Comparison of Dilution, Filtration, and Microwave Digestion Sample Pretreatments in Elemental Profiling of Wine by ICP-MS


Abstract
:1. Introduction
2. Results and Discussion
2.1. Detection Limits and Method Blanks
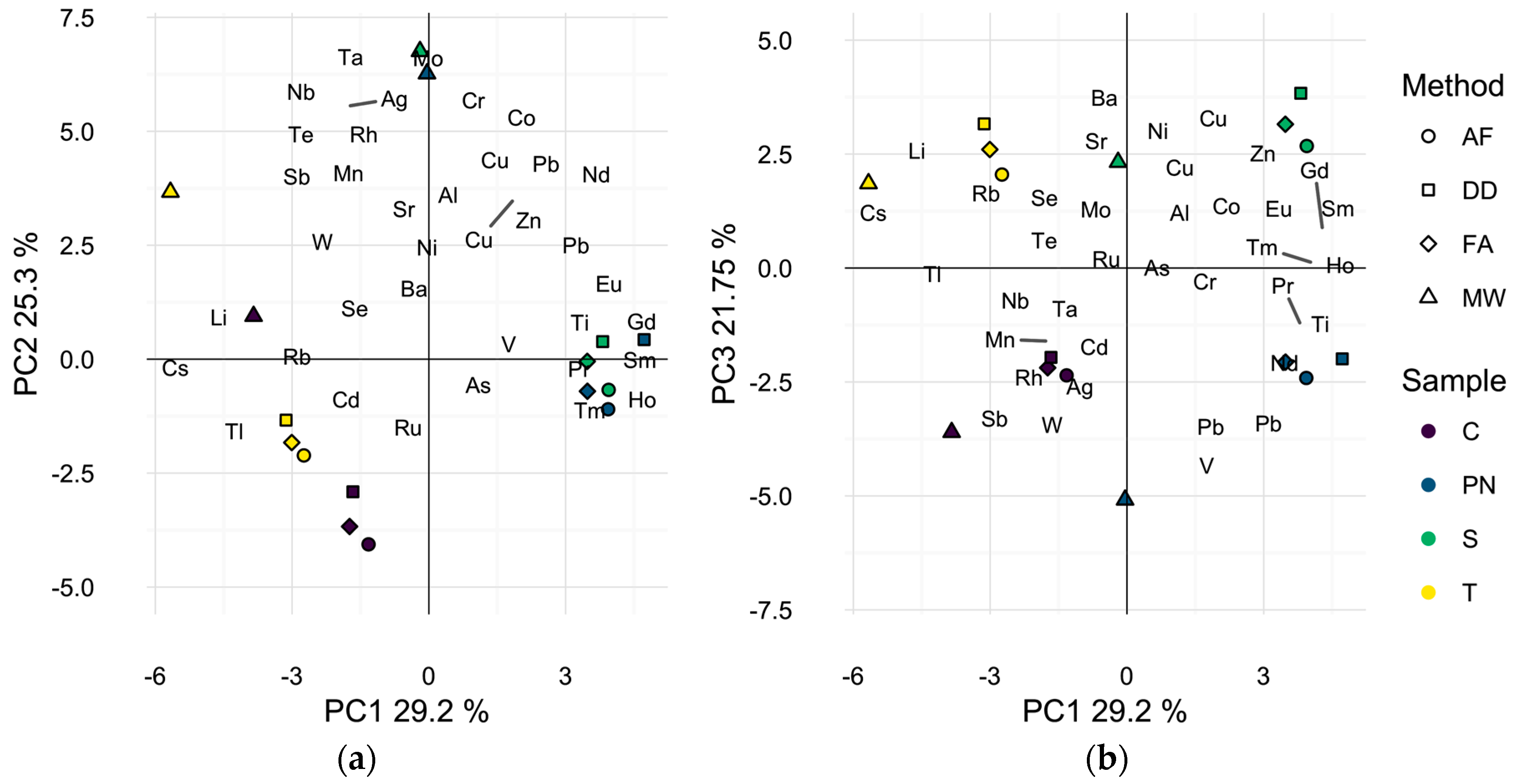
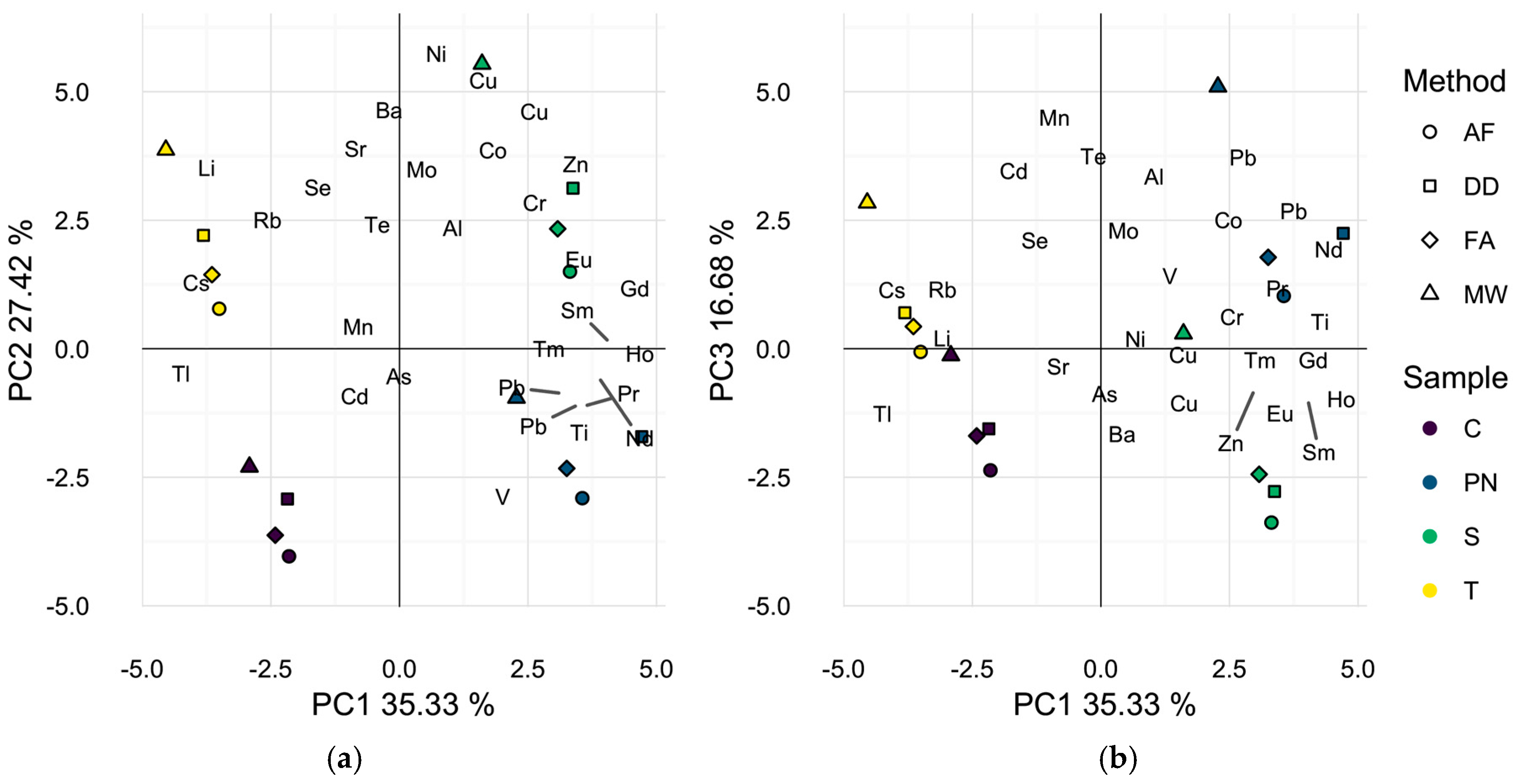
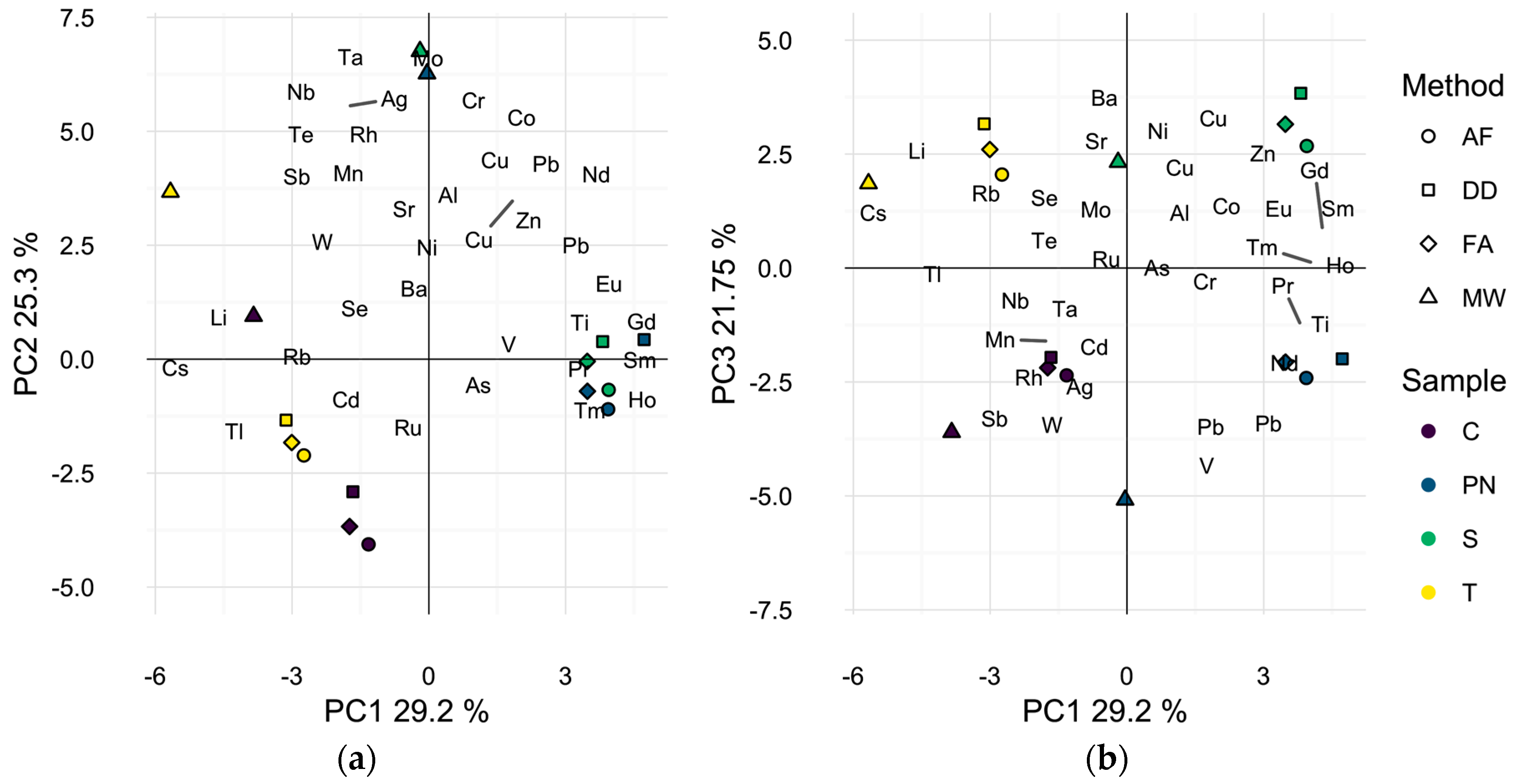
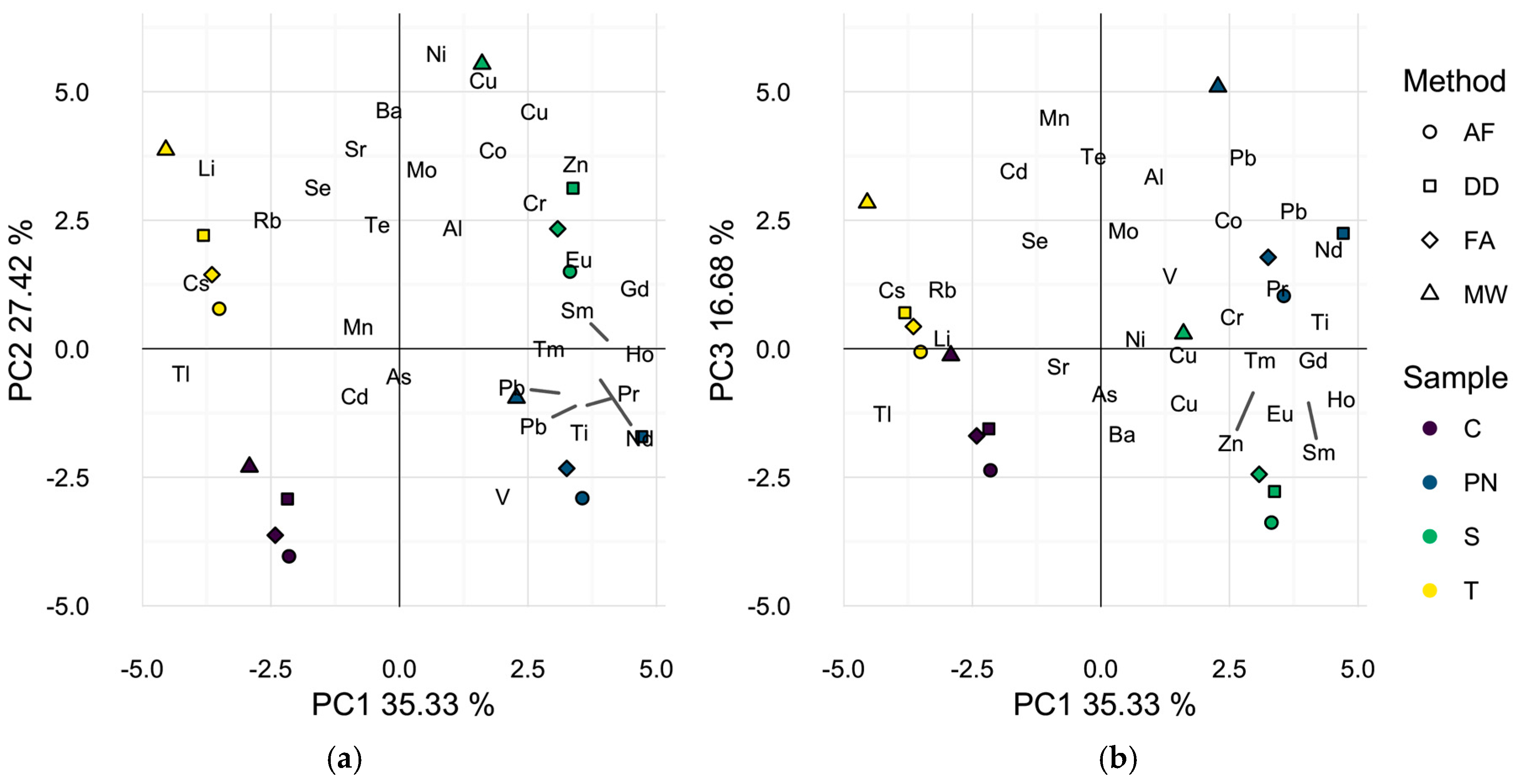
2.2. Profiling of Wines
