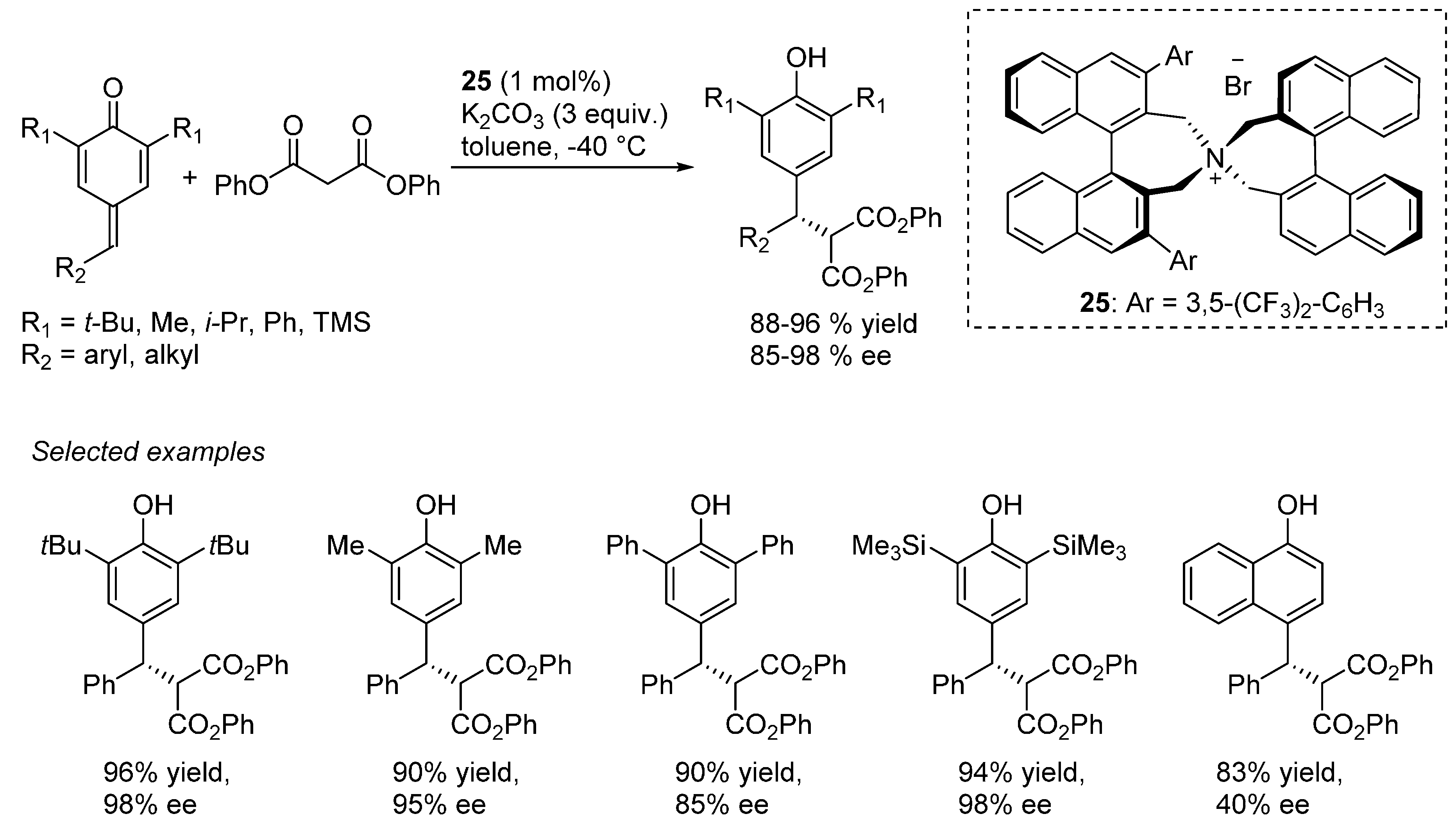
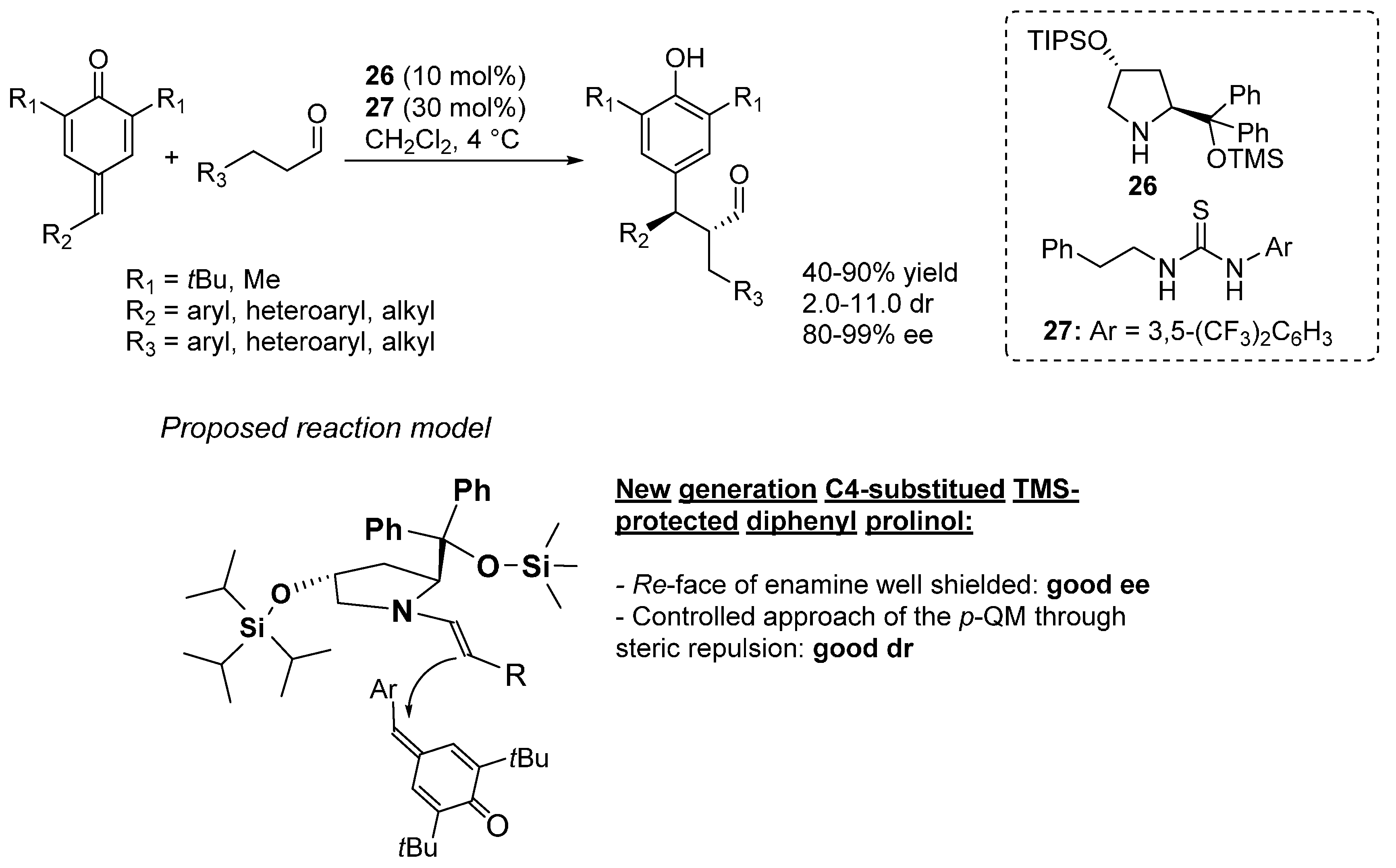
3.6. o-QMs Generated in Situ by Sulfinic Acid Elimination from 2-Sulfonylalkyl Phenols under Brønsted Basic Conditions




4. Catalytic Asymmetric Reactions with p-QMs




5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Toteva, M.M.; Richard, J.P. The generation and reactions of quinone methides. Adv. Phys. Org. Chem. 2011, 45, 39–91. [Google Scholar] [PubMed]
- Fries, K.; Kann, K. Ueber die Einwirkung von Brom und von Chlor auf Phenole: Substitutionsproducte, Pseudobromide und Pseudochloride. Ueber o-Pseudohalogenide und o-Methylenchinone aus o-Oxymesitylalkohol. Liebigs Ann. Chem. 1907, 353, 335–356. [Google Scholar] [CrossRef]
- Chapman, O.L.; McIntosh, C.L. Photochemical decarbonylation of unsaturated lactones and carbonates. J. Chem. Soc. D 1971, 383–384. [Google Scholar] [CrossRef]
- Amouri, H.; Besace, Y.; Le Bras, J.; Vaisserman, J. General synthesis, first crystal structure, and reactivity of stable o-quinone methide complexes of Cp*Ir. J. Am. Chem. Soc. 1998, 120, 6171–6172. [Google Scholar] [CrossRef]
- Rokita, S.E. Quinone Methides; Rokita; Wiley: New York, NY, USA, 2009. [Google Scholar]
- Di Antonio, M. Thematic issue: Quinone methides generation: Application in chemical biology. Curr. Org. Chem. 2014, 18. [Google Scholar] [CrossRef]
- Van De Water, R.W.; Pettus, T.R.R. o-Quinone methides: Intermediates underdeveloped and underutilized in organic synthesis. Tetrahedron 2002, 58, 5367–5405. [Google Scholar] [CrossRef]
- Singh, M.S.; Nagaraju, A.; Anand, N.; Chowdhury, S. ortho-Quinone methide (o-QM): A highly reactive, ephemereal and versatile intermediate in organic synthesis. RSC Adv. 2014, 4, 55924–55959. [Google Scholar] [CrossRef]
