
Novel Selective and Potent EGFR Inhibitor that Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer

Abstract
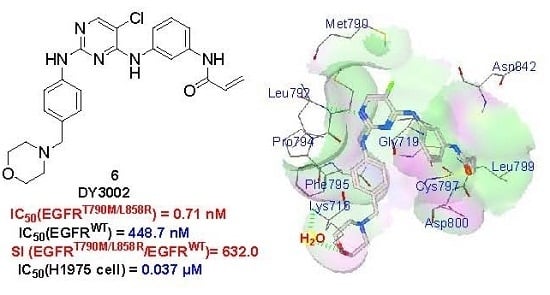
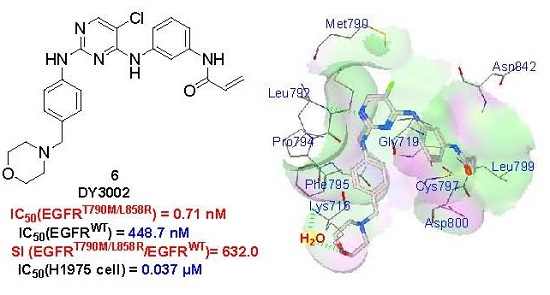
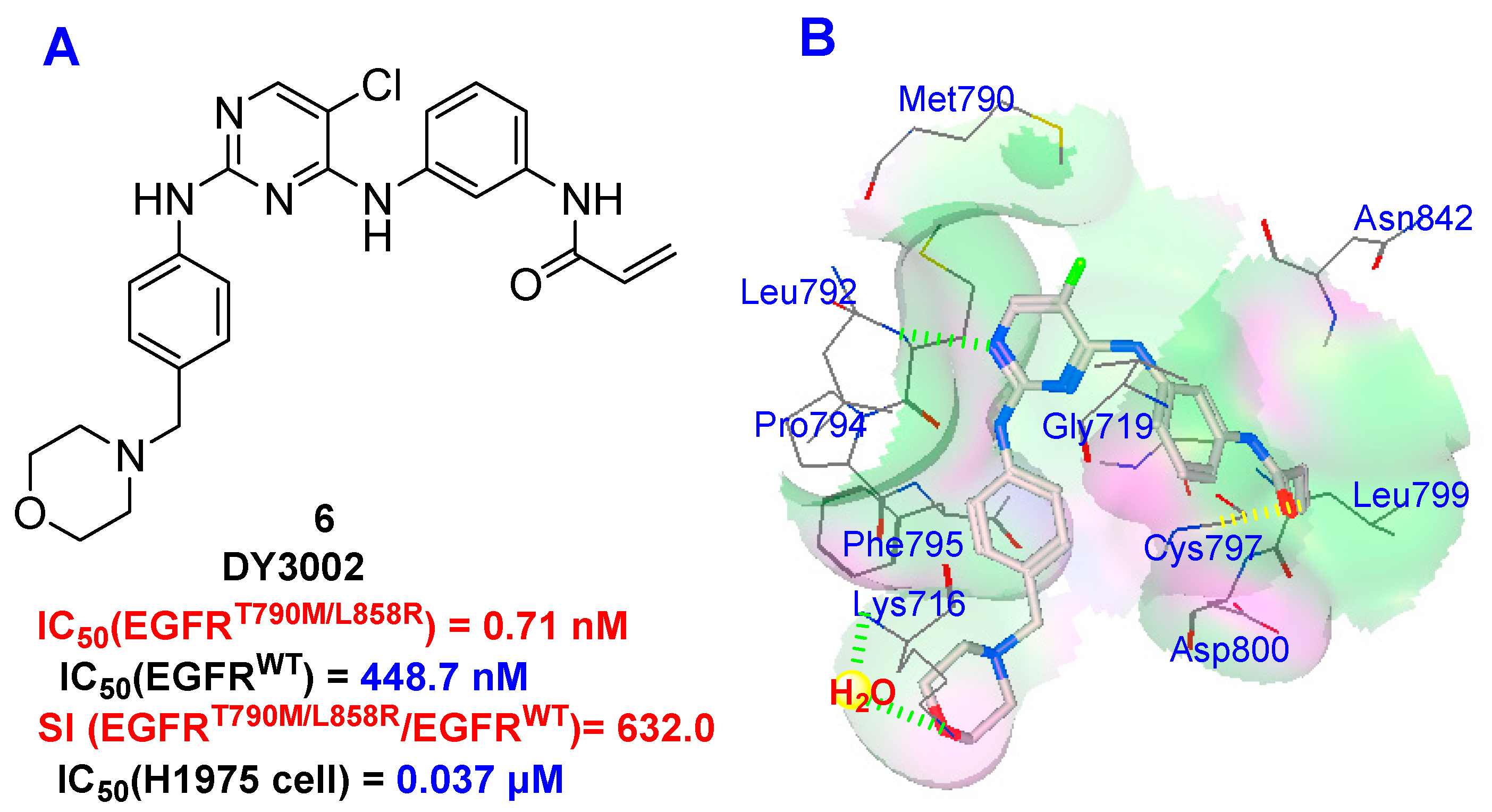
:
1. Introduction
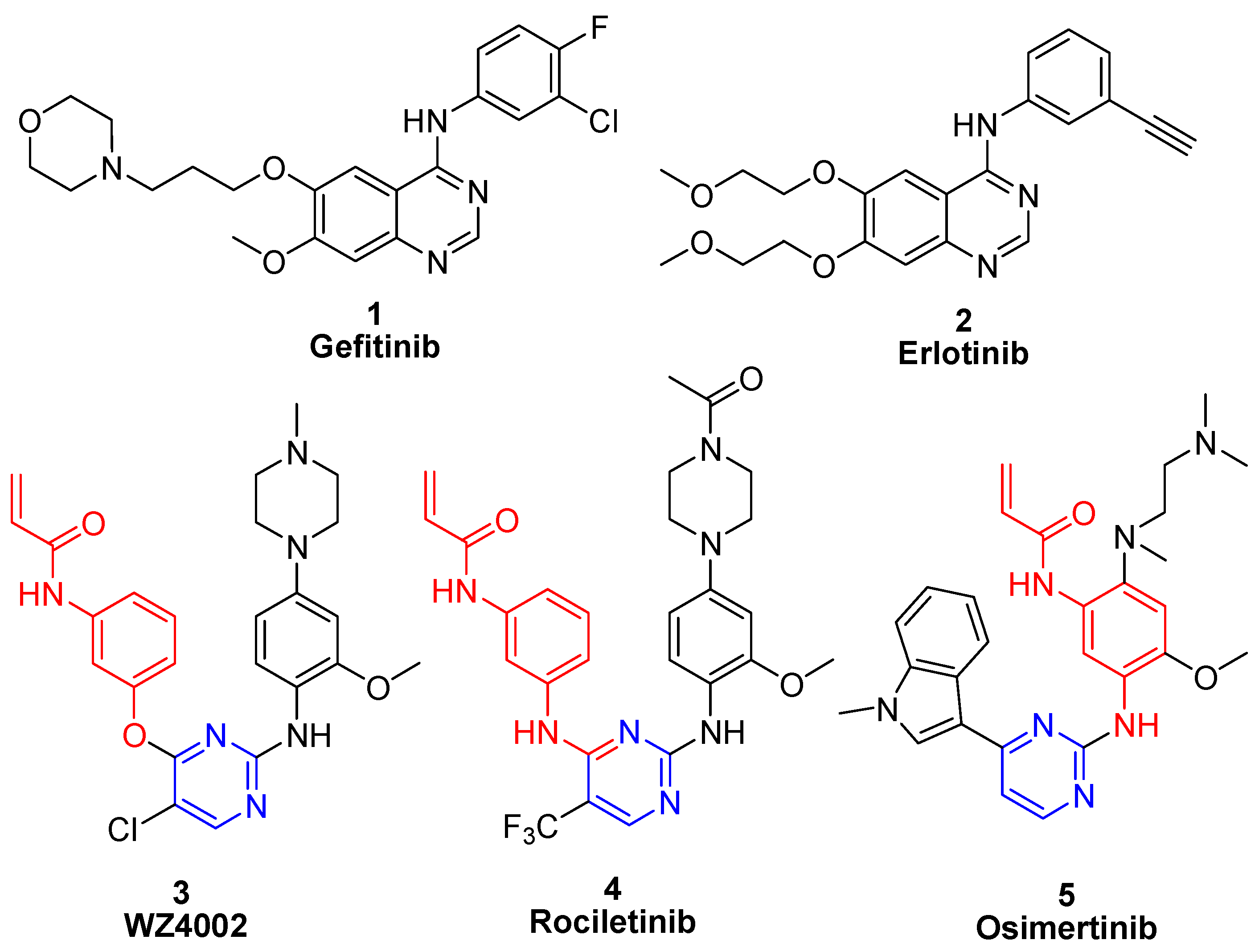
2. Results and Discussion
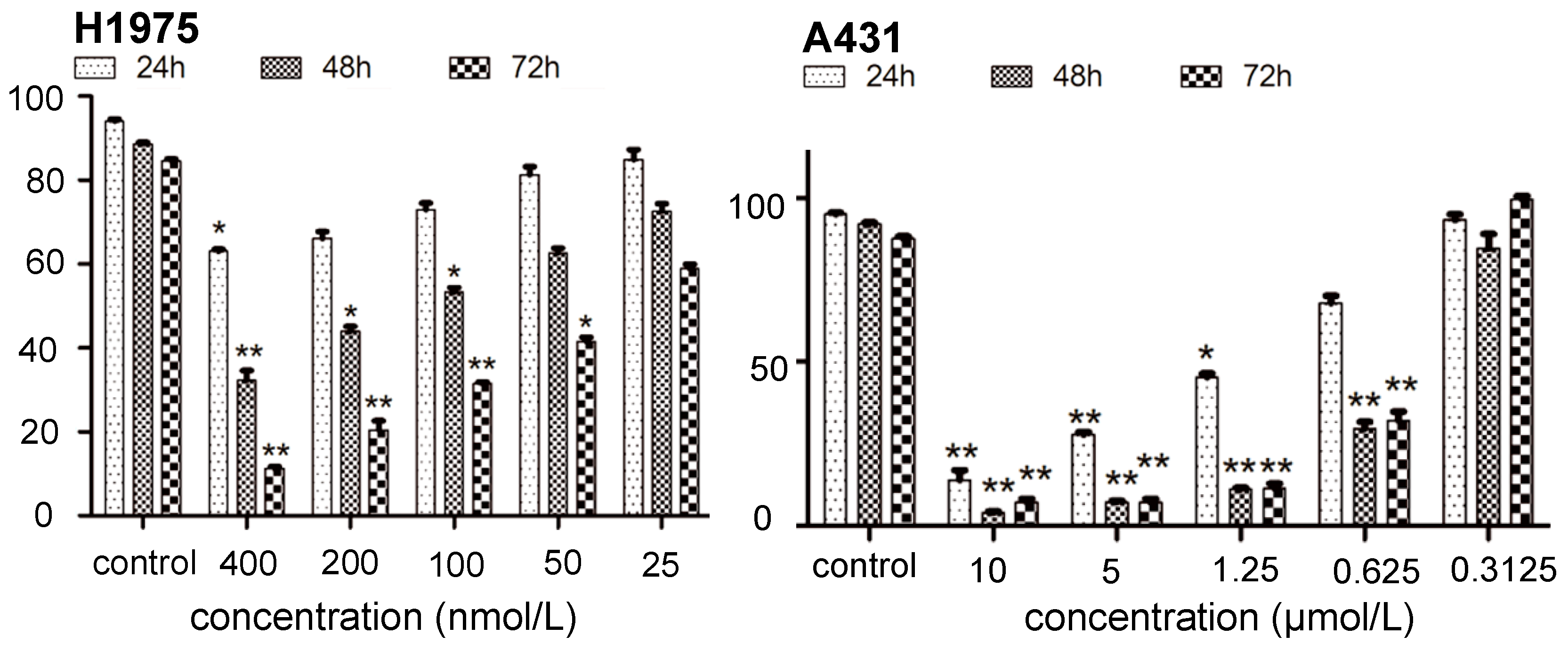
2.1. Inhibition of Kinase and Cancer Cell Viability
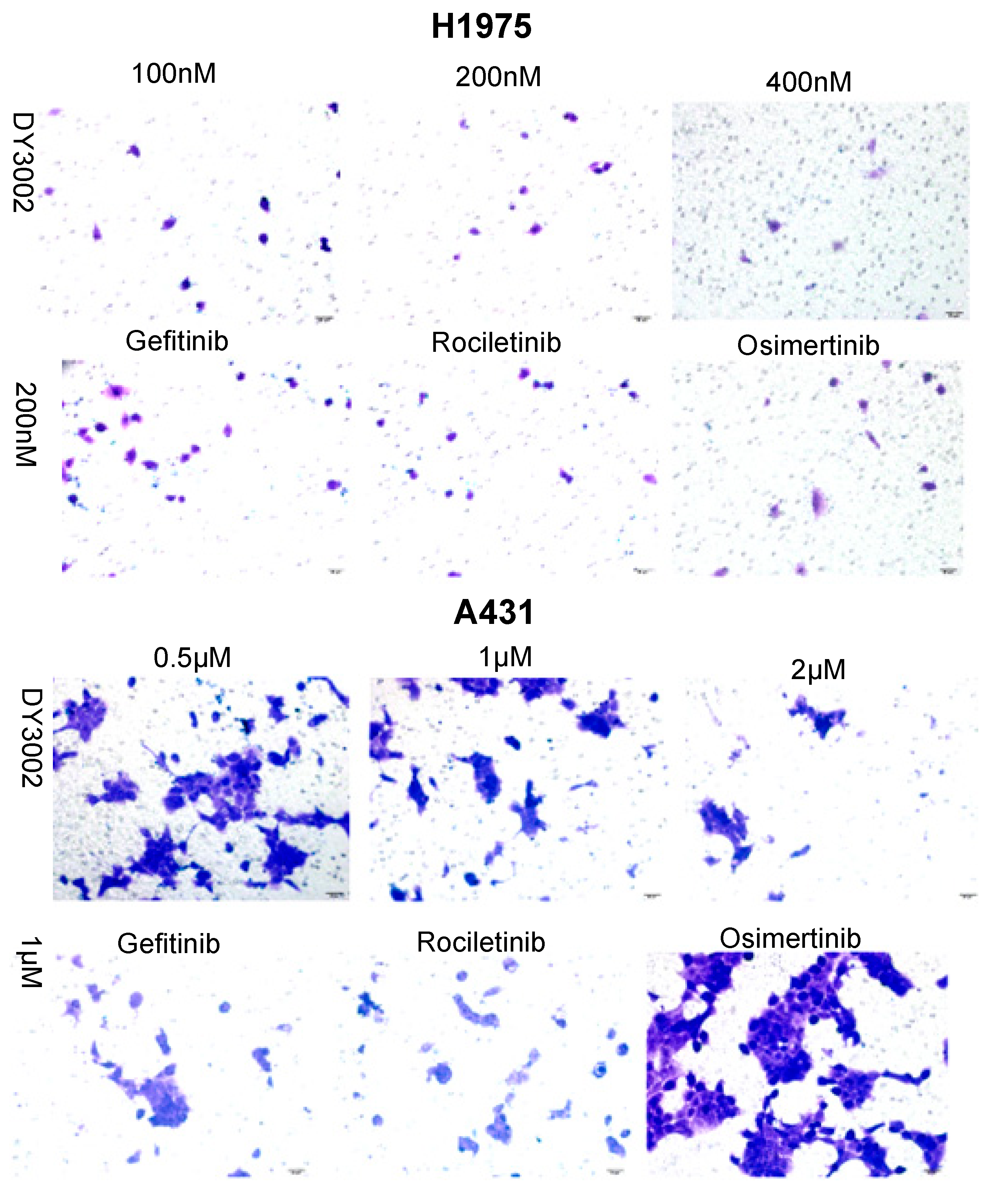
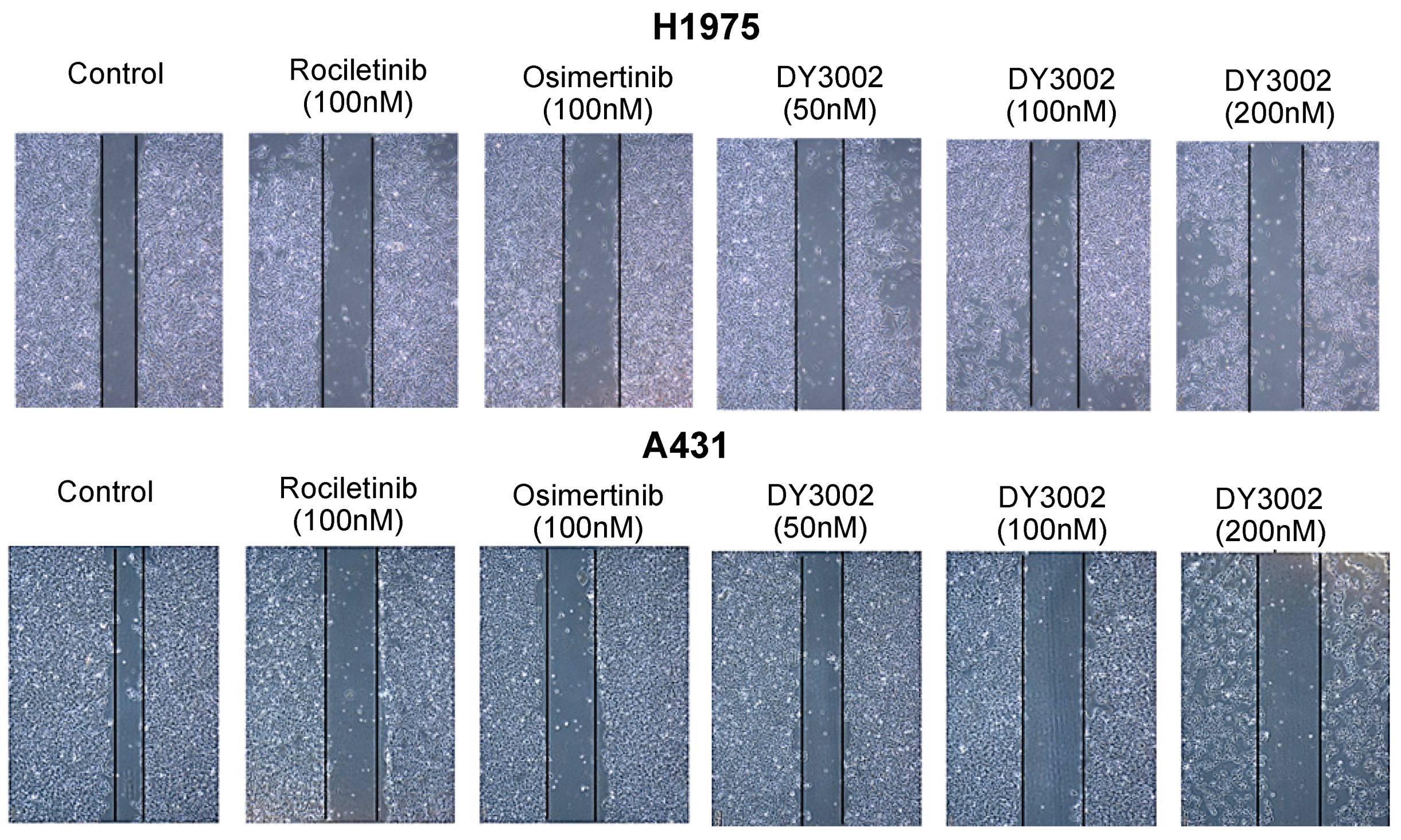
2.2. Wound Healing and Transwell Assays
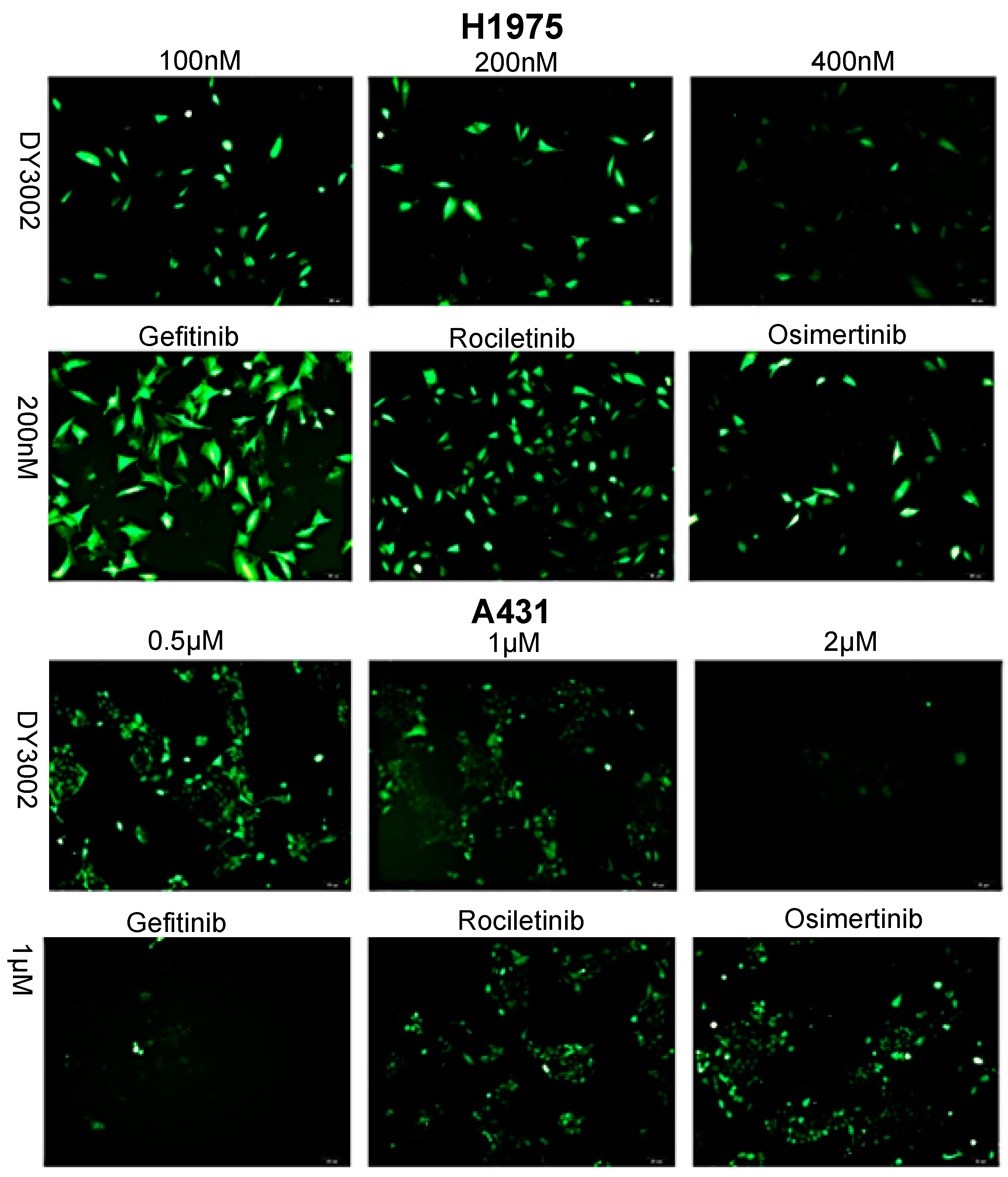
2.3. Effects on ROS Levels
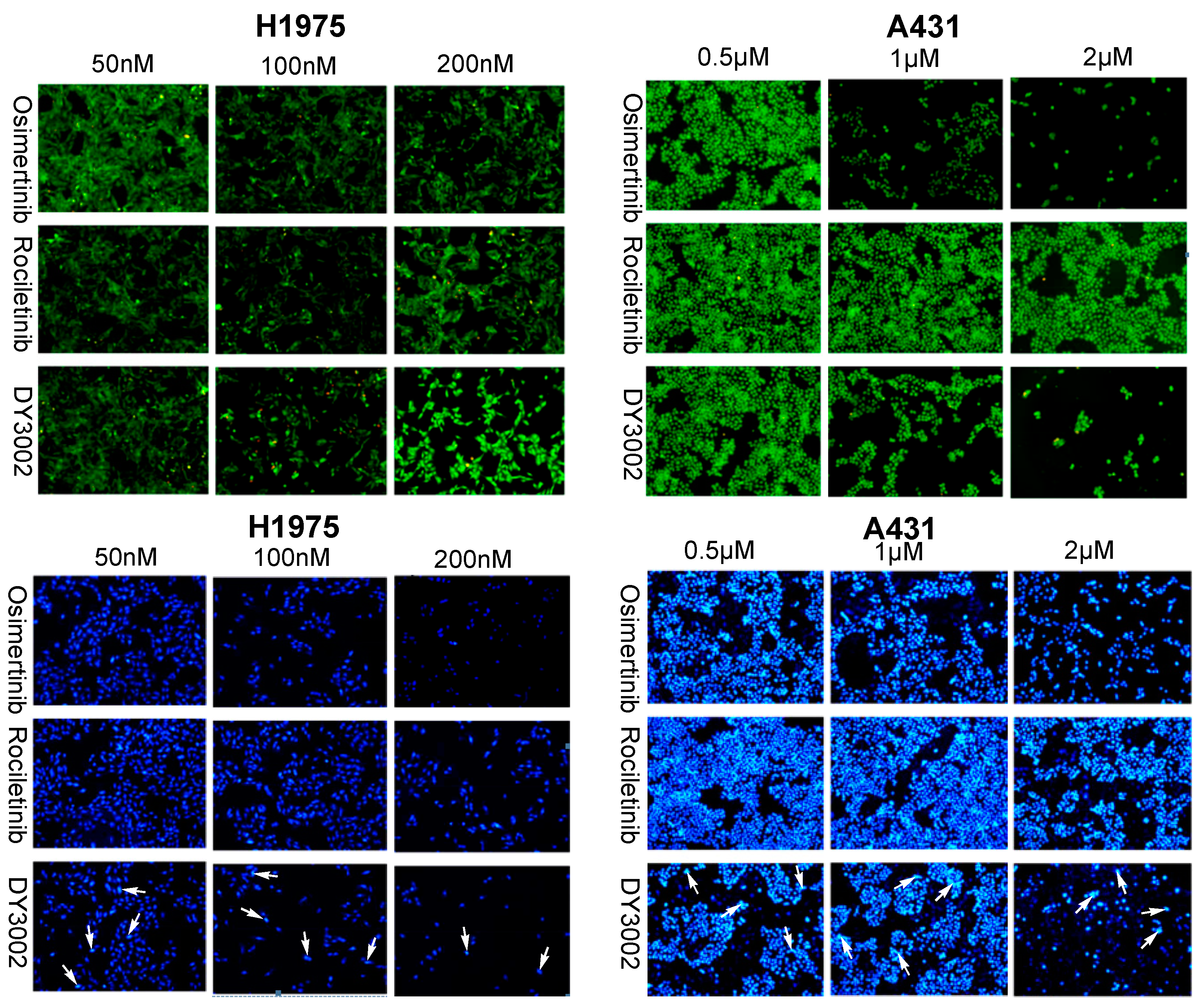