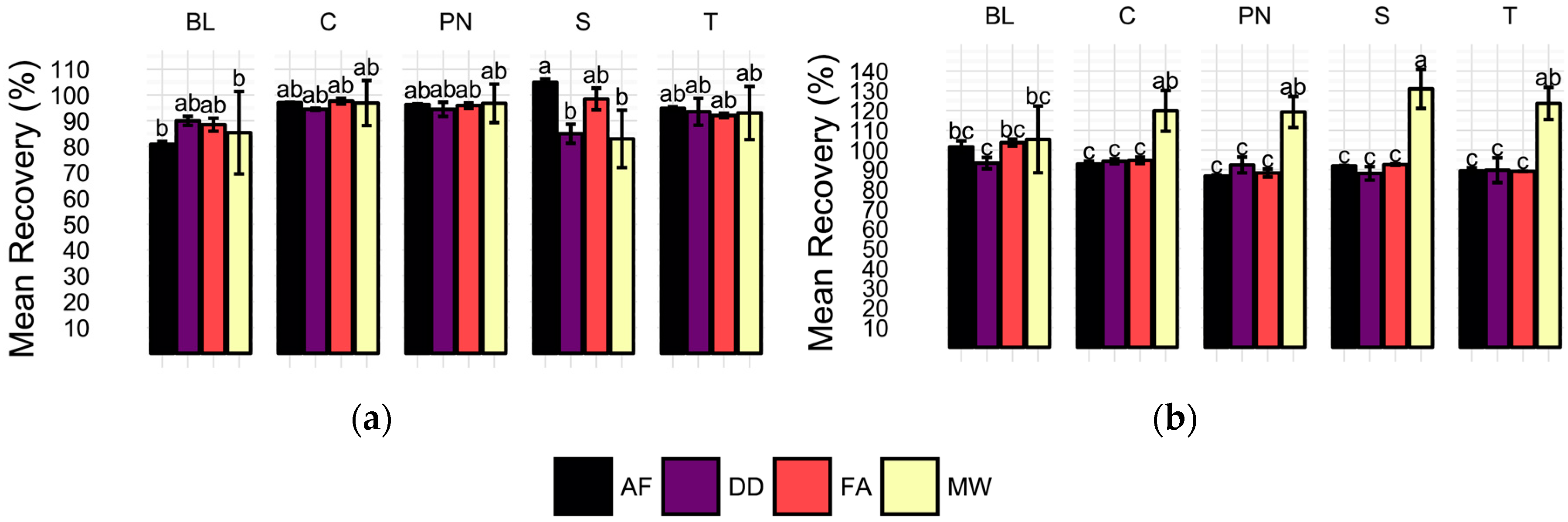
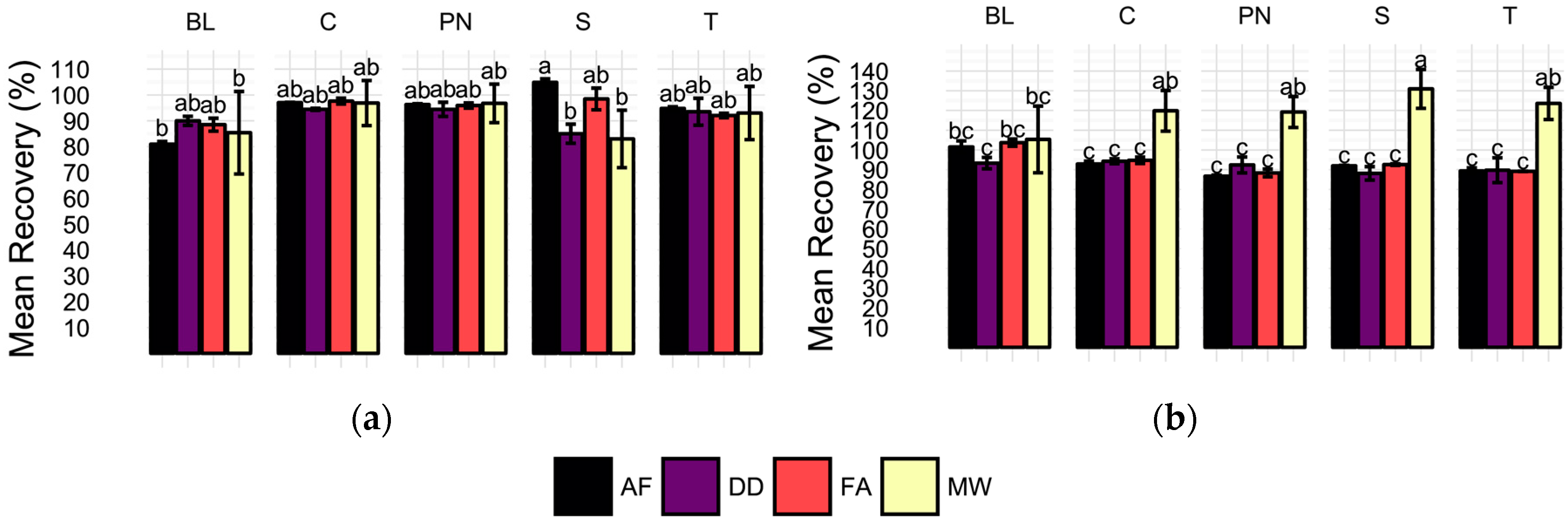
2.3. Spike Recovery
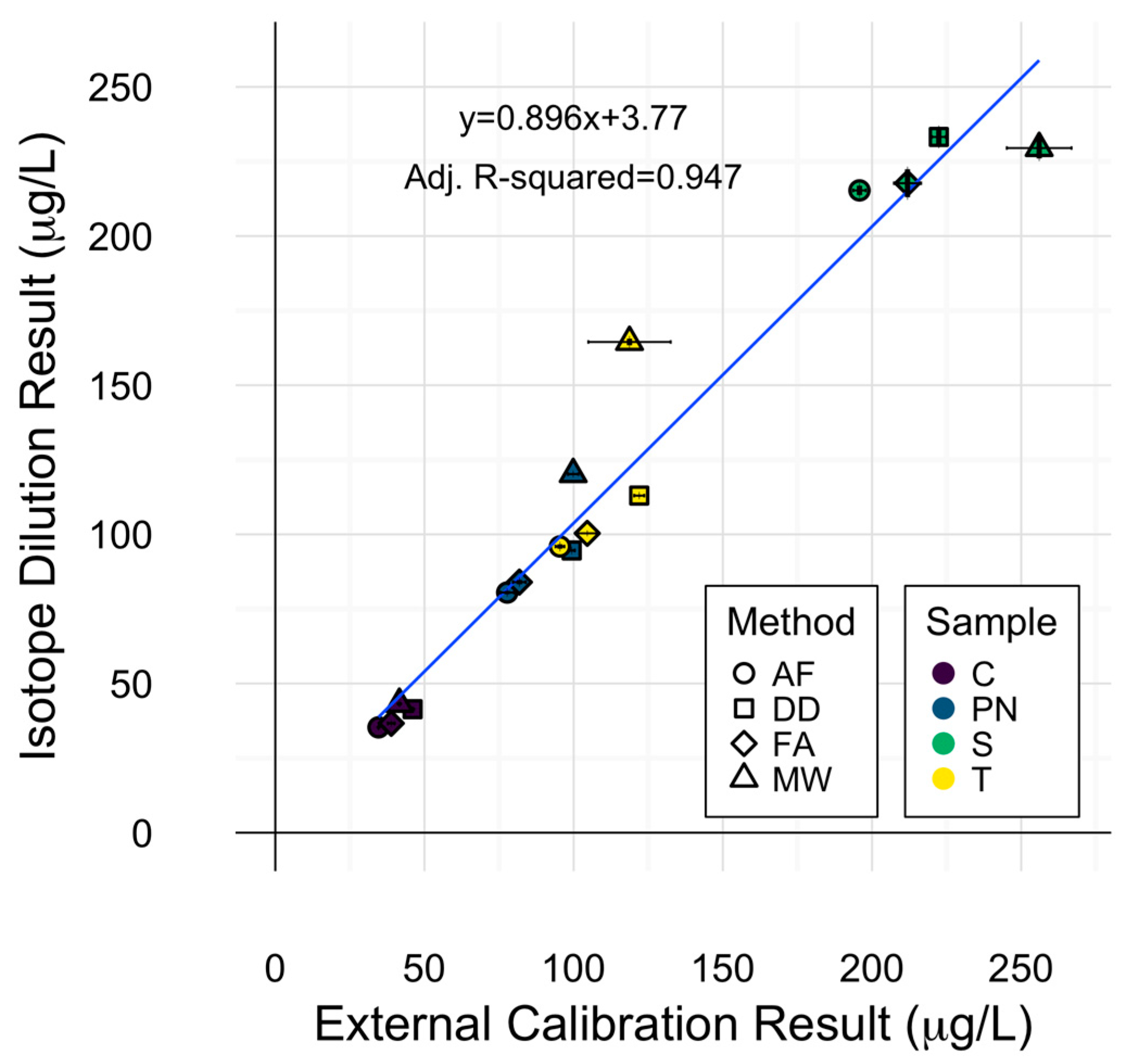
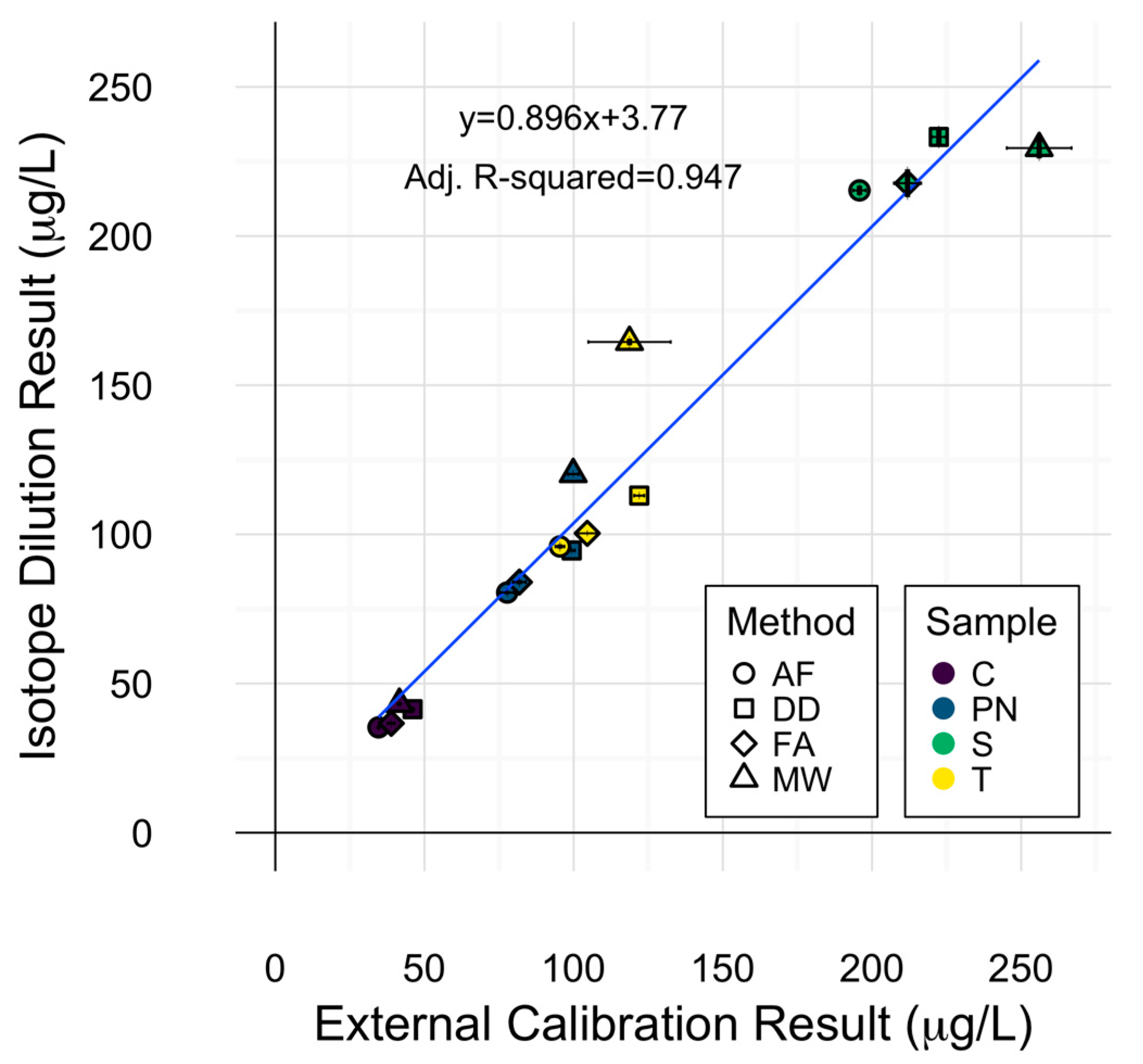
2.4. Cu Isotope Dilution
3. Conclusions
4. Materials and Methods
4.1. Reagents and Samples
4.2. Sample Preparation
- Direct Dilution (DD): samples were diluted 1:3 with 5% HNO3 (v/v) prior to analysis. Method blanks contained 4% ethanol and 5% HNO3 (v/v).
