Identification of DNA-Binding Proteins Using Mixed Feature Representation Methods


Abstract
:1. Introduction
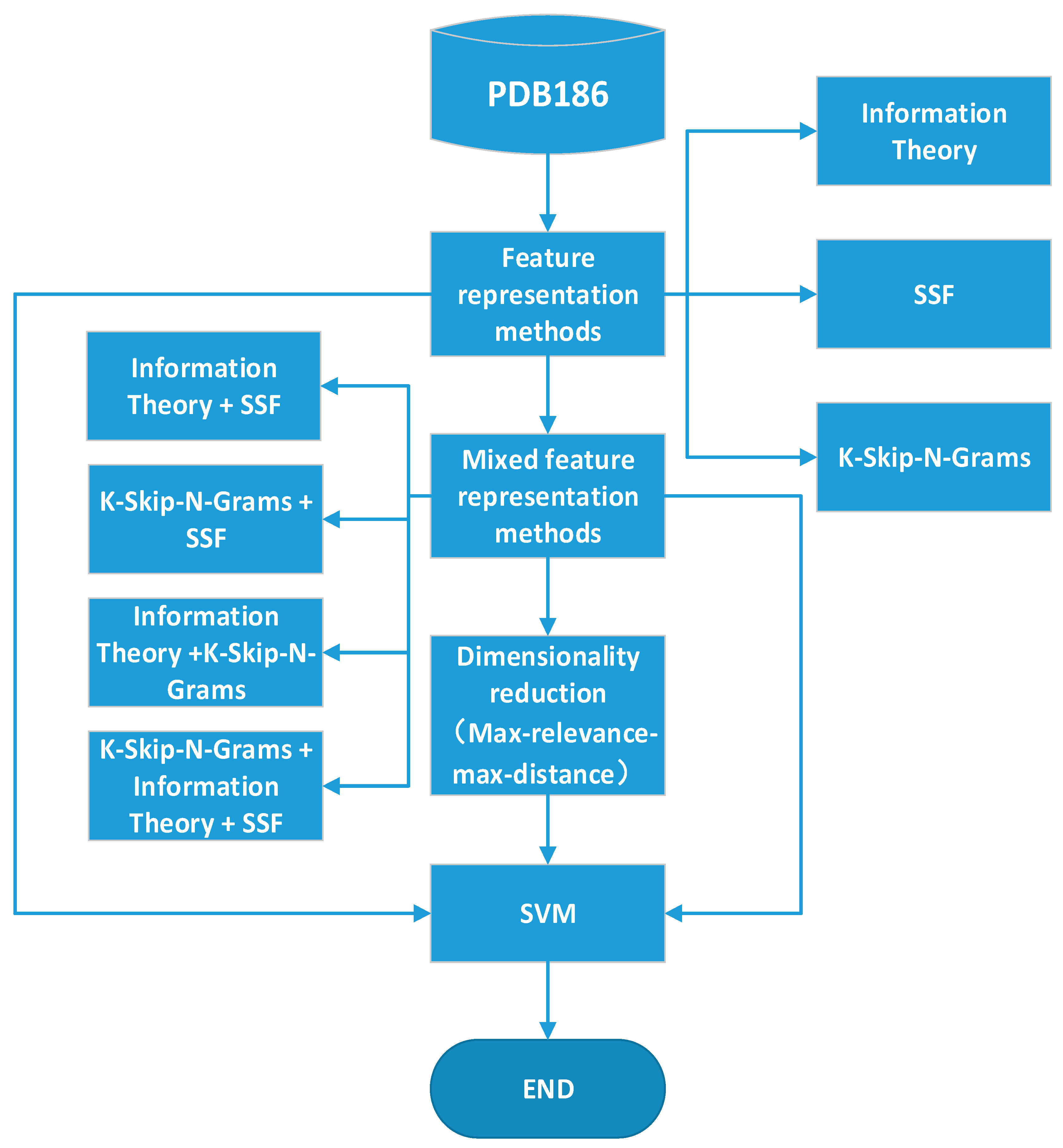
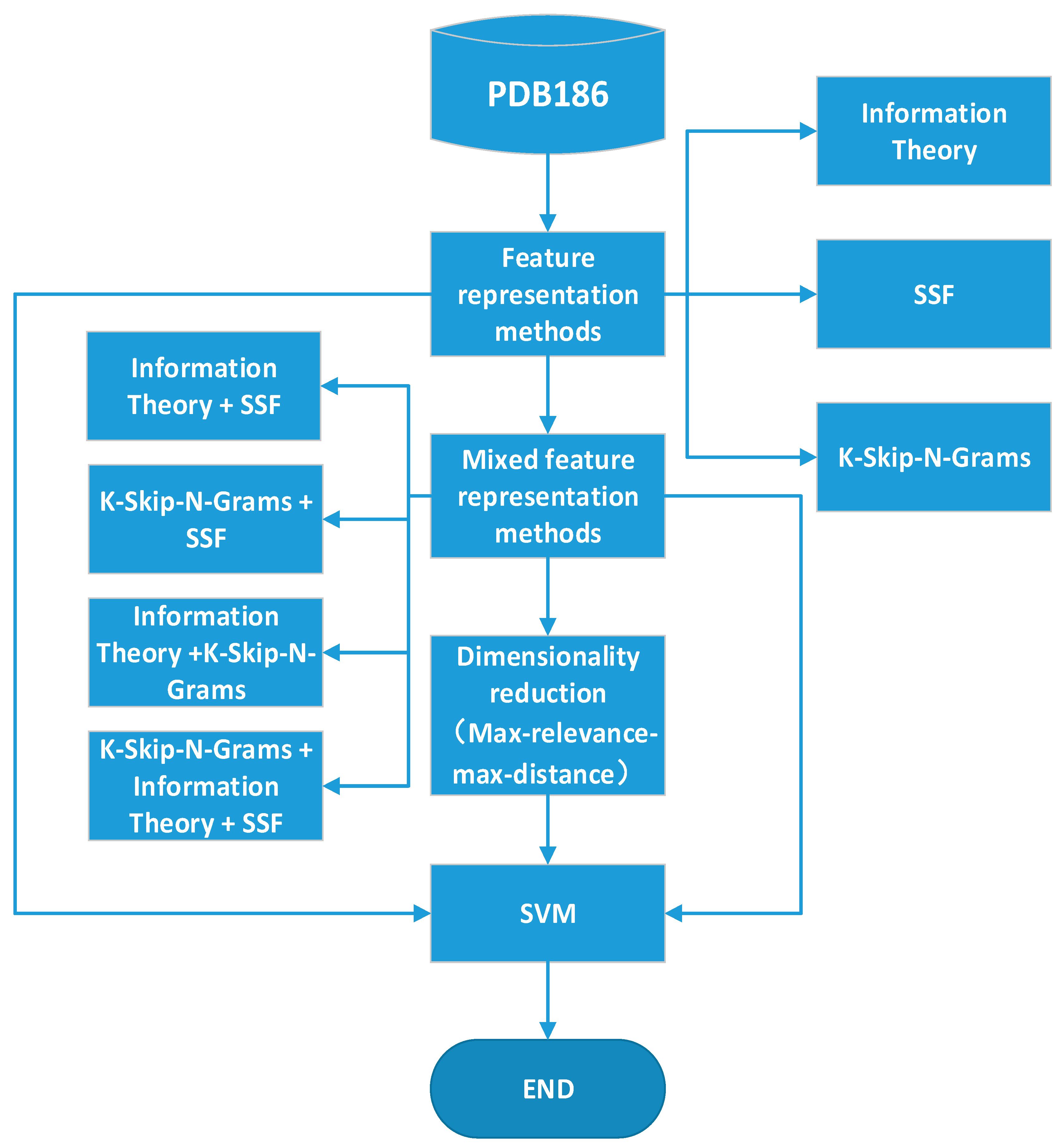
2. Methods
2.1. Dataset
2.2. Classifier
2.3. Single Feature Representation Methods
2.3.1. Information Theory (IT)
2.3.2. Sequential and structural features (SSF)
2.3.3. K-Skip-N-Grams (KSNG)
2.4. Mixed Feature Representation Methods and Feature Selection
3. Experiment
3.1. Measurement
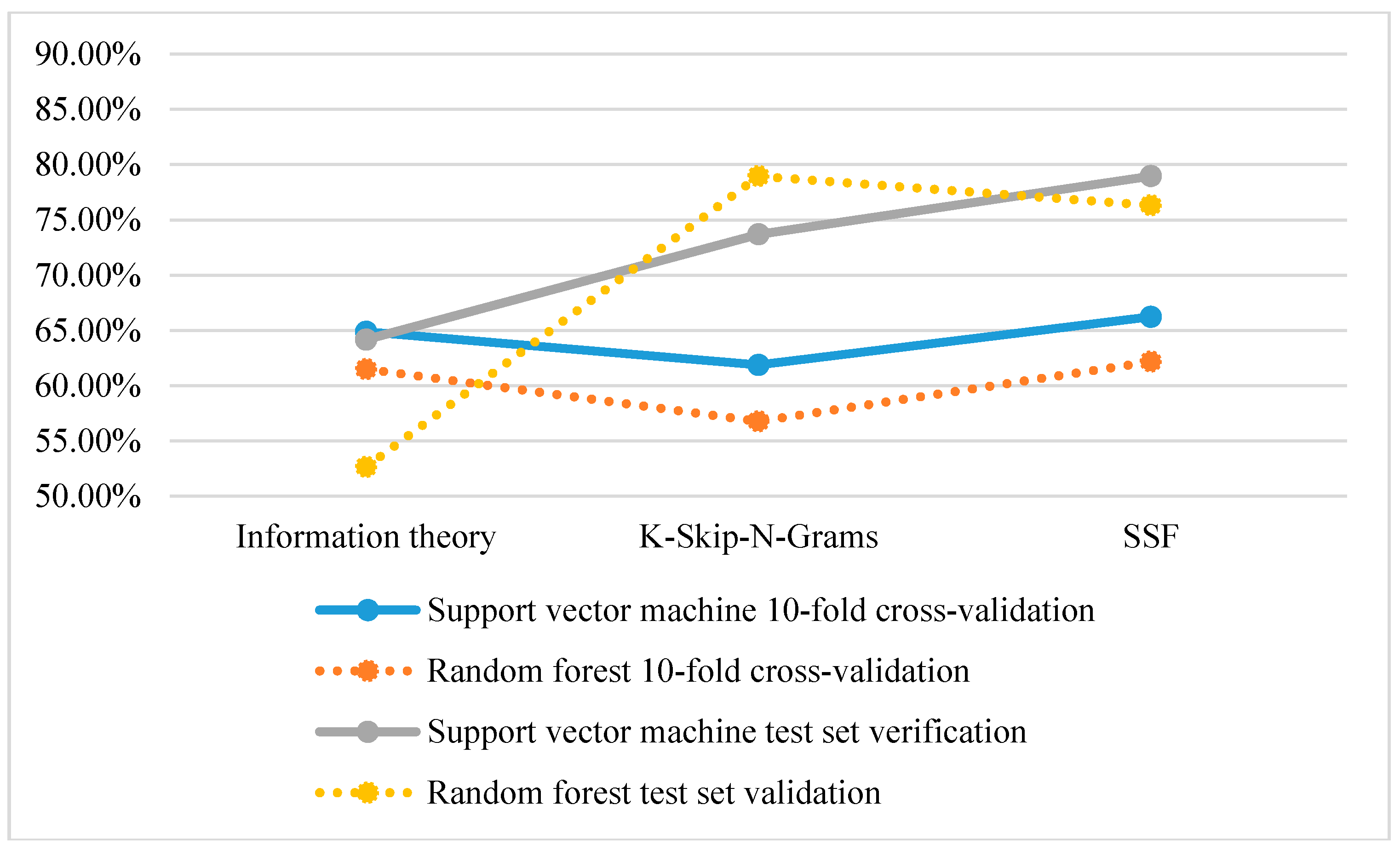
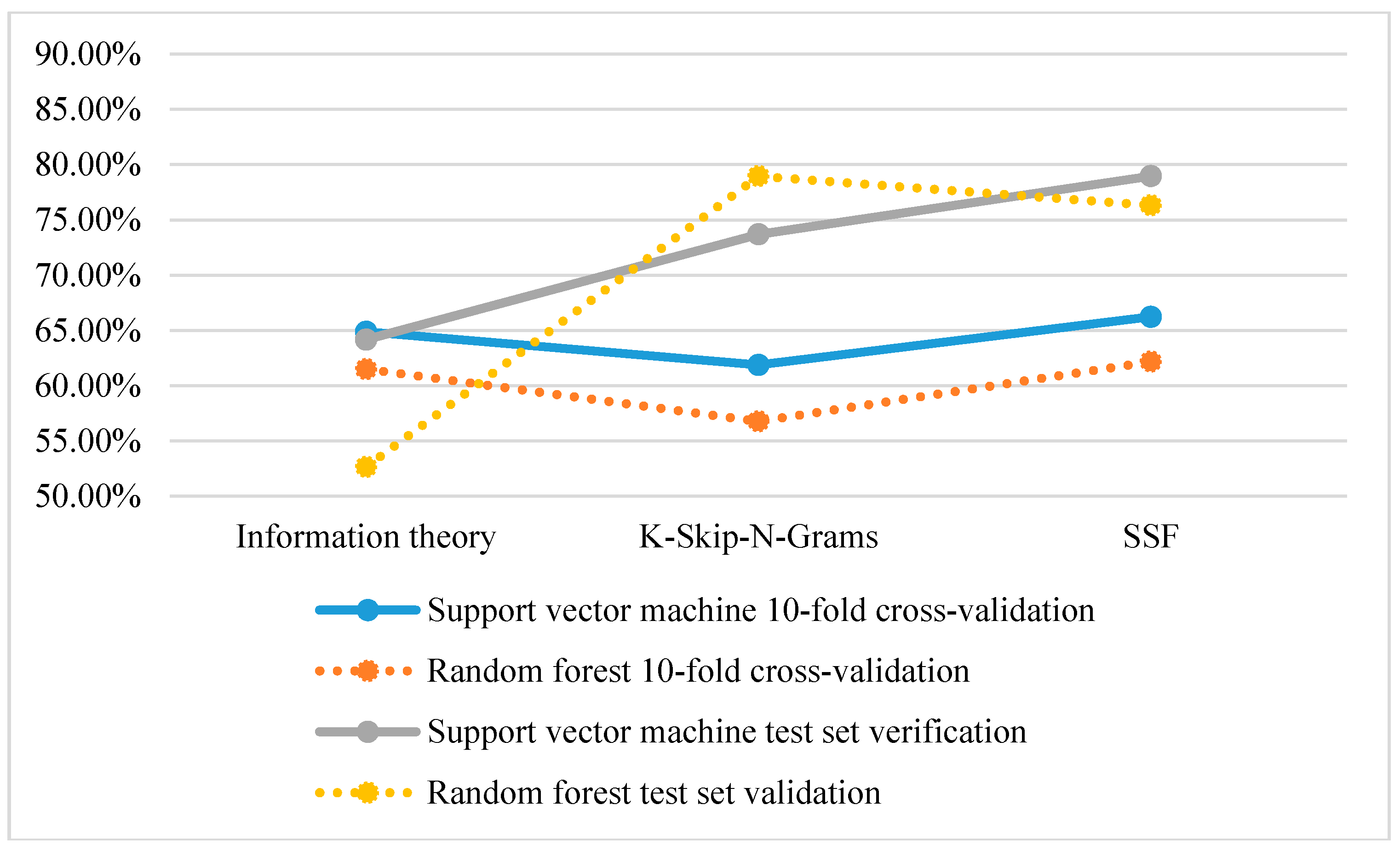
3.2. Performance of Different Features
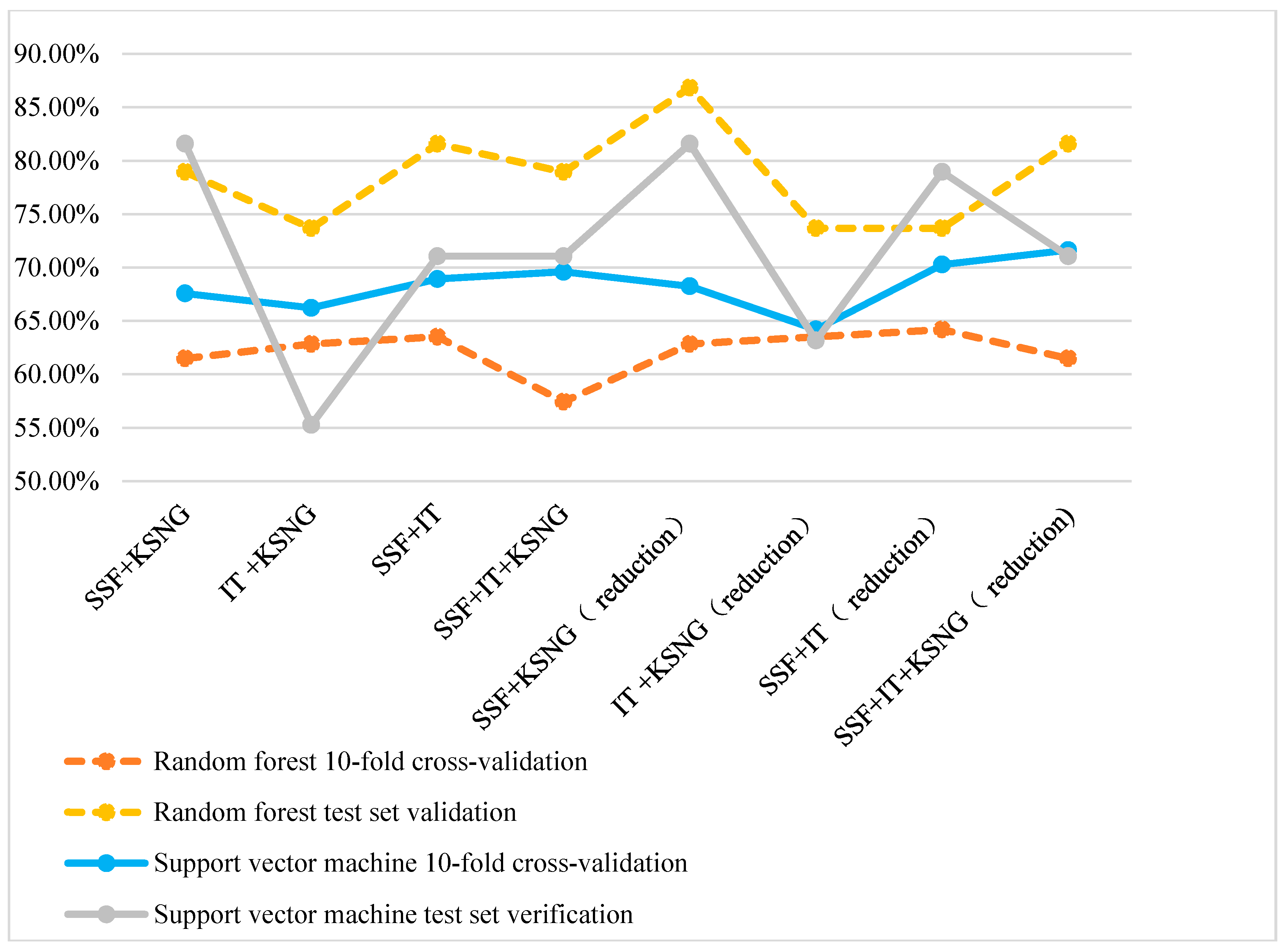
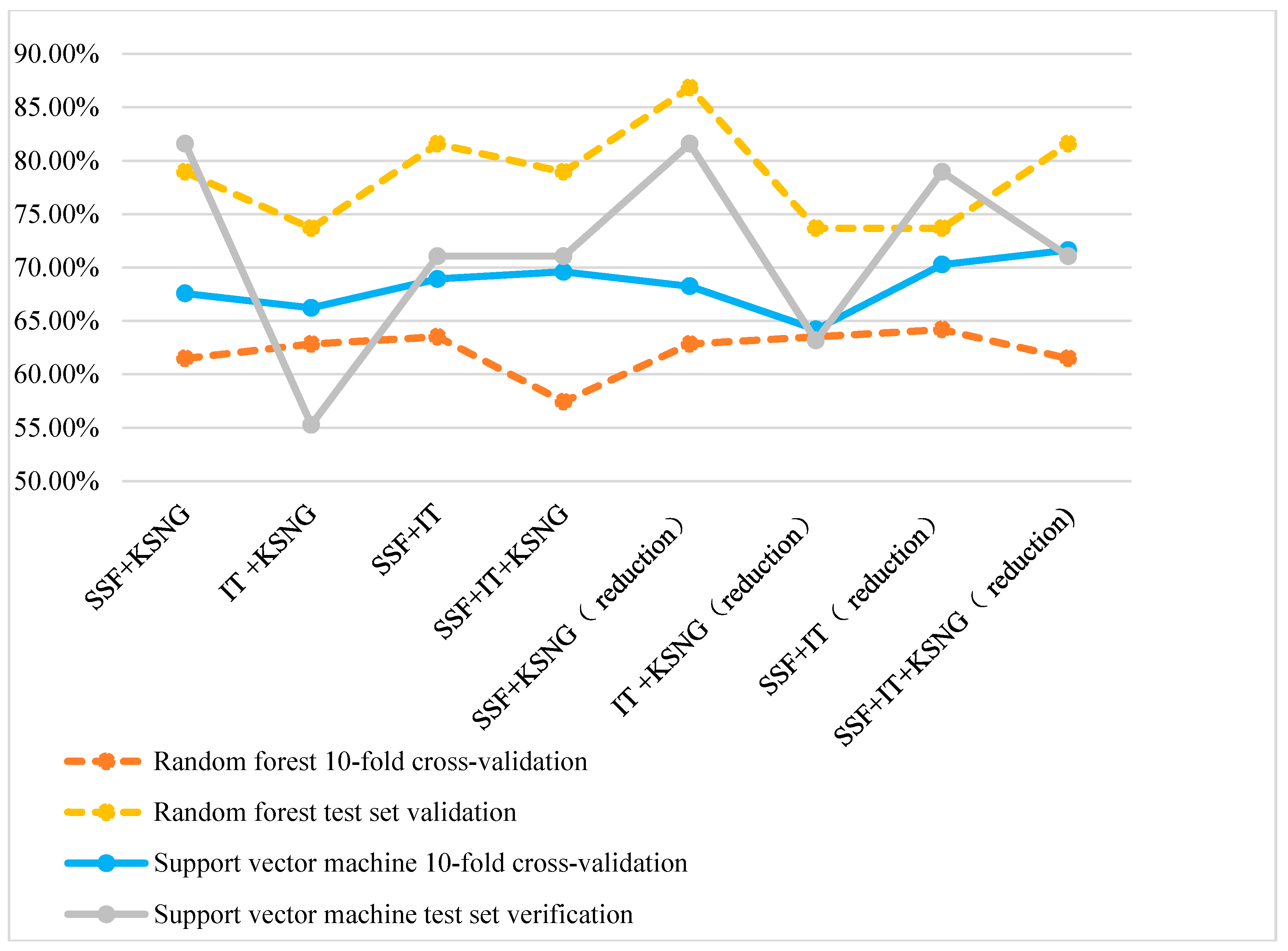
3.3. Performance of the Mixed Features
3.4. Comparison with State-of-the-Art Methods
3.5. Comparison with Other Classifiers
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gualberto, J.M.; Kühn, K. DNA-binding proteins in plant mitochondria: Implications for transcription. Mitochondrion 2014, 19, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Dong, Q.; Li, S.; Liu, X. Identification of DNA-binding proteins by combining auto-cross covariance transformation and ensemble learning. IEEE Trans. Nanobiosci. 