
Synthesis of a Spirocyclic Oxetane-Fused Benzimidazole

Abstract
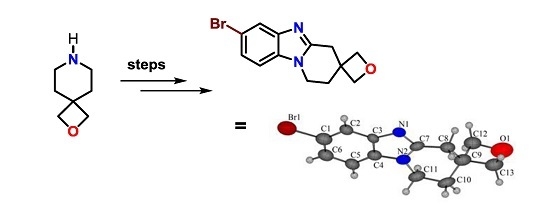
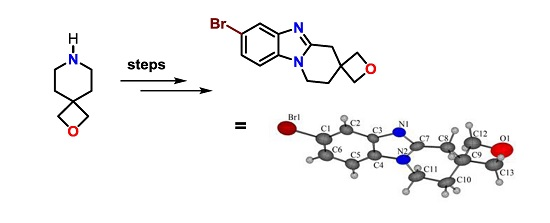
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
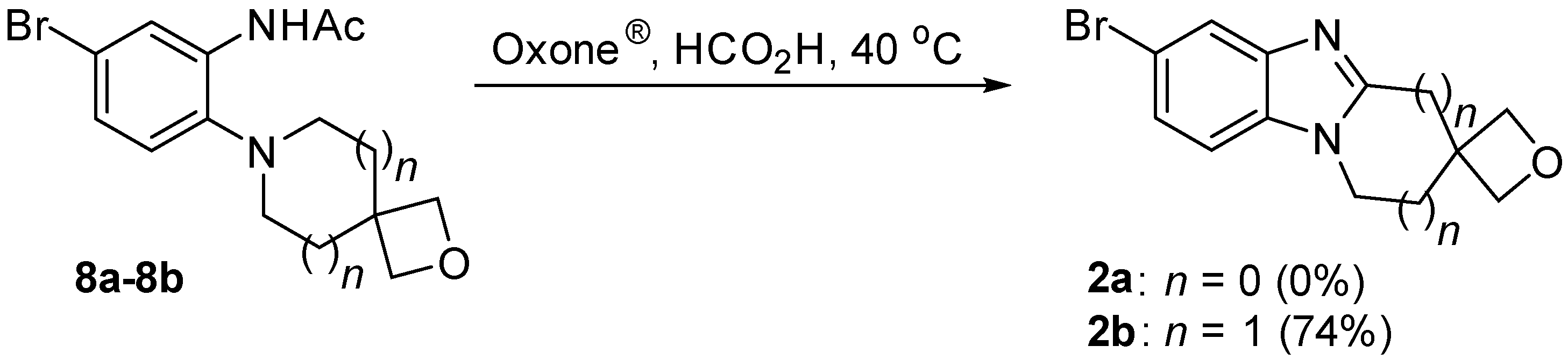{kind=link}
1. Introduction

2. Results and Discussion
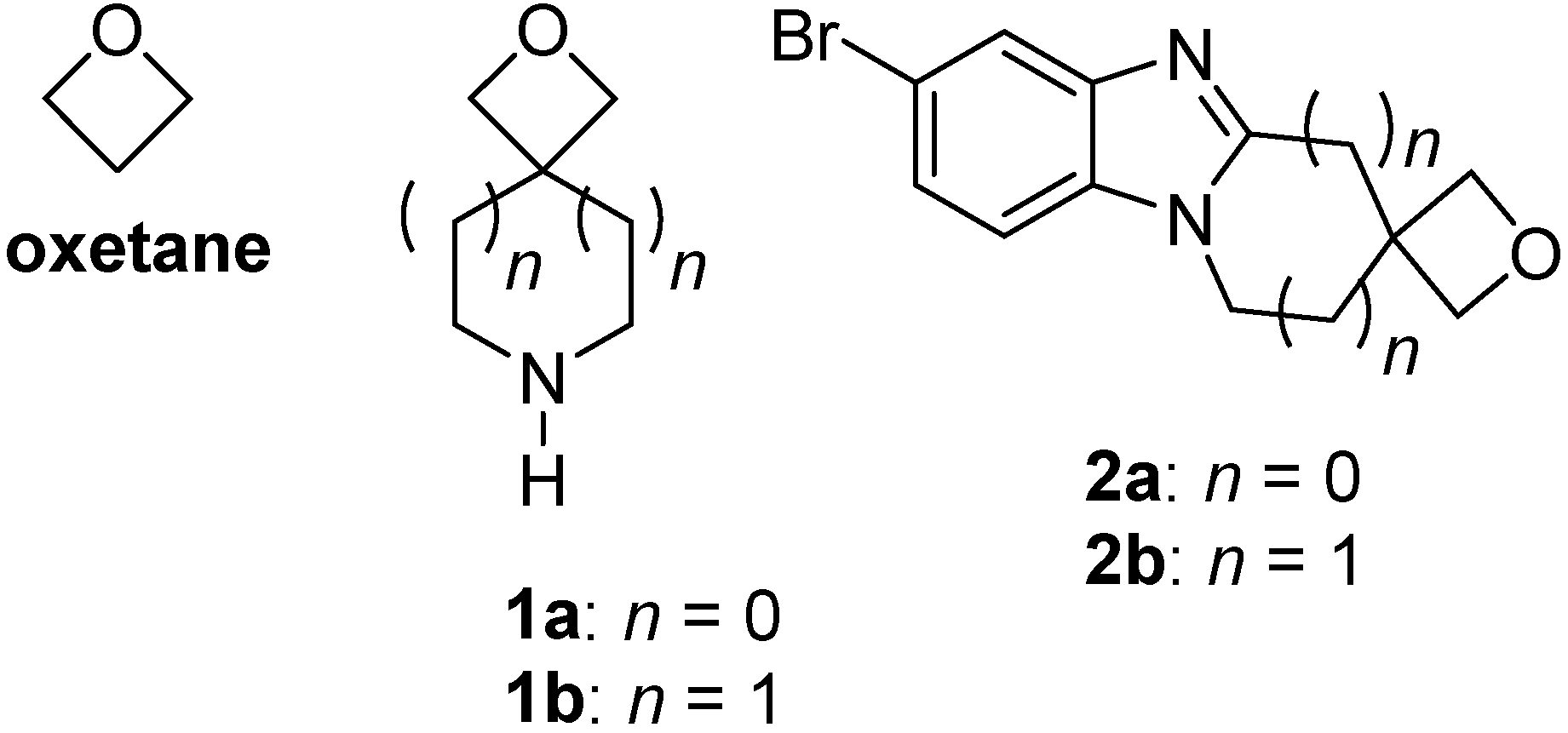
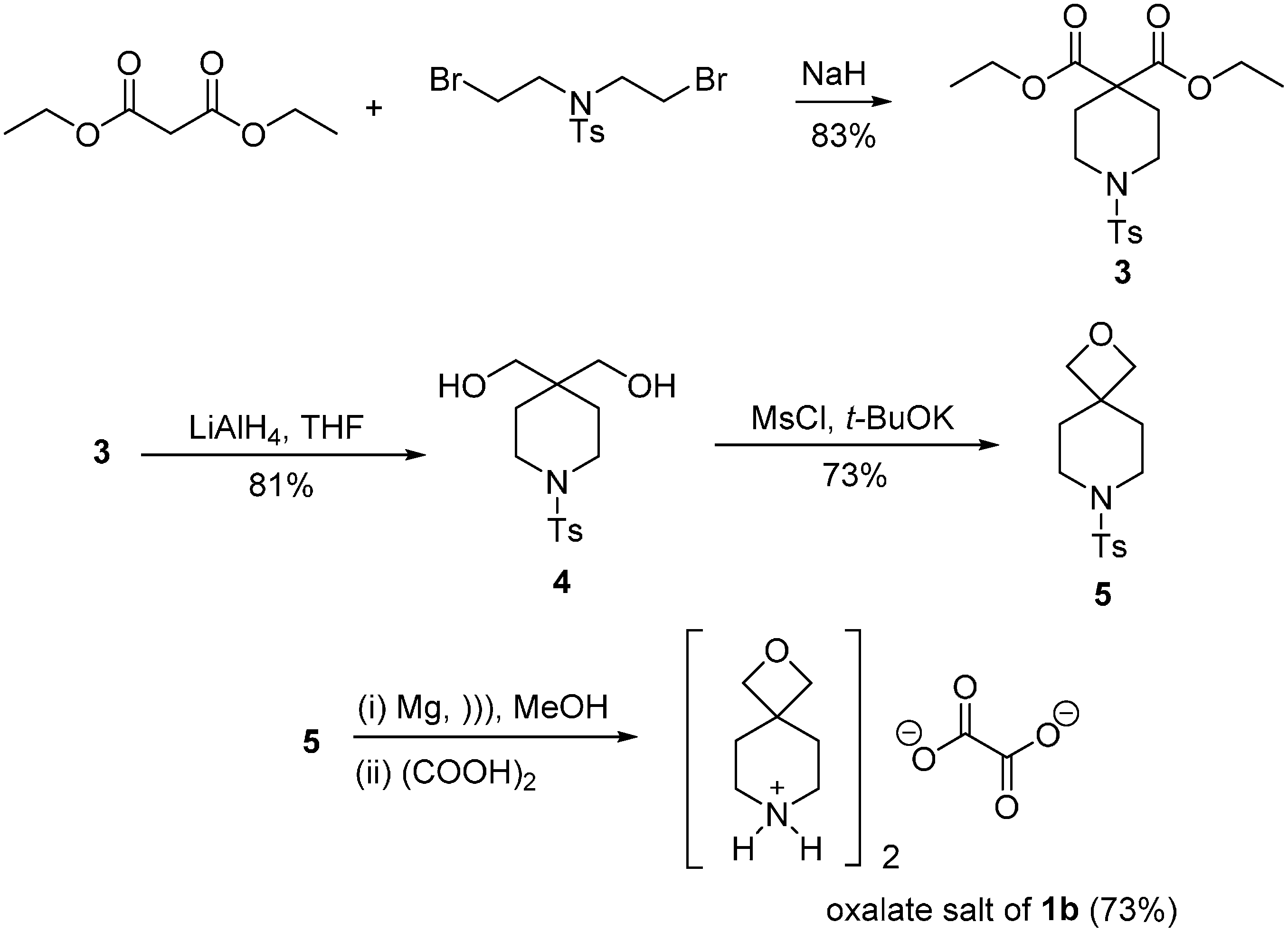
2.1. Synthesis of Spirocyclic Oxetanes

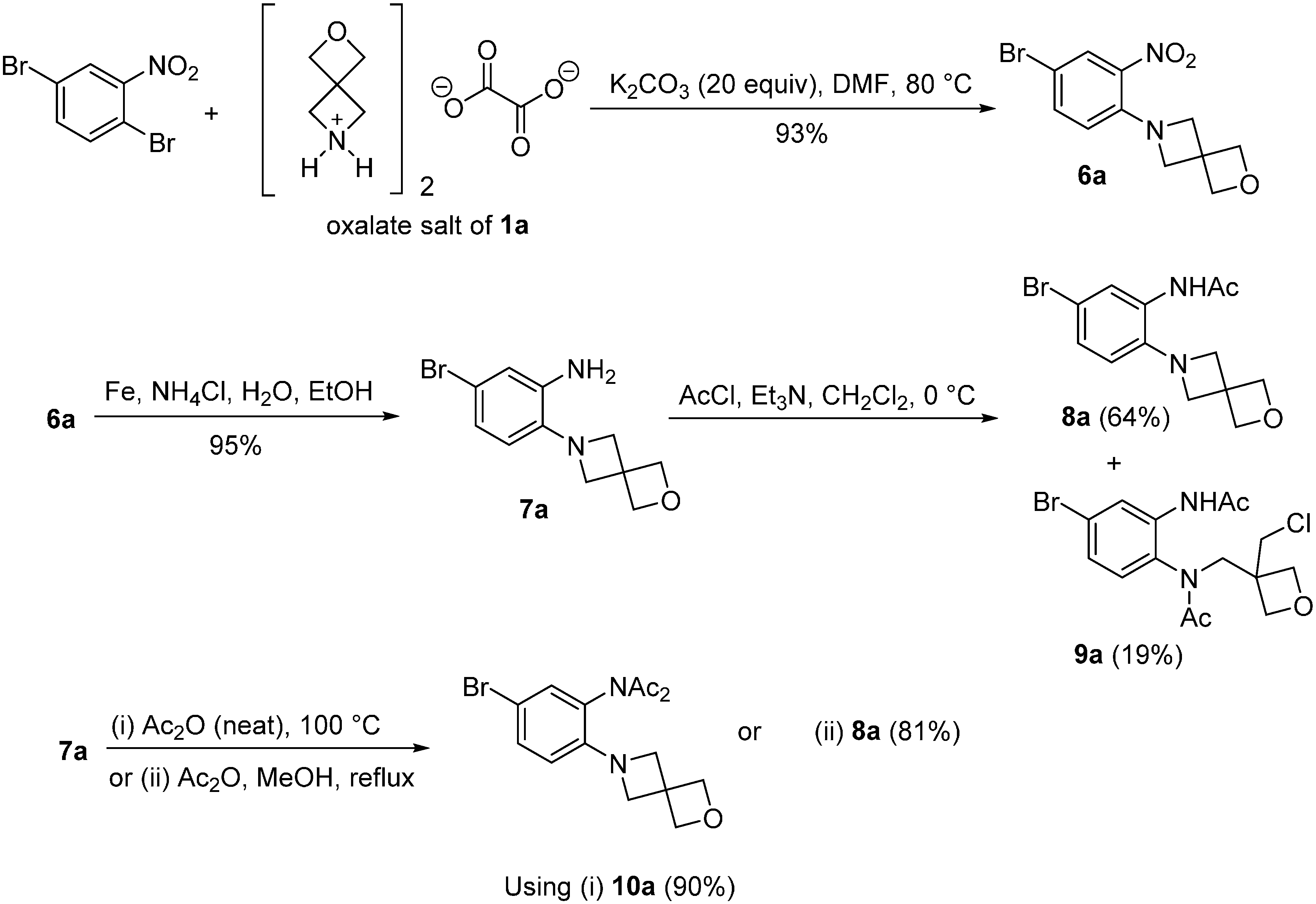
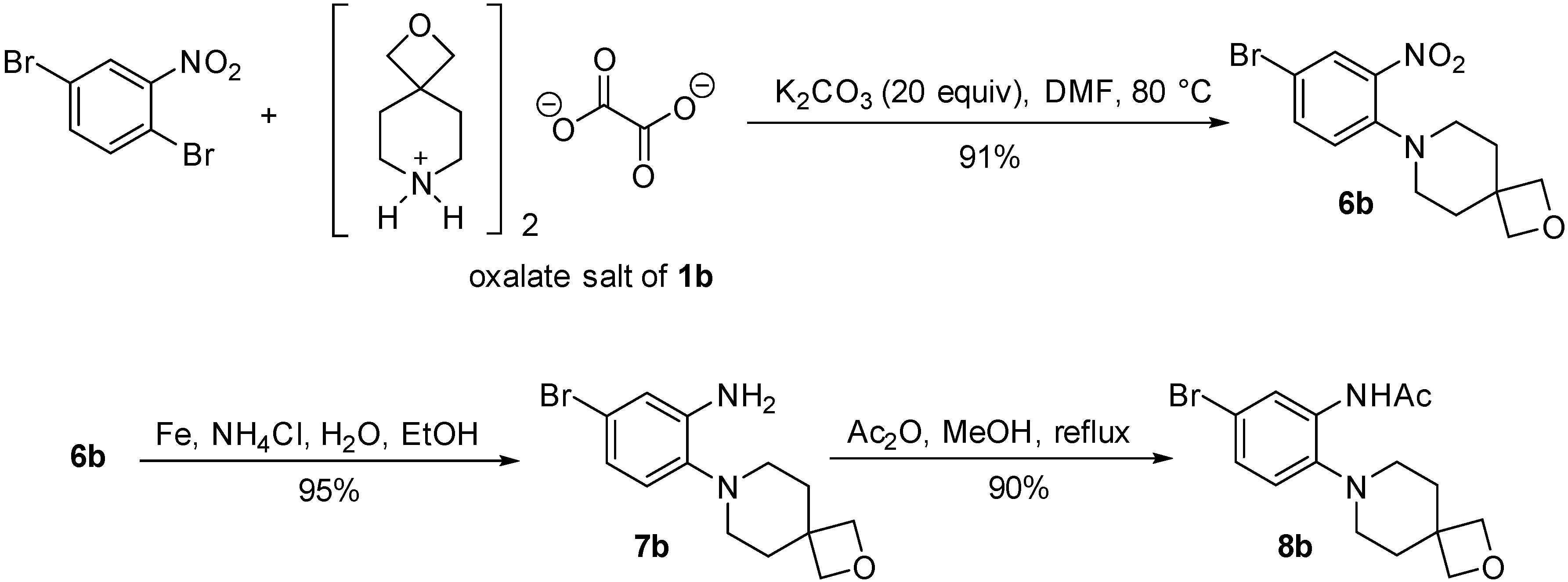
2.2. Synthesis of o-Cycloalkylaminoacetanilides



2.3. Fusion of the Spirocyclic Oxetanes


3. Experimental Section
3.1. General
3.2. Experimental Procedures
3.2.1. Synthesis of Spirocyclic Oxetanes
3.2.2. Synthesis of Acetanilide Cyclization Precursors
General Procedure for the Synthesis of Nitrobenzenes 6a and 6b
General Procedure for the Synthesis of Anilines 7a and 7b