- Willis, N.J.; Bray, C.D. ortho-Quinone methides in natural product synthesis. Chem. Eur. J. 2012, 18, 9160–9173. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.J.; David, J.G.; Feng, Z.G.; Weaver, M.G.; Wu, K.L.; Pettus, T.R.R. The domestication of ortho-quinone methides. Acc. Chem. Res. 2014, 47, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Pathak, T.P.; Sigman, M.S. Applications of ortho-quinone methide intermediates in catalysis and asymmetric synthesis. J. Org. Chem. 2011, 76, 9210–9215. [Google Scholar] [CrossRef] [PubMed]
- Jurd, L. Quinones and quinone-methides—I: Cyclization and dimerisation of crystalline ortho-quinone methides from phenol oxidation reactions. Tetrahedron 1977, 33, 163–168. [Google Scholar] [CrossRef]
- Alden-Danforth, E.; Scerba, M.T.; Lectka, T. Asymmetric cycloadditions of o-quinone methides employing chiral ammonium fluoride precatalysts. Org. Lett. 2008, 10, 4951–4953. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; You, L.; Ye, S. Enantioselective synthesis of dihydrocoumarins via N-heterocyclic carbene-catalyzed cycloaddition of ketenes and o-quinone methides. Adv. Synth. Catal. 2009, 351, 2822–2826. [Google Scholar] [CrossRef]
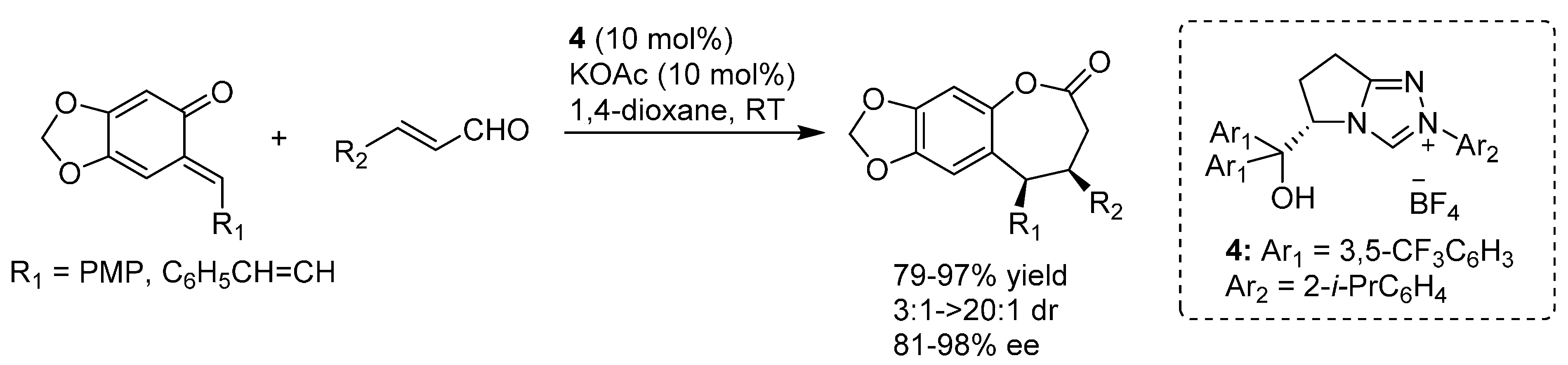
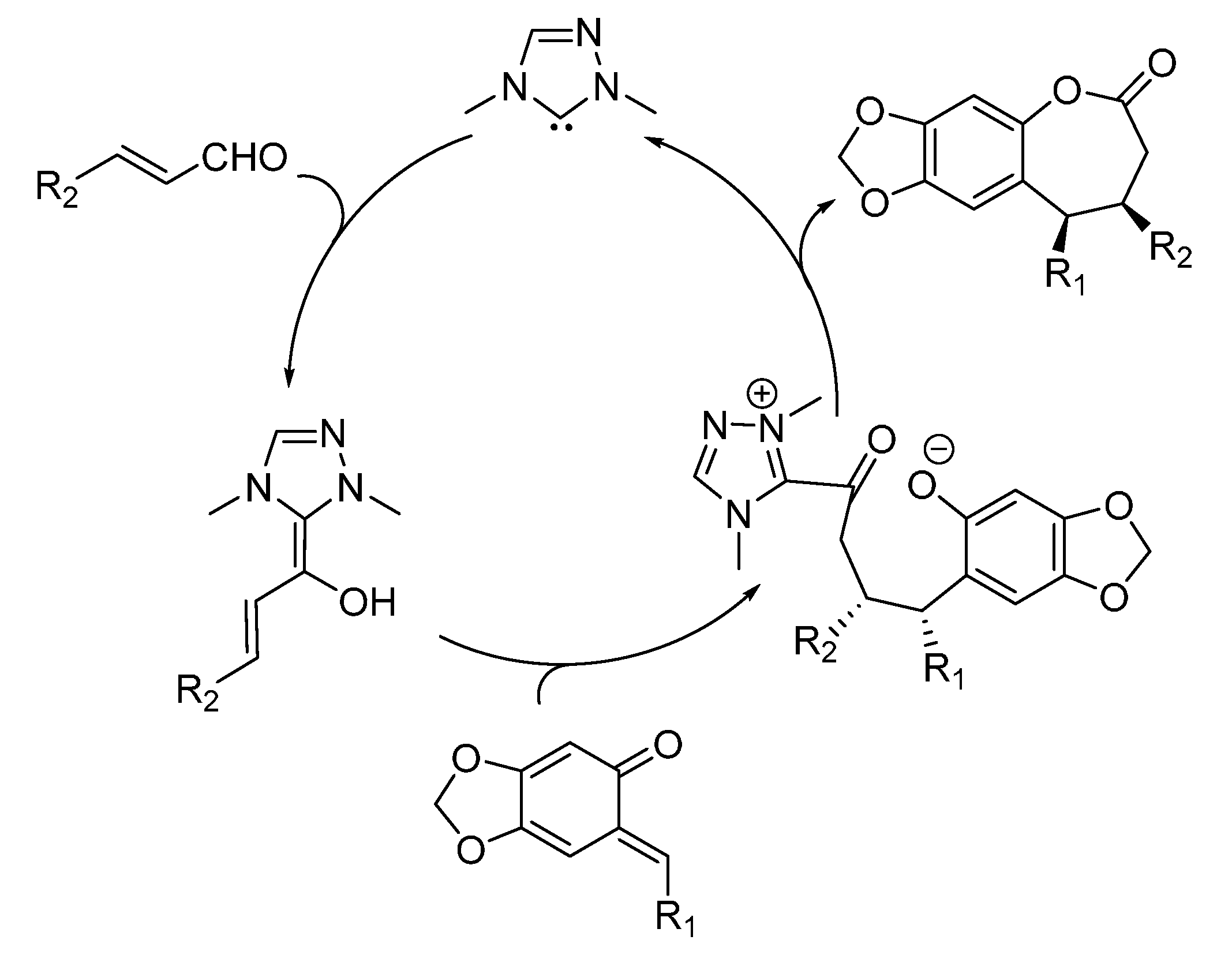
- Lv, H.; Jia, W.Q.; Sun, L.H.; Ye, S. N-Heterocyclic carbene catalyzed [4 + 3] annulation of enals and o-quinone methides: Highly enantioselective synthesis of benzo-ε-lactones. Angew. Chem. Int. Ed. 2013, 52, 8607–8610. [Google Scholar] [CrossRef] [PubMed]
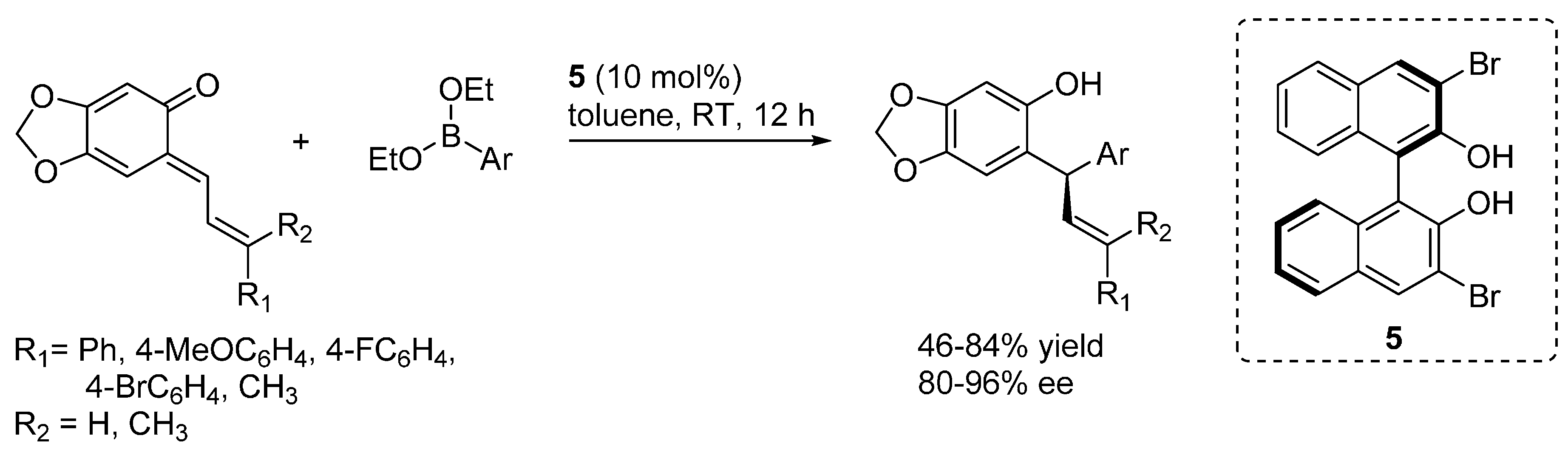
- Luan, Y.; Schaus, S.E. Enantioselective addition of boronates to o-quinone methides catalyzed by chiral biphenols. J. Am. Chem. Soc. 2012, 134, 19965–19968. [Google Scholar] [CrossRef] [PubMed]
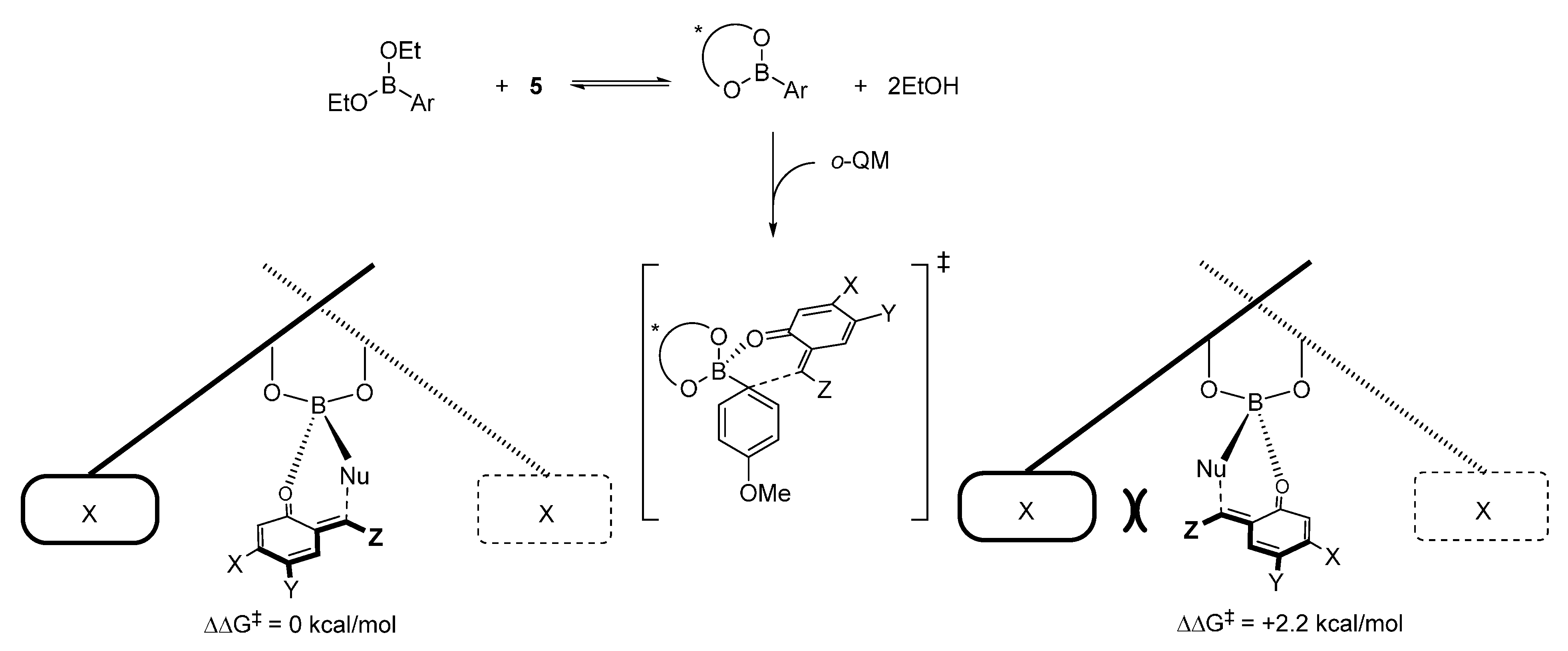
- Grayson, M.N.; Goodman, J.M. Asymmetric boronate addition to o-quinone methides: Ligand exchange, solvent effects, and Lewis acid catalysis. J. Org. Chem. 2015, 80, 2056–2061. [Google Scholar] [CrossRef] [PubMed]