- Filtration prior to acidification (FA): A 7 mL aliquot of sample was filtered through an Agilent Captiva Premium syringe filter (PTFE, 15 mm, 0.45 µm; Wilmington, DE, USA), then an aliquot of each filtered sample was diluted 1:3 with 5% HNO3 (v/v) prior to analysis. Method blank was the same as for DD.
- Acidification prior to filtration (AF): A 15 mL aliquot of diluted sample (1:3 with 5% HNO3 (v/v)) was filtered through an Agilent Captiva Premium syringe filter (PTFE, 15 mm, 0.45 µm) prior to analysis. Method blank was the same as for DD.
- Microwave-assisted acid digestion (MW): 2 × 1 mL concentrated HNO3 was added to 2 mL of sample in PTFE microwave tubes that were capped and digested using a Milestone UltraWAVE microwave system (Sorisole, Italy) according to manufacturer’s recommendations (20 min ramp to 240 °C and 150 bar, 15 min hold; E = 1500 W). Two milliliter aliquots of digested samples were then diluted 1:5 with ultrapure water prior to analysis. PTFE microwave vessels were cleaned using a cleaning program (15 min ramp to 230 °C and 150 bar, 5 min hold; E = 1500 W) with concentrated nitric acid according to manufacturer’s recommendations, subsequently rinsed (5% HNO3 and ultrapure water) and allowed to dry between digestions.