Procedures for the Synthesis of N-[5-Bromo-2-(2-oxa-6-azaspiro[3.3]heptan-6-yl)phenyl]acetamide (8a)
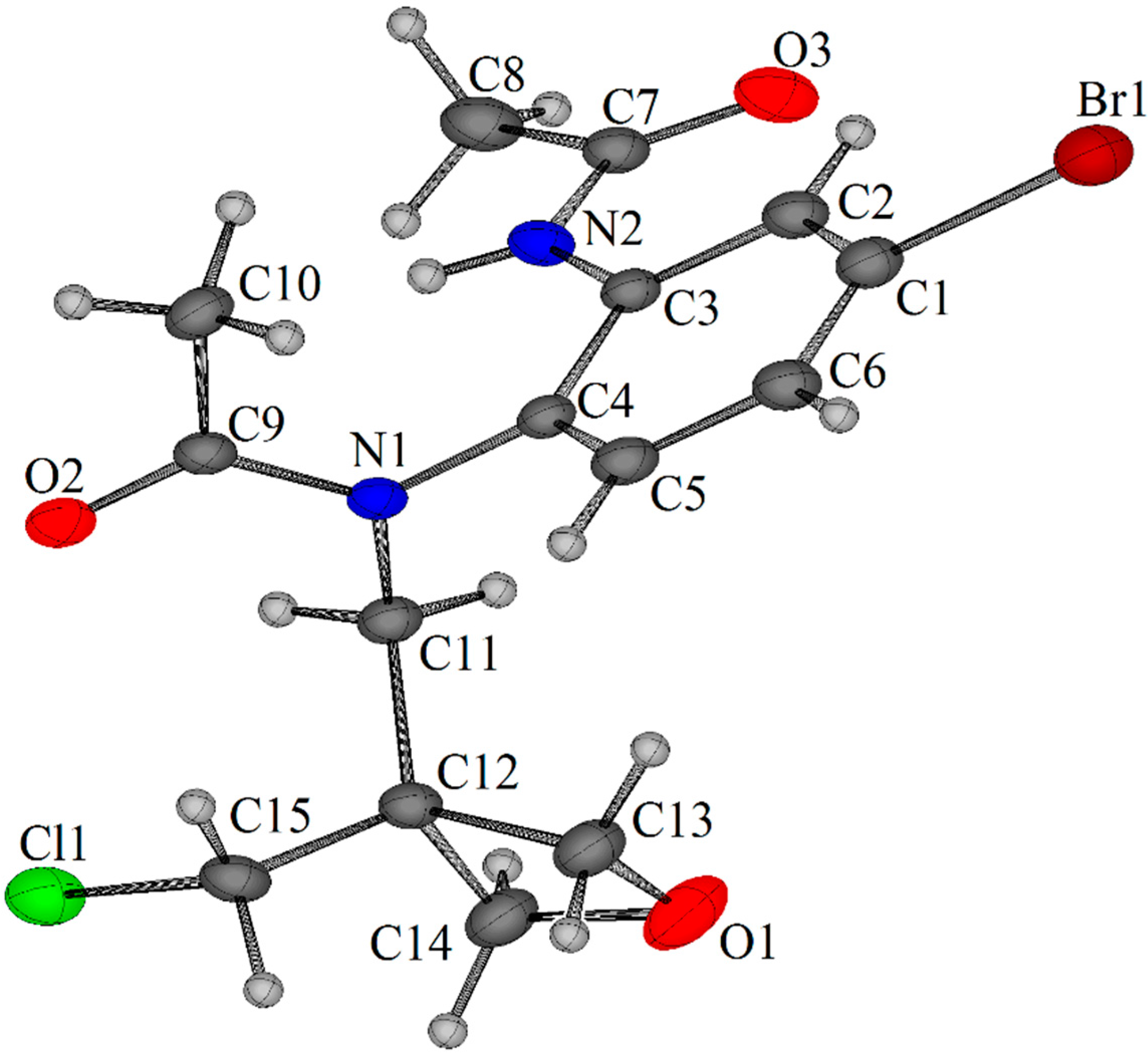
3.2.3. Oxidative Cyclization to Give the Spirocyclic Oxetane Fused Benzimidazole
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E.M. Oxetane as promising modules in drug discovery. Angew. Chem. Int. Ed. 2006, 45, 7736–7739. [Google Scholar] [CrossRef] [PubMed]
- Wuitschik, G.; Carreira, E.M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. Oxetanes in drug discovery: Structural and synthetic insights. J. Med. Chem. 2010, 53, 3227–3246. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, J.A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E.M. Oxetanes as versatile elements in drug discovery and synthesis. Angew. Chem. Int. Ed. 2010, 49, 9052–9067. [Google Scholar] [CrossRef] [PubMed]