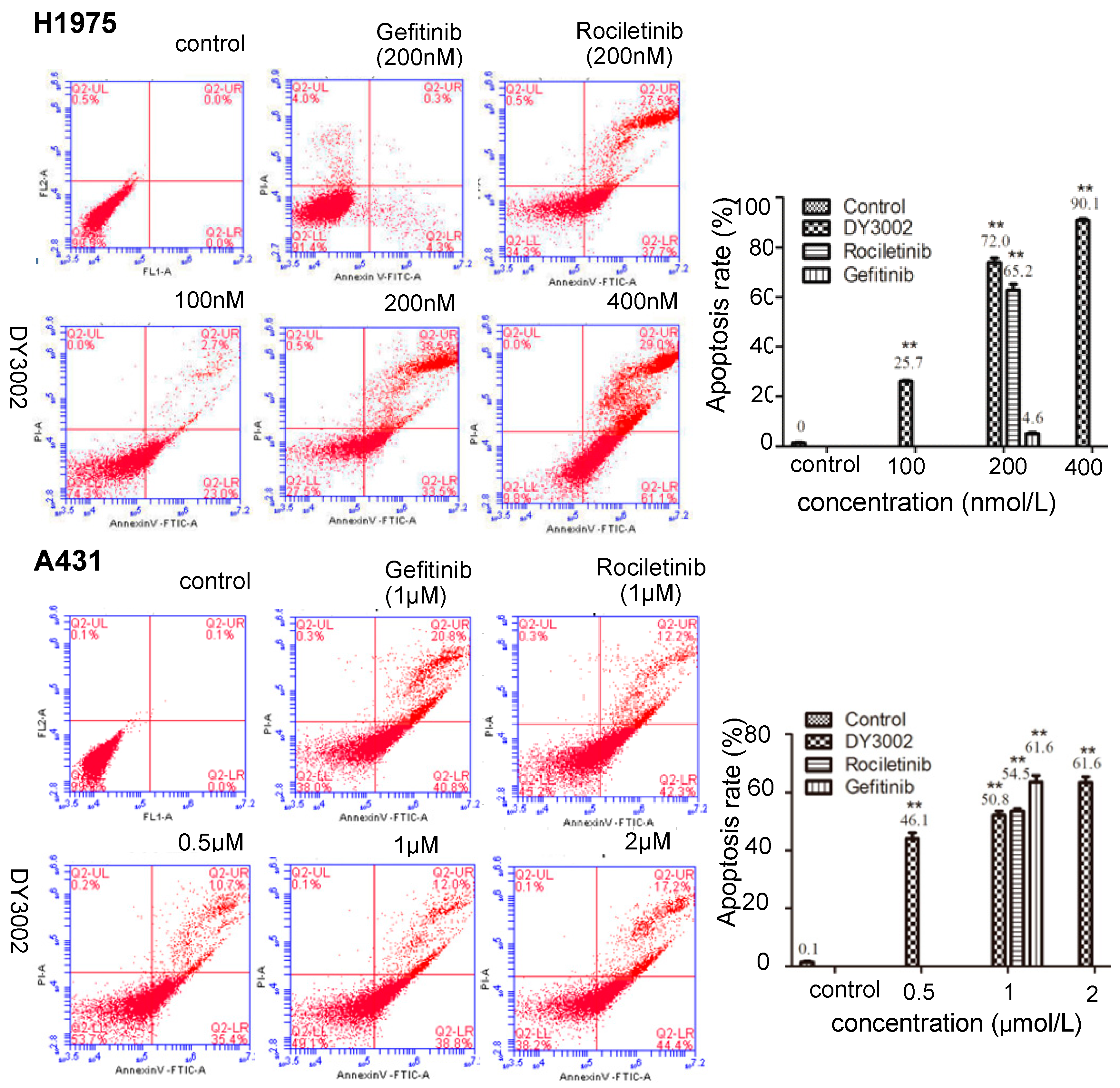
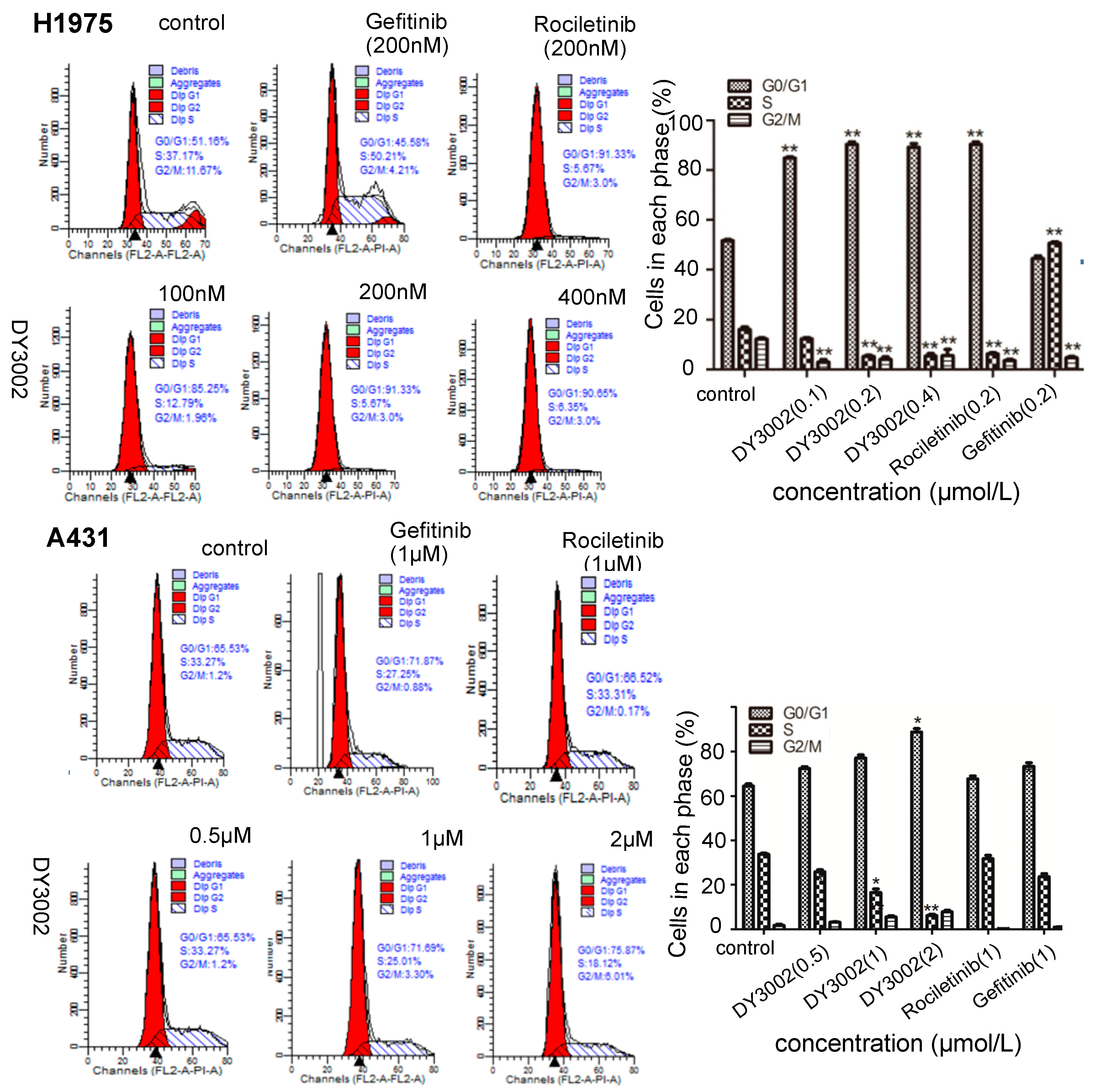
2.4. Flow Cytometer Analysis
2.5. Molecular Simulation
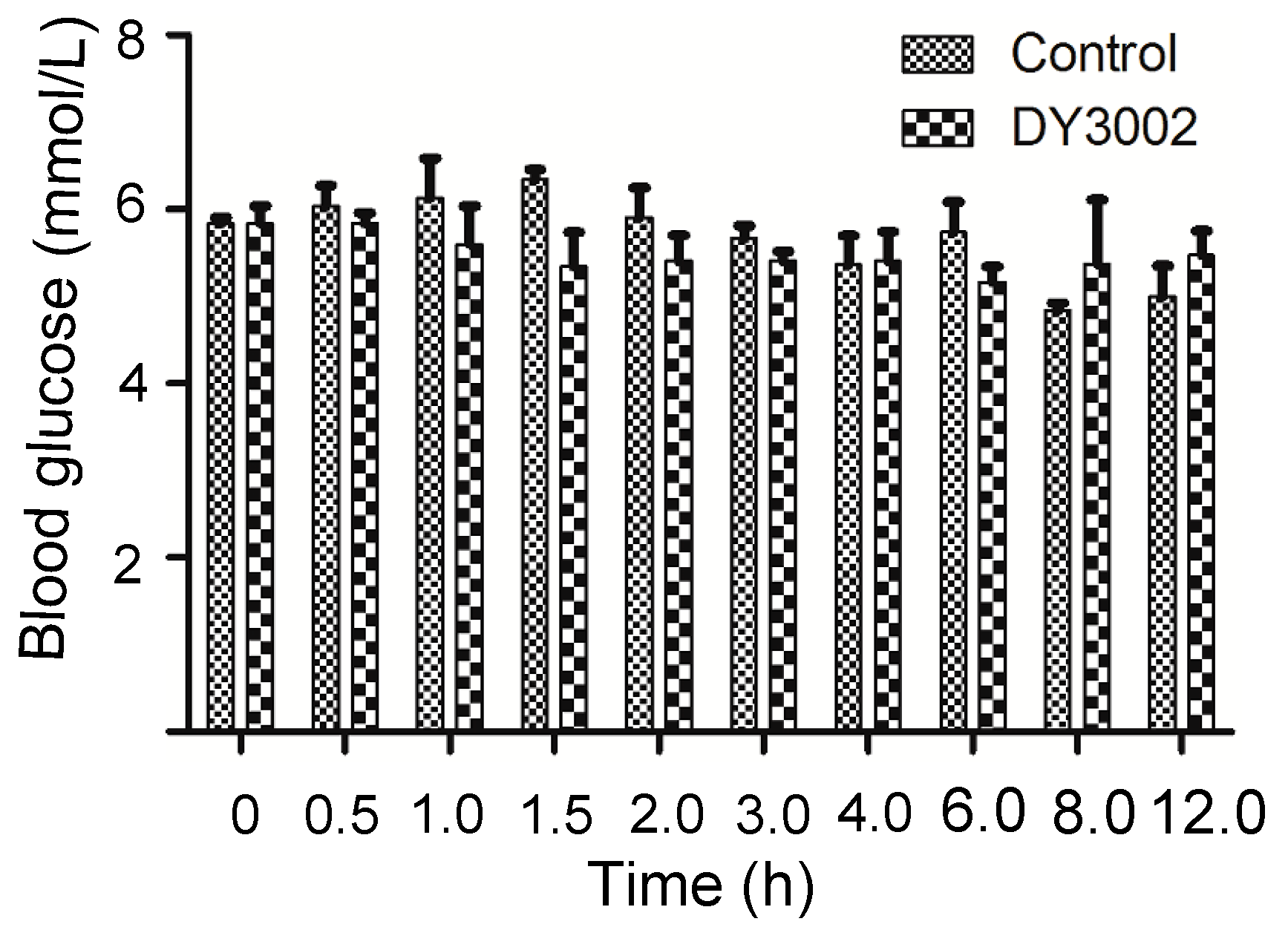
2.6. In Vivo Blood Sugar Test
3. Materials and Methods
3.1. Cell Culture and Reagents
3.2. Statistical Analysis Data
3.3. Biological Test Method
3.3.1. Kinase Enzymatic Assays
3.3.2. Cell Growth Inhibitory Activity
CCK-8 Assay
AO/EB and DAPI Staining Assay
3.3.3. Wound-Healing Assay
3.3.4. Transwell Invasion Assay
3.3.5. Detection of Intracellular ROS Generation
3.3.6. Cell Apoptosis
3.3.7. Cell Cycle Analysis
3.3.8. Glucose Concentration Determination
3.4. Molecular Modeling