- Adili, A.; Tao, Z.L.; Chen, D.F.; Han, Z.Y. Quinine-catalyzed highly enantioselective cycloannulation of o-quinone methides with malononitrile. Org. Biomol. Chem. 2015, 13, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Weinert, E.E.; Dondi, R.; Colloredo-Melz, S.; Frankenfield, K.N.; Mitchell, C.H.; Freccero, M.; Rokita, S.E. Substituents on quinone methides strongly modulate formation and stability of their nucleophilic adducts. J. Am. Chem. Soc. 2006, 128, 11940–11947. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.; Kresge, A.J.; Zhu, J. Reactive intermediates. Some chemistry of quinone methides. Pure Appl. Chem. 2000, 72, 2299–2308. [Google Scholar] [CrossRef]
- Wilcke, D.; Herdtweck, E.; Bach, T. Enantioselective Brønsted acid catalysis in the Friedel-Crafts reaction of indoles with secondary ortho-hydroxybenzylic alcohols. Synlett 2011, 1235–1238. [Google Scholar] [CrossRef]
- Rueping, M.; Uria, U.; Lin, M.Y.; Atodiresei, I. Chiral organic contact ion pairs in metal-free catalytic asymmetric allylic substitutions. J. Am. Chem. Soc. 2011, 133, 3732–3735. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, S.J.; Sathiyanarayanan, A.M.; Vuram, P.K. Hetero-Diels-Alder reaction of olefin with o-quinone methides generated using (±)-binolphosphoric acid for the stereoselective synthesis of 2,4-diarylbenzopyrans: Application to the formal synthesis of myristinin B/C. RSC Adv. 2013, 3, 18279–18282. [Google Scholar] [CrossRef]
- El-Sepelgy, O.; Haseloff, S.; Alamsetti, S.K.; Schneider, C. Brønsted acid catalyzed, conjugate addition of β-dicarbonyls to in situ generated ortho-quinone methides-enantioselective synthesis of 4-aryl-4H-chromenes. Angew. Chem. Int. Ed. 2014, 53, 7923–7927. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.C.; Liao, H.H.; Rueping, M. Enantio- and diastereoselective access to distant stereocenters embedded within tetrahydroxanthenes: Utilizing ortho-quinone methides as reactive intermediates in asymmetric Brønsted acid catalysis. Angew. Chem. Int. Ed. 2014, 53, 13258–13263. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Freccero, M.; Zanaletti, R.; Sarzi-Amadè, M. o-Quinone methide as alkylating agent of nitrogen, oxygen, and sulfur nucleophiles. The role of H-bonding and solvent effects on the reactivity through a DFT computational study. J. Am. Chem. Soc. 2001, 123, 8366–8377. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Alamasetti, S.K.; Schneider, C. Chiral Brønsted acid-catalyzed Friedel-Crafts alkylation of electron-rich arenes with in situ-generated ortho-quinone methides: Highly enantioselective synthesis of diarylindolylmethanes and triarylmethanes. Chem. Commun. 2015, 51, 1461–1464. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Schneider, C. Brønsted acid-catalyzed, highly enantioselective addition of enamides to in situ-generated ortho-quinone methides: A domino approach to complex acetamidotetrahydroxanthenes. Chem. Eur. J. 2015, 21, 2348–2352. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Schneider, C. Directing group assisted nucleophilic substitution of propargylic alcohols via o-quinone methide intermediates: Brønsted acid catalyzed, highly enantio- and diastereoselective synthesis of 7-alkynyl-12a-acetamido-substututed benzoxanthenes. Org. Lett. 2015, 17, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, Z.; Chu, B.; Sun, J. Enantioselective formation of all-carbon quaternary stereocenters from indoles and tertiary alcohols bearing a directing group. Angew. Chem. Int. Ed. 2015, 54, 1461–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Sun, S.B.; He, S.H.; Wu, Q.; Shi, F. Catalytic asymmetric inverse-electron-demand oxa-Diels-Alder reaction of in situ generated ortho-quinone methides with 3-methyl-2-vinylindoles. Angew. Chem. Int. Ed. 2015, 54, 5460–5464. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.C.; Raja, S.; Liao, H.H.; Atodiresei, I.; Rueping, M. Ortho-quinone methides as reactive intermediates in asymmetric Brønsted acid catalyzed cycloadditions with unactivated alkenes by exclusive activation of the electrophile. Angew. Chem. Int. Ed. 2015, 54, 5762–5765. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ai, F.; Wang, Z.; Zhao, W.; Zhu, G.; Lin, Z.; Sun, J. Organocatalytic asymmetric synthesis of 1,1-diaryethanes by transfer hydrogenation. J. Am. Chem. Soc. 2015, 137, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Chen, D.F.; Luo, S.W.; Wu, X. Chiral Brønsted acid catalyzed intermolecular Friedel-Crafts alkylation of styrenes with indoles: Construction of all-carbon quaternary sterecenters. Tetrahedron Asymmetry 2015, 26, 219–224. [Google Scholar] [CrossRef]
- Dai, W.; Lu, H.; Jiang, X.L.; Gao, T.T.; Shi, F. Organocatalytic asymmetric hydroarylation of o-hydroxyl styrenes via remote activation of phenylhydrazolnes. Tetrahedron Asymmetry 2015, 26, 109–117. [Google Scholar] [CrossRef]
- Fochi, M.; Caruana, L.; Bernardi, L. Catalytic asymmetric aza-Diels-Alder reactions: The Povarov cycloaddition reaction. Synthesis 2014, 135–157. [Google Scholar] [CrossRef]
- Li, T.; Rokita, S.E. Selective modification of DNA controlled by an ionic signal. J. Am. Chem. Soc. 1991, 113, 7771–7773. [Google Scholar] [CrossRef]
- Heslin, J.C.; Moody, C.J. Rhodium carbenoid mediated cyclisations. Part 2. Synthesis of cyclic ethers. J. Chem. Soc. Perkin Trans. 1 1988, 1417–1423. [Google Scholar] [CrossRef]