2016, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, Y.; Zhou, Y. Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function. Bioinformatics 2010, 26, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Leng, F. Protein-induced DNA linking number change by sequence-specific DNA binding proteins and its biological effects. Biophys. Rev. 2016, 8 (Suppl. 1), 197–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Wang, X. DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation. Sci. Rep. 2015, 5, 15479. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lu, J.; Kang, T. Human single-stranded DNA binding proteins: Guardians of genome stability. Acta Biochim. Biophys. Sin. 2016, 48, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Broderick, S.; Rehmet, K.; Concannon, C.; Nasheuer, H.P. Eukaryotic Single-Stranded DNA Binding Proteins: Central Factors in Genome Stability; Springer: Dordrecht, The Netherlands, 2010; p. 143. [Google Scholar]
- Lou, W.; Wang, X.; Chen, F.; Chen, Y.; Jiang, B.; Zhang, H. Sequence based prediction of DNA-binding proteins based on hybrid feature selection using random forest and Gaussian naive Bayes. PLoS ONE 2014, 9, e86703. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, D.; Zeng, X.X.; Wu, Y.F.; Guo, L.; Zou, Q. nDNA-prot: Identification of DNA-binding Proteins Based on Unbalanced Classification. BMC Bioinform. 2014, 15, 298. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.H.; Lan, X.; Xu, R.F.; Zhou, J.Y.; Wang, X.L.; Chou, K.-C. iDNA-Prot|dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS ONE 2014, 9, e106691. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, A.; Skolnick, J. Efficient Prediction of Nucleic Acid Binding Function from Low-resolution Protein Structures. J. Mol. Biol. 2006, 358, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinform. 2007, 8, 463. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Z.; Fang, J.A.; Xiao, X.; Chou, K.C. iDNA-Prot: Identification of DNA binding proteins using random forest with grey model. PLoS ONE 2011, 6, e24756. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Gong, J.; Li, H. An improved sequence based prediction protocol for DNA-binding proteins using SVM and comprehensive feature analysis. BMC Bioinform. 