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Alex, A.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J. Gefitinib (Iressa): A novel treatment for non-small cell lung cancer. Expert Rev. Anticancer Ther. 2004, 4, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Tiseo, M.; Loprevite, M.; Ardizzoni, A. Epidermal growth factor receptor inhibitors: A new prospective in the treatment of lung cancer. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, P. Erlotinib: A new therapeutic approach for non-small cell lung cancer. Expert Opin. Investig. Drugs 2003, 12, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Bridjes, A.J. Chemical inhibitors of protein kinases. Chem. Rev. 2001, 101, 2541–2572. [Google Scholar] [CrossRef]
- Sos, M.L.; Rode, H.B.; Heynck, S.; Peifer, M.; Fischer, F.; Klüter, S.; Pawar, V.G.; Reuter, C.; Heuckmann, J.M.; Weiss, J.; et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res. 2010, 70, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ko, J.; Cui, Z.; Abolhoda, A.; Ahn, J.S.; Ou, S.H.; Ahn, M.J.; Park, K. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol. Cancer Ther. 2012, 11, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wag, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Finlay, M.R.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57, 8249–8267. [Google Scholar] [CrossRef] [PubMed]
- Yver, A. Osimertinib (AZD9291)—A science-driven, collaborative approach to rapid drug design and development. Ann. Oncol. 2016, 27, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, Y.; Ge, Y.; Song, Z.; Wang, C.; Huang, S.; Jin, Y.; Han, X.; Zhen, Y.; Liu, K.; et al. Novel 4-anilinoquinazoline derivatives featuring an 1-adamantyl moiety as potent EGFR inhibitors with enhanced activity against NSCLC cell lines. Eur. J. Med. Chem. 2016, 110, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Jin, Y.; Ge, Y.; Wang, C.; Zhang, J.; Tang, Z.; Peng, J.; Liu, K.; Li, Y.; Ma, X. Synthesis