- Mattson, A.E.; Scheidt, K.A. Nucleophilic acylation of o-quinone methides: An umpolung strategy for the synthesis of α-aryl ketones and benzofurans. J. Am. Chem. Soc. 2007, 129, 4508–4509. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.; Orue, A.; Scheidt, K.A. A dual Lewis base activation strategy for enantioselective carbene-catalyzed annulations. J. Am. Chem. Soc. 2013, 135, 10634–10637. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Scheidt, K.A. N-Heterocyclic carbene-catalyzed enantioselective annulations: A dual activation strategy for a formal [4 + 2] addition for dihydrocoumarins. Chem. Commun. 2015, 51, 3407–3410. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, R.E.; Tan, S.J.; Kam, T.S.; Porco, J.A., Jr. Development of an alkaloid-pyrone annulation: Synthesis of pleiomaltinine. Angew. Chem. Int. Ed. 2012, 51, 9348–9351. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Ziegler, R.E.; Porco, J.A., Jr.; Jacobsen, E.N. Thiourea catalyzed enantioselective addition of indoles to pyrones: Alkaloid cores with quaternary carbons. J. Am. Chem. Soc. 2014, 136, 13614–13617. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, A.; Petrini, M.; Shaik, R.R. Synthesis of 3-substituted indoles via reactive alkylideneindolenine intermediates. Org. Biomol. Chem. 2010, 8, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Petrini, M. α-Amido sulfones as stable precursors of reactive N-acylimino derivatives. Chem. Rev. 2005, 105, 3949–3977. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Cao, L.L.; Ye, Z.S.; Jiang, G.F.; Zhou, Y.G. A mild method for generation of o-quinone methides under basic conditions. The facile synthesis of trans-2,3-dihydrobenzofurans. Chem. Commun. 2013, 49, 1660–1662. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Gao, X.; Chen, M.W.; Zhou, Y.G. The straightforward preparation of 2-(2-hydroxyphenyl)acetonitriles via the o-quinone methides generated from 2-(1-tosylalkyl)phenols. Chin. J. Chem. 2014, 32, 981–984. [Google Scholar] [CrossRef]
- Wu, B.; Gao, X.; Chen, M.W.; Zhou, Y.G. Direct amination of 2-(1-tosylalkyl)phenols with aqueous ammonia: A metal-free synthesis of primary amines. Tetrahedron Lett. 2015, 56, 1135–1137. [Google Scholar] [CrossRef]