- Wuitschik, G.; Rogers-Evans, M.; Buckl, A.; Bernasconi, M.; Märki, M.; Godel, T.; Fischer, H.; Wagner, B.; Parrilla, I.; Schuler, F.; et al. Spirocyclic oxetanes: Synthesis and properties. Angew. Chem. Int. Ed. 2008, 47, 4512–4515. [Google Scholar] [CrossRef] [PubMed]
- Fagan, V.; Bonham, S.; Carty, M.P.; Saenz-Méndez, P.; Eriksson, L.A.; Aldabbagh, F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorg. Med. Chem. 2012, 20, 3223–3232. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, M.; Xiang, F. Preparation of 4-substituted and N-substituted piperidine-4-carboxylic acid ethyl ester. CN 101525313, 09 September 2009. Scifinder Scholar 2009:1117956. [Google Scholar]
- Capet, M.; Levoin, N.; Berrebi-Bertrand, I.; Robert, P.; Schwartz, J.C.; Lecomte, J.M.; Aradhye, J.D.; Pillai, M.N.; Panchal, B.M.; Jivani, J.K.; et al. Dicarboxylic Acid Derivatives, Processes for Preparing Them, Pharmaceutical Compositions Containing Them, and Their Use as Sphingosine-1-Phosphate Receptor Agonists. WO 2008/152149, 18 December 2008. Scifinder Scholar 2008:1507568. [Google Scholar]
- Allen, J.R.; Chen, J.J.; Frohn, M.J.; Hu, E.; Liu, Q.; Pickrell, A.J.; Rumfelt, S.; Rzasa, R.M.; Zhong, W. Preparation of Nitrogen Heterocyclic Compounds Useful as 3ʹ,5ʹ-Cyclic Nucleotide-Specific Phosphodiesterase 10 (PDE10) Inhibitors. WO 2011/143365, 17 November 2011. Scifinder Scholar 2011:145609. [Google Scholar]
- Kenis, S.; D’hooghe, M.; Verniest, G.; Thi, T.A.D.; The, C.P.; Nguyen, T.V.; de Kimpe, N. Synthesis of 1-alkyl-2-(trifluoromethyl)azetidines and their regiospecific ring opening toward diverse α-(trifluoromethyl)amines via intermediate azetidinium salts. J. Org. Chem. 2012, 77, 5982–5992. [Google Scholar] [CrossRef] [PubMed]
- Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC 1408035 for 9a and CCDC 1408036 for 2b. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif (or from 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-Mail: deposit@ccdc.cam.ac.uk) .
- Meth-Cohn, O.; Suschitzky, H. Syntheses of heterocyclic compounds. Part IV. Oxidative cyclisation of aromatic amines and their N-acyl derivatives. J. Chem. Soc. 1963, 4666–4669. [Google Scholar] [CrossRef]
- Fahey, K.; Aldabbagh, F. Synthesis of seven- and eight-membered [1,2-a] alicyclic ring-fused benzimidazoles and 3-aziridinylazepino[1,2-a]benzimidazolequinone as a potential antitumour agent. Tetrahedron Lett. 2008, 49, 5235–5237. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; McArdle, P.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity of new ring-fused imidazo[5,4-f]benzimidazolequinones and mechanism using amine N-oxide cyclizations. Eur. J. Org. Chem. 2012, 1967–1975. [Google Scholar] [CrossRef]
- Chak, B.C.M.; McAuley, A. The synthesis and characterization of the pendant-armed ligand N,N′-bis(2′-pyridylmethyl)-1,7-dithia-4,11-diazacyclotetradecane (L4) and crystal structures of L4 and the copper(II) complex [Cu(L4)](ClO4)2—Crystal structure of the nickel(II)complex of N-(2′-pyridylmethyl)-1,4,7-trithia-11-azacyclotetradecane (L2), [Ni(L2)(CH3CN)](ClO4)2·CH3CN. Can. J. Chem. 2006, 84, 187–195. [Google Scholar]
- Gurry, M.; Sweeney, M.; McArdle, P.; Aldabbagh, F. One-pot hydrogen peroxide and hydrohalic acid induced ring closure and selective aromatic halogenation to give new ring-fused benzimidazoles. Org. Lett. 2015, 17, 2856–2859. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of all compounds are available from the authors upon request.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurry, M.; McArdle, P.; Aldabbagh, F. Synthesis of a Spirocyclic Oxetane-Fused Benzimidazole. Molecules 2015, 20, 13864-13874. https://doi.org/10.3390/molecules200813864
Gurry M, McArdle P, Aldabbagh F. Synthesis of a Spirocyclic Oxetane-Fused Benzimidazole. Molecules. 2015; 20(8):13864-13874. https://doi.org/10.3390/molecules200813864
Chicago/Turabian StyleGurry, Michael, Patrick McArdle, and Fawaz Aldabbagh. 2015. "Synthesis of a Spirocyclic Oxetane-Fused Benzimidazole" Molecules 20, no. 8: 13864-13874. https://doi.org/10.3390/molecules200813864
APA StyleGurry, M., McArdle, P., & Aldabbagh, F. (2015). Synthesis of a Spirocyclic Oxetane-Fused Benzimidazole. Molecules, 20(8), 13864-13874. https://doi.org/10.3390/molecules200813864