and biological evaluation of azole-diphenylpyrimidine derivatives (AzDPPYs) as potent T790M mutant form of epidermal growth factor receptor inhibitors. Bioorg. Med. Chem. 2016, 24, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ge, Y.; Wang, C.; Huang, S.; Shu, X.; Liu, K.; Zhou, Y.; Ma, X. Challenges and Perspectives on the development of small-molecule EGFR inhibitors against T790M-mediated resistance in non-small-cell lung cancer. J. Med. Chem. 2016, 59, 6580–6594. [Google Scholar] [CrossRef] [PubMed]
- The spectrum data for compound N-(3-((5-chloro-2-(4-((1-morpholino)methyl)phenylamino)-4-pyrimidinyl)amino)phenyl)acrylamide (DY3002): Yield 56.6%; off-white solid. 1H NMR (400 MHz, DMSO-d6): δ 3.32 (s, 2H), 3.36 (t, J = 7.6 Hz, 2H), 3.54–3.56 (m, 6H), 5.76 (dd, J = 2.0, 10.4 Hz, 1H), 6.27 (dd, J = 2.0, 16.8 Hz, 1H), 6.46 (dd, J = 10.4, 16.8 Hz, 1H), 7.03 (d, J = 8.4 Hz, 2H), 7.31 (t, J = 6.0 Hz, 2H), 7.56 (m, 3H), 7.91 (s, 1H), 8.15 (s, 1H), 8.96 (s, 1H), 9.32 (s, 1H), 10.20 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): δ 163.6, 158.2, 156.7, 155.2, 139.8, 139.6, 139.3, 132.4, 130.6, 129.5 (2C), 129.0, 127.3, 119.8, 119.1 (2C), 115.9, 115.8, 104.2, 66.7 (2C), 62.6, 53.6 (2C). HRMS (ESI) for C24H25ClN6O2, [M+H]+ calcd: 465.1801, found: 465.1808.