- Guo, W.; Wu, B.; Zhou, X.; Chen, P.; Wang, X.; Zhou, Y.G.; Liu, Y.; Li, C. Formal asymmetric catalytic thiolation with a bifunctional catalyst at a water-oil interface: Synthesis of benzyl thiols. Angew. Chem. Int. Ed. 2015, 54, 4522–4526. [Google Scholar] [CrossRef] [PubMed]
- Caruana, L.; Mondatori, M.; Corti, V.; Morales, S.; Mazzanti, A.; Fochi, M.; Bernardi, L. Catalytic asymmetric addition of Meldrum’s acid, malononitrile and 1,3-dicarbonyls to ortho-quinone methides generated in situ under basic conditions. Chem. Eur. J. 2015, 21, 6037–6041. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.J.; Magauer, T.; Mulzer, J. In pursuit of a competitive target: Total synthesis of the antibiotic Kendomycin. Angew. Chem. Int. Ed. 2010, 49, 5614–5626. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Gerth, K.; Steinmetz, H.; Reinecke, S.; Kessler, W.; Kirschning, A.; Müller, R. Elansolid A3, a unique p-quinone methide antibiotic from Chitinophaga sancti. Chem. Eur. J. 2011, 17, 7739–7744. [Google Scholar] [CrossRef] [PubMed]
- Dehn, R.; Katsuyama, Y.; Weber, A.; Gerth, K.; Jansen, R.; Steinmetx, H.; Höfle, G.; Müller, R.; Kirschning, A. Molecular basis of Elansolid biosynthesis: Evidence for an unprecedented quinone methide initiated intramolecular Diels–Alder cycloaddition/macrolactonization. Angew. Chem. Int. Ed. 2011, 50, 3882–3887. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Hampel, N.; Singer, T.; Ofial, R.A.; Mayr, H. Synthesis and characterizaion of novel quinone methides: References electrophiles for the construction of nucleophilicity scales. Eur. J. Org. Chem. 2009, 3203–3211. [Google Scholar] [CrossRef]