2013, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, H.P.; Garcia, M.A.; Jones, S.; Thornton, J.M. Identifying DNA-binding proteins using structural motifs and the electrostatic potential. Nucleic Acids Res. 2004, 32, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Langlois, R.E.; Zhao, G.; Lu, H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005, 33, 6486–6493. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; He, J.; Li, X.; Lu, L.; Yang, X.; Feng, K.; Lu, W.; Kong, X. A Novel Computational Approach to Predict Transcription Factor DNA Binding Preference. J. Proteome Res. 2009, 8, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liang, Z.Y.; Tang, H.; Chen, W. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, H.; Chen, X.X.; Zhang, C.J.; Zhu, P.P.; Ding, H.; Chen, W.; Lin, H. Identification of Secretory Proteins in Mycobacterium tuberculosis Using Pseudo Amino Acid Composition. bioMed Res. Int. 2016, 2016, 5413903. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Mol. Biosyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Tang, H.; Li, W.-C.; Wu, H.; Chen, W.; Ding, H.; Lin, H. Identification of Bacterial Cell Wall Lyases via Pseudo Amino Acid Composition. BioMed Res. Int. 2016, 2016, 1654623. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Feng, P.M.; Chen, W.; Lin, H. Identification of bacteriophage virion proteins by the ANOVA feature selection and analysis. Mol. Biosyst. 2014, 10, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, Q.C.; Chen, Z.; Meng, Y.; Guan, J.; Zhou, S. PredHS: A web server for predicting protein—Protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014, 42, W290–W295. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Z.; Han, L.Y.; Ji, Z.L.; Chen, X.; Chen, Y.Z. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003, 31, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. Nanobiosci. 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liao, M.; Gao, X.; Zou, Q. Enhanced Protein Fold Prediction Method Through a Novel Feature Extraction Technique. IEEE Trans. Nanobiosci. 2015, 14, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Liu, D.W.; Huang, R.; Chen, Z.G.; Deng, L. PredRSA: A gradient boosted regression trees approach for predicting protein solvent accessibility. BioMed Central Ltd. BMC Bioinform. 2016, 17 (Suppl. 1), 8. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, D.; Deng, L. Accurate prediction of functional effects for variants by combining gradient tree boosting with optimal neighborhood properties. PLoS ONE 2017, 12, e0179314. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.C.; Cao, L.J.; Ji, R.R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou’s PseAAC and Physicochemical Distance Transformation. Mol. Inform. 2015, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.K.; Pugalenthi, G.; Suganthan, P.N. DNA-Prot: Identification of DNA Binding Proteins from Protein Sequence Information using Random Forest. J. Biomol. Struct. Dyn. 2009, 26, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The data and methods are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
| Method | Ten-Cross Validation Accuracy (%) | Test Set Validation Accuracy (%) | |||
|---|---|---|---|---|---|
| SN | SP | MCC | ACC | ||
| Information theory | 64.86 | 68.42 | 57.89 | 0.26 | 64.16 |
| K-Skip-N-Grams | 61.86 | 68.42 | 78.95 | 0.48 | 73.68 |
| SSF | 66.22 | 73.68 | 84.21 | 0.58 | 78.95 |
| Method | Non-Dimensionality-Reduction | Dimensionality-Reduction | ||
|---|---|---|---|---|
| Ten-Cross Validation Accuracy (%) | Test Set Validation Accuracy (%) | Ten-Cross Validation Accuracy (%) | Test Set Validation Accuracy (%) | |
| SSF + K-Skip-N-Grams | 67.57 | 81.58 | 68.24 | 81.58 |
| Information theory + K-Skip-N-Grams | 66.22 | 55.26 | 64.19 | 63.16 |
| SSF + Information theory | 68.92 | 71.05 | 70.27 | 78.95 |
| SSF + Informationtheory + K-Skip-N-Grams | 69.59 | 71.05 | 71.62 | 71.05 |
| Method | References | ACC (%) | MCC | SN (%) | SP (%) |
|---|---|---|---|---|---|
| SSF + Informationtheory + K-Skip-N-Grams (reduction) | This paper | 77.43 | 0.55 | 77.84 | 77.05 |
| SSF + Informationtheory + K-Skip-N-Grams | This paper | 75.19 | 0.51 | 76.88 | 73.59 |
| PseDNA-Pro | [31] | 76.55 | 0.53 | 79.61 | 73.63 |
| DNAbinder (P400) | [12] | 73.58 | 0.47 | 66.47 | 80.36 |
| DNAbinder (P21) | [12] | 73.95 | 0.48 | 68.57 | 79.09 |
| DNA-Prot | [32] | 72.55 | 0.44 | 82.67 | 59.76 |
| iDNA-Prot | [13] | 75.40 | 0.50 | 83.81 | 64.73 |
| Method | Ten-Cross Validation Accuracy (%) | Test Set Validation Accuracy (%) |
|---|---|---|
| Information theory | 61.49 | 52.63 |
| K-Skip-N-Grams | 56.76 | 78.95 |
| SSF | 62.16 | 76.32 |
| Method | Non-Dimensionality-Reduction | Dimensionality-Reduction | ||
|---|---|---|---|---|
| Ten-Cross Validation Accuracy (%) | Test Set Validation Accuracy (%) | Ten-Cross Validation Accuracy (%) | Test Set Validation Accuracy (%) | |