- Huo, Y.; Qiu, W.; Pan, Q.; Yao, Y.; Xing, K.; Loua, M.F. Reactive oxygen species (ROS) are essential mediators in epidermal growth factor (EGF)-stimulated corneal epithelial cell proliferation, adhesion, migration, and wound healing. Exp. Eye Res. 2009, 89, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.A.; Zheng, X.; Sept, D.; Auletta, J.T.; Rosenberry, T.L.; Marshall, G.R. Drug-like leads for steric discrimination between substrate and inhibitors of human acetylcholinesterase. Chem. Biol. Drug Des. 2011, 78, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Sequist, L.V.; Goldman, J.; Wakelee, H.A.; Neal, J.; Camidge, R.; Gadgeel, S.; Papadimitrakopoulou, V.; Dziadziuszko, R.; Piotrowska, Z.; et al. Rociletinib: An oral, irreversible, highly selective small molecule inhibitor of mutant EGFR including T790M. Ann. Oncol. 2015, 26, ii14. [Google Scholar] [CrossRef]
- Sequist, L.V.; Soria, J.C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds DY3002 are available from the authors.










| Time (h) | 0 | 0.5 | 1 | 1.5 | 2 | 3 | 4 | 6 | 8 | 12 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blood Glucose (mmol/L) | Blank | 1 | 5.7 | 5.6 | 7.0 | 6.5 | 5.2 | 5.7 | 4.9 | 5.2 | 4.7 | 4.6 |
| 2 | 5.9 | 6.4 | 5.9 | 6.1 | 6.7 | 5.4 | 6.0 | 6.4 | 4.8 | 4.7 | ||
| 3 | 5.9 | 6.1 | 5.5 | 5.4 | 6.3 | 5.9 | 5.2 | 5.6 | 5.0 | 5.7 | ||
| DY3002 | 1 | 6.2 | 5.9 | 6.3 | 6.0 | 4.8 | 5.4 | 4.8 | 5.5 | 6.3 | 5.8 | |
| 2 | 5.8 | 5.6 | 5.7 | 5.4 | 5.8 | 5.6 | 6.0 | 4.9 | 5.9 | 5.7 | ||
| 3 | 5.5 | 6.0 | 4.8 | 4.6 | 5.6 | 5.2 | 5.4 | 5.1 | 3.9 | 4.9 | ||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EGFR (IC50, nM) b | SI(WT:L858R/T790M) | |
|---|---|---|---|
| WT | L858R/T790M | ||
| DY3002 | 448.7 | 0.71 | 632.0 |
| Rociletinib | 460.2 | 21.5 | 21.4 |
| Osimertinib | 482.3 | 11.8 | 40.9 |
| Gefitinib | 15.5 | 823.3 | -- |
| Compounds | Cellular Antiproliferative Activity (IC50, μM) b | |||||||
|---|---|---|---|---|---|---|---|---|
| H1975 | HCC827 | A431 | H1299 | LoVo | A549 | LO-2 | HBE | |
| DY3002 | 0.037 | 0.0104 | 0.382 | 4.12 | 2.46 | 4.45 | 4.24 | >40.0 |
| Rociletinib | 0.137 | 0.0181 | 1.29 | 20.1 | 20.1 | 6.50 | 8.57 | >40.0 |
| Osimertinib | 0.087 | 0.0102 | 0.915 | 7.49 | 4.85 | 7.28 | 4.56 | 8.39 |
| Gefitinib | 10.8 | 0.0106 | 3.30 | 10.8 | 13.2 | 29.4 | 14.5 | 23.8 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Song, Z.; Jin, Y.; Tang, Z.; Kang, J.; Ma, X. Novel Selective and Potent EGFR Inhibitor that Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Molecules 2016, 21, 1462. https://doi.org/10.3390/molecules21111462
Li Y, Song Z, Jin Y, Tang Z, Kang J, Ma X. Novel Selective and Potent EGFR Inhibitor that Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Molecules. 2016; 21(11):1462. https://doi.org/10.3390/molecules21111462
Chicago/Turabian StyleLi, Yanxia, Zhendong Song, Yue Jin, Zeyao Tang, Jian Kang, and Xiaodong Ma. 2016. "Novel Selective and Potent EGFR Inhibitor that Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer" Molecules 21, no. 11: 1462. https://doi.org/10.3390/molecules21111462
APA StyleLi, Y., Song, Z., Jin, Y., Tang, Z., Kang, J., & Ma, X. (2016). Novel Selective and Potent EGFR Inhibitor that Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Molecules, 21(11), 1462. https://doi.org/10.3390/molecules21111462