4.3. ICP-MS Analysis
4.3.1. External Calibration
4.3.2. Isotope Dilution
4.4. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isotope | AF | DD | FA | MW |
|---|---|---|---|---|
| 7Li | 12.3 ± 0.209 b | 18.8 ± 0.483 a | 14.3 ± 0.614 b | 20 ± 1.25 a |
| 27Al | 247 ± 6.97 c | 281 ± 9.13 b | 267 ± 12.3 b | 325 ± 13.2 a |
| 47Ti | 6.04 ± 0.577 ab | 5.02 ± 0.736 ab | 6.58 ± 0.621 a | 4.7 ± 2.04 b |
| 51V | 1.16 ± 0.033 b | 1.33 ± 0.061 a | 1.26 ± 0.05 ab | 1.36 ± 0.12 a |
| 52Cr | 14.0 ± 0.33 b | 16.1 ± 0.308 ab | 15.1 ± 0.564 ab | 20.8 ± 6.15 a |
| 55Mn | 1100 ± 24.4 d | 1190 ± 27.7 b | 1150 ± 26.3 c | 1310 ± 41.2 a |
| 59Co | 4.26 ± 0.074 c | 5.23 ± 0.103 b | 4.5 ± 0.103 c | 5.63 ± 0.139 a |
| 60Ni | 30.0 ± 0.44 c | 36.4 ± 0.356 a | 32.8 ± 1.06 b | 36.9 ± 1.99 a |
| 63Cu | 101 ± 1.5 b | 122 ± 1.36 a | 109 ± 2.74 b | 129 ± 6.65 a |
| 65Cu | 31.9 ± 0.47 b | 35.5 ± 0.46 b | 35.1 ± 0.979 b | 44.0 ± 2.28 a |
| 66Zn | 634 ± 8.68 c | 682 ± 8.25 b | 686 ± 17 b | 717 ± 25.1 a |
| 71Ga | 0.109 ± 0.023 | 0.107 ± 0.025 | 0.113 ± 0.03 | 0.125 ± 0.125 |
| 75As | 0.954 ± 0.032 c | 1.65 ± 0.525 a | 1.25 ± 0.095 b | 1.03 ± 0.108 bc |
| 78Se | 1.42 ± 0.066 b | 2.05 ± 0.457 a | 1.99 ± 0.557 a | 1.85 ± 0.533 ab |
| 85Rb | 1740 ± 28.3 b | 1970 ± 49.8 a | 1690 ± 11.9 b | 1950 ± 46.7 a |
| 88Sr | 578 ± 7.13 b | 784 ± 13.4 a | 576 ± 6.95 b | 779 ± 19.9 a |
| 93Nb | <LOD b | <LOD b | <LOD b | 0.346 ± 0.098 a |
| 98Mo | 1.52 ± 0.052 b | 1.28 ± 0.044 b | 1.64 ± 0.057 b | 3.18 ± 0.524 a |
| 101Ru | <LOD b | <LOD b | 0.012 ± 0.005 a 1 | <LOD b |
| 103Rh | <LOD b | 0.004 ± 0.003 b 1 | <LOD b | 0.019 ± 0.009 a |
| 107Ag | <LOD b | <LOD b | 0.016 ± 0.002 b 1 | 0.091 ± 0.054 a |
| 111Cd | 0.189 ± 0.01 ab | 0.183 ± 0.006 ab | 0.198 ± 0.01 a | 0.177 ± 0.017 b |
| 123Sb | 0.188 ± 0.015 b | 0.11 ± 0.013 b 1 | 0.202 ± 0.012 b | 0.6 ± 0.296 a |
| 125Te | 0.018 ± 0.003 c | 0.037 ± 0.009 b | 0.033 ± 0.016 bc | 0.061 ± 0.031 a |
| 133Cs | 3.36 ± 0.075 c | 4.01 ± 0.094 b | 3.37 ± 0.112 c | 4.42 ± 0.14 a |
| 137Ba | 167 ± 1.94 b | 155 ± 2.19 c | 170 ± 2.86 b | 177 ± 5.15 a |
| 140Ce | 0.047 ± 0.002 | 0.047 ± 0.003 | 0.049 ± 0.002 | 0.047 ± 0.008 |
| 141Pr | 0.004 ± 0.001 b | 0.008 ± 0.001 a | 0.004 ± 0.001 b | <LOD b |
| 146Nd | 0.034 ± 0.006 b | 0.029 ± 0.005 b | 0.034 ± 0.005 b | 0.048 ± 0.018 a |
| 147Sm | 0.011 ± 0.003 a | 0.011 ± 0.006 a | 0.01 ± 0.004 a | 0.003 ± 0.003 b 1 |
| 153Eu | 0.003 ± 0.002 ab | 0.004 ± 0.001 a | 0.003 ± 0.001 b | <LOD b |
| 157Gd | 0.013 ± 0.003 a | 0.016 ± 0.004 a | 0.012 ± 0.003 a | 0.006 ± 0.004 b 1 |
| 163Dy | 0.024 ± 0.005 | 0.019 ± 0.004 | 0.023 ± 0.003 | 0.018 ± 0.008 |
| 165Ho | 0.006 ± 0.001 a | 0.006 ± 0.001 a | 0.006 ± 0.001 a | <LOD b |
| 166Er | 0.027 ± 0.003 | 0.024 ± 0.003 | 0.024 ± 0.002 | 0.022 ± 0.006 |
| 169Tm | 0.004 ± 0.001 b | 0.007 ± 0.001 a | 0.003 ± 0.001 b | <LOD c |
| 172Yb | 0.049 ± 0.008 | 0.045 ± 0.005 | 0.047 ± 0.007 | 0.044 ± 0.008 |
| 181Ta | <LOD b | 0.18 ± 0.166 b | 0.046 ± 0.035 b 1 | 1.06 ± 0.429 a |
| 182W | 0.257 ± 0.041 c 1 | 0.509 ± 0.114 b | 0.298 ± 0.033 c 1 | 0.831 ± 0.436 a |
| 205Tl | 0.143 ± 0.004 b | 0.189 ± 0.005 a | 0.15 ± 0.006 b | 0.182 ± 0.016 a |
| 206Pb | 0.612 ± 0.019 c | 0.738 ± 0.021 b | 0.538 ± 0.013 c | 0.963 ± 0.051 a |
| 208Pb | 2.36 ± 0.06 b | 2.89 ± 0.06 a | 2.26 ± 0.032 b | 3.13 ± 0.155 a |
| 238U | 0.025 ± 0.002 | 0.029 ± 0.003 | 0.026 ± 0.001 | 0.031 ± 0.004 |
References
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological profile for Copper. Available online: http://www.atsdr.cdc.gov/toxprofiles/TP.asp?id=206&tid=37 (accessed on 23 March 2017).