- Chu, W.D.; Zhang, L.F.; Bao, X.; Zhao, X.H.; Zeng, C.; Du, J.Y.; Zhang, G.B.; Wang, F.X.; Ma, X.Y.; Fan, C.A. Asymmetric catalytic 1,6-conjugated addition/aromatization of para-quinone methides: Enantioselective introduction of functionalized diarylmethine stereogenic centers. Angew. Chem. Int. Ed. 2013, 52, 9229–9233. [Google Scholar] [CrossRef] [PubMed]
- Caruana, L.; Kniep, F.; Johansen, T.K.; Poulsen, H.P.; Jørgensen, K.A. A new organocatalytic concept for asymmetric α-alkylation of aldehydes. J. Am. Chem. Soc. 2014, 136, 15929–15932. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Younai, A.; Price, C.K.; Izquierdo, J.; Mishra, R.K.; Scheidt, K.A. Enantioselective annulations for dihydroquinolones by in situ generation of azolium enolates. J. Am. Chem. Soc. 2014, 136, 10589–10592. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Q.; Wang, Q.; An, J.; Chen, J.R.; Lu, L.Q.; Xiao, W.J. Construction of optically active indolines by formal [4 + 1] annulation of sulfur ylides and N-(ortho-chloromethyl)aryl amides. Chem. Eur. J. 2013, 19, 8401–8404. [Google Scholar] [CrossRef] [PubMed]
- Parra, A.; Tortosa, M. para-Quinone methide: A new player in asymmetric catalysis. ChemCatChem 2015, 7, 1524–1526. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruana, L.; Fochi, M.; Bernardi, L. The Emergence of Quinone Methides in Asymmetric Organocatalysis. Molecules 2015, 20, 11733-11764. https://doi.org/10.3390/molecules200711733
Caruana L, Fochi M, Bernardi L. The Emergence of Quinone Methides in Asymmetric Organocatalysis. Molecules. 2015; 20(7):11733-11764. https://doi.org/10.3390/molecules200711733
Chicago/Turabian StyleCaruana, Lorenzo, Mariafrancesca Fochi, and Luca Bernardi. 2015. "The Emergence of Quinone Methides in Asymmetric Organocatalysis" Molecules 20, no. 7: 11733-11764. https://doi.org/10.3390/molecules200711733
APA StyleCaruana, L., Fochi, M., & Bernardi, L. (2015). The Emergence of Quinone Methides in Asymmetric Organocatalysis. Molecules, 20(7), 11733-11764. https://doi.org/10.3390/molecules200711733