| SSF + K-Skip-N-Grams | 61.49 | 78.95 | 62.84 | 86.84 |
| Information theory + K-Skip-N-Grams | 62.84 | 73.68 | 63.51 | 73.68 |
| SSF + Informationtheory | 63.51 | 81.58 | 64.19 | 73.68 |
| SSF + Informationtheory + K-Skip-N-Grams | 57.43 | 78.95 | 61.49 | 81.58 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, K.; Han, K.; Wu, S.; Wang, G.; Wei, L. Identification of DNA-Binding Proteins Using Mixed Feature Representation Methods. Molecules 2017, 22, 1602. https://doi.org/10.3390/molecules22101602
Qu K, Han K, Wu S, Wang G, Wei L. Identification of DNA-Binding Proteins Using Mixed Feature Representation Methods. Molecules. 2017; 22(10):1602. https://doi.org/10.3390/molecules22101602
Chicago/Turabian StyleQu, Kaiyang, Ke Han, Song Wu, Guohua Wang, and Leyi Wei. 2017. "Identification of DNA-Binding Proteins Using Mixed Feature Representation Methods" Molecules 22, no. 10: 1602. https://doi.org/10.3390/molecules22101602
APA StyleQu, K., Han, K., Wu, S., Wang, G., & Wei, L. (2017). Identification of DNA-Binding Proteins Using Mixed Feature Representation Methods. Molecules, 22(10), 1602. https://doi.org/10.3390/molecules22101602