- Castiñeira, M.D.M.; Brandt, R.; Jakubowski, N.; Andersson, J.T. Changes of the metal composition in German white wines through the winemaking process. A study of 63 elements by inductively coupled plasma−mass spectrometry. J. Agric. Food Chem. 2004, 52, 2953–2961. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.M.R.; Vasconcelos, M.T.S.D. Multielement composition of wines and their precursors including provenance soil and their potentialities as fingerprints of wine origin. J. Agric. Food Chem. 2003, 51, 4788–4798. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, M.; Besalú, E.; Anticó, E. Internal standardization−atomic spectrometry and geographical pattern recognition techniques for the multielement analysis and classification of Catalonian red wines. J. Agric. Food Chem. 2007, 55, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Rossano, E.C.; Szilágyi, Z.; Malorni, A.; Pocsfalvi, G. Influence of winemaking practices on the concentration of rare earth elements in white wines studied by inductively coupled plasma mass spectrometry. J. Agric. Food Chem. 2007, 55, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Hopfer, H.; Nelson, J.; Collins, T.S.; Heymann, H.; Ebeler, S.E. The combined impact of vineyard origin and processing winery on the elemental profile of red wines. Food Chem. 2015, 172, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Gonzálvez, A.; Armenta, S.; Pastor, A.; de la Guardia, M. Searching the most appropriate sample pretreatment for the elemental analysis of wines by inductively coupled plasma-based techniques. J. Agric. Food Chem. 2008, 56, 4943–4954. [Google Scholar] [CrossRef] [PubMed]
- Grindlay, G.; Mora, J.; Maestre, S.; Gras, L. Application of a microwave-based desolvation system for multi-elemental analysis of wine by inductively coupled plasma based techniques. Anal. Chim. Acta 2008, 629, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Grindlay, G.; Mora, J.; Gras, L.; de Loos-Vollebregt, M.T.C. Atomic spectrometry methods for wine analysis: A critical evaluation and discussion of recent applications. Anal. Chim. Acta 2011, 691, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Dams, R.F.J.; Goossens, J.; Moens, L. Spectral and non-spectral interferences in inductively coupled plasma mass-spectrometry. Microchim. Acta 1995, 119, 277–286. [Google Scholar] [CrossRef]
- Martin, A.E.; Watling, R.J.; Lee, G.S. The multi-element determination and regional discrimination of Australian wines. Food Chem. 2012, 133, 1081–1089. [Google Scholar] [CrossRef]
- Hopfer, H.; Nelson, J.; Mitchell, A.E.; Heymann, H.; Ebeler, S.E. Profiling the trace metal composition of wine as a function of storage temperature and packaging type. J. Anal. At. Spectrom. 2013, 28, 1288–1291. [Google Scholar] [CrossRef]
- International Organisation of Vine and Wine (OIV). Compendium of International Methods of Wine and Must Analysis. Available online: http://www.oiv.int/public/medias/4231/compendium-2016-en-vol1.pdf (accessed on 23 March 2017).
- Goossens, J.; De Smaele, T.; Moens, L.; Dams, R. Accurate determination of lead in wines by inductively coupled plasma mass spectrometry. Fresenius J. Anal. Chem. 1993, 347, 119–125. [Google Scholar] [CrossRef]
- Grindlay, G.; Mora, J.; de Loos-Vollebregt, M.T.C.; Vanhaecke, F. Evaluation of the multi-element capabilities of collision/reaction cell inductively coupled plasma–mass spectrometry in wine analysis. Talanta 2014, 128, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulton, R. The copigmentation of anthocyanins and its role in the color of red wine: A critical review. Am. J. Enol. Vitic. 2001, 52, 67–87. [Google Scholar]
- Vasconcelos, M.T.; Azenha, M.; de Freitas, V. Role of polyphenols in copper complexation in red wines. J. Agric. Food Chem. 1999, 47, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Pohl, P. What do metals tell us about wine? Trends Anal. Chem. 2007, 26, 941–949. [Google Scholar] [CrossRef]
- Materials Authorized for the Treatment of Wine and Juice, 27 CFR § 24.246. Available online: https://www.gpo.gov/fdsys/granule/CFR-2012-title27-vol1/CFR-2012-title27-vol1-sec24–246/content-detail.html (accessed on 23 March 2017).
- Clark, A.C.; Grant-Preece, P.; Cleghorn, N.; Scollary, G.R. Copper(II) addition to white wines containing hydrogen sulfide: Residual copper concentration and activity. Aust. J. Grape Wine Res. 2015, 21, 30–39. [Google Scholar] [CrossRef]
- McKinnon, A.J.; Scollary, G.R. Size fractionation of metals in wine using ultrafiltration. Talanta 1997, 44, 1649–1658. [Google Scholar] [CrossRef]
- Almeida, C.M.; Vasconcelos, M.T.S.D. ICP-MS determination of strontium isotope ratio in wine in order to be used as a fingerprint of its regional origin. J. Anal. At. Spectrom. 2001, 16, 607–611. [Google Scholar] [CrossRef]
- Coetzee, P.P.; Vanhaecke, F. Classifying wine according to geographical origin via quadrupole-based ICP–mass spectrometry measurements of boron isotope ratios. Anal. Bioanal. Chem. 2005, 383, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Augagneur, S.; Medina, B.; Grousset, F. Measurement of lead isotope ratios in wine by ICP-MS and its applications to the determination of lead concentration by isotope dilution. Fresenius J. Anal. Chem. 1997, 357, 1149–1152. [Google Scholar] [CrossRef]
- Dean, J.R.; Ebdon, L.; Massey, R.C. Isotope ratio and isotope dilution analysis of lead in wine by inductively coupled plasma-mass spectrometry. Food Addit. Contamin. 1990, 7, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Quetel, C.R.; Nelms, S.M.; Van Nevel, L.; Papadakis, I.; Taylor, P.D.P. Certification of the lead mass fraction in wine for comparison 16 of the International Measurement Evaluation Programme. J. Anal. At. Spectrom. 2001, 16, 1091–1100. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Otero, M.; Alves, A.A.; Coimbra, J.; Coimbra, M.A.; Pereira, E.; Duarte, A.C. Elemental analysis for categorization of wines and authentication of their certified brand of origin. J. Food Comp. Anal. 2011, 24, 548–562. [Google Scholar] [CrossRef]
- Thomsen, V.; Schatzlein, D.; Mercuro, D. Limits of detection in spectroscopy. Spectroscopy 2003, 18, 112–114. [Google Scholar]
- Meija, J.; Coplen, T.B.; Berglund, M.; Brand, W.A.; De Bièvre, P.; Gröning, M.; Holden, N.E.; Irrgeher, J.; Loss, R.D.; Walczyk, T.; et al. Isotopic compositions of the elements 2013 (IUPAC Technical Report). Pure Appl. Chem. 2016, 88, 293–306. [Google Scholar] [CrossRef]
- Croghan, C.; Egeghy, P.P. Methods of Dealing with Values Below the Limit of Detection Using SAS. Southeastern SAS User Group. Available online: http://analytics.ncsu.edu/sesug/2003/SD08-Croghan.pdf (accessed on 23 March 2017).
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Mendiburu, F. Agricolae: Statistical Procedures for Agricultural Research. Available online: http://cran.r-project.org/web/packages/agricolae/index.html (accessed on 23 March 2017).
- Wickham, H. ggplot2: Elegant Graphic for Data Analysis. Available online: http://ggplot2.org/ (accessed on 23 March 2017).
Sample Availability: Samples of the compounds are not available. |
| Isotope | Mode 1 | LOD 2 | Method Blank Concentration [µg/L] | ||||
|---|---|---|---|---|---|---|---|
| AF-DD-FA | MW | AF | DD | FA | MW | ||
| 7Li | NG | 0.103 | 0.029 | 1.23 | 1.77 | 1.50 | 0.962 |
| 27Al | He | 0.802 | 0.988 | <LOD | 1.84 | <LOD | 13.7 |
| 47Ti | He | 0.236 | 0.174 | 0.611 | <LOD | <LOD | 2.27 |
| 51V | He | 0.010 | 0.008 | 0.013 | 0.020 | 0.015 | 0.029 |
| 52Cr | He | 0.220 | 0.049 | <LOD | 1.04 | <LOD | 5.07 |
| 55Mn | He | 0.068 | 0.041 | <LOD | 0.772 | <LOD | 20.1 |
| 59Co | He | 0.005 | 0.003 | <LOD | 0.026 | <LOD | 1.36 |
| 60Ni | He | 0.086 | 0.042 | <LOD | 0.600 | <LOD | 5.22 |
| 63Cu | He | 0.049 | 0.042 | <LOD | <LOD | <LOD | 0.248 |
| 65Cu | He | 0.018 | 0.010 | <LOD | <LOD | <LOD | 0.045 |
| 66Zn | He | 0.280 | 0.184 | <LOD | <LOD | <LOD | 1.16 |
| 71Ga | He | 0.005 | 0.003 | 0.007 | 0.006 | 0.010 | <LOD |
| 75As | HEHe | 0.013 | 0.011 | 0.056 | 0.154 | 0.116 | 0.106 |
| 78Se | HEHe | 0.080 | 0.018 | <LOD | <LOD | <LOD | 0.280 |
| 85Rb | He | 0.043 | 0.065 | 0.467 | 0.558 | 0.279 | 0.130 |
| 88Sr | He | 0.021 | 0.020 | 0.065 | <LOD | <LOD | 0.050 |
| 93Nb | He | 0.007 | 0.007 | <LOD | 0.018 | <LOD | 0.524 |
| 98Mo | NG | 0.075 | 0.017 | <LOD | <LOD | 0.099 | 1.96 |
| 101Ru | He | 0.008 | 0.009 | 0.009 | <LOD | <LOD | <LOD |
| 103Rh | He | 0.002 | 0.001 | <LOD | <LOD | <LOD | 0.034 |
| 107Ag | He | 0.015 | 0.002 | <LOD | <LOD | <LOD | 0.138 |
| 111Cd | He | 0.007 | 0.009 | 0.040 | <LOD | 0.040 | <LOD |
| 123Sb | He | 0.043 | 0.012 | <LOD | 0.060 | <LOD | 0.732 |
| 125Te | NG | 0.003 | 0.003 | <LOD | 0.007 | 0.004 | 0.005 |
| 133Cs | He | 0.018 | 0.014 | 0.029 | 0.033 | 0.074 | 0.033 |
| 137Ba | He | 0.041 | 0.041 | <LOD | <LOD | <LOD | 0.124 |
| 140Ce | He | 0.002 | 0.004 | 0.003 | <LOD | <LOD | <LOD |
| 141Pr | He | 0.0005 | 0.003 | <LOD | 0.003 | <LOD | <LOD |
| 146Nd | He | 0.002 | 0.003 | 0.005 | 0.002 | 0.002 | 0.003 |
| 147Sm | He | 0.002 | 0.002 | 0.002 | 0.004 | <LOD | <LOD |
| 153Eu | He | 0.0005 | 0.003 | <LOD | 0.001 | <LOD | <LOD |
| 157Gd | He | 0.001 | 0.004 | <LOD | 0.004 | <LOD | <LOD |
| 163Dy | He | 0.001 | 0.005 | <LOD | <LOD | <LOD | <LOD |
| 165Ho | He | 0.0004 | 0.003 | 0.001 | 0.0004 | <LOD | <LOD |
| 166Er | He | 0.001 | 0.003 | <LOD | 0.003 | <LOD | <LOD |
| 169Tm | He | 0.001 | 0.002 | <LOD | 0.001 | <LOD | <LOD |
| 172Yb | He | 0.001 | 0.003 | 0.008 | 0.004 | 0.005 | 0.005 |
| 181Ta | He | 0.022 | 0.033 | <LOD | 0.032 | <LOD | 1.94 |
| 182W | He | 0.117 | 0.013 | <LOD | 0.189 | 0.122 | 1.27 |
| 205Tl | He | 0.002 | 0.003 | 0.005 | 0.007 | 0.008 | <LOD |
| 206Pb | He | 0.009 | 0.003 | <LOD | 0.140 | <LOD | 0.030 |
| 208Pb | He | 0.007 | 0.004 | 0.017 | 0.130 | <LOD | 0.043 |
| 238U | He | 0.004 | 0.001 | 0.009 | <LOD | 0.009 | <LOD |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godshaw, J.; Hopfer, H.; Nelson, J.; Ebeler, S.E. Comparison of Dilution, Filtration, and Microwave Digestion Sample Pretreatments in Elemental Profiling of Wine by ICP-MS. Molecules 2017, 22, 1609. https://doi.org/10.3390/molecules22101609
Godshaw J, Hopfer H, Nelson J, Ebeler SE. Comparison of Dilution, Filtration, and Microwave Digestion Sample Pretreatments in Elemental Profiling of Wine by ICP-MS. Molecules. 2017; 22(10):1609. https://doi.org/10.3390/molecules22101609
Chicago/Turabian StyleGodshaw, Joshua, Helene Hopfer, Jenny Nelson, and Susan E. Ebeler. 2017. "Comparison of Dilution, Filtration, and Microwave Digestion Sample Pretreatments in Elemental Profiling of Wine by ICP-MS" Molecules 22, no. 10: 1609. https://doi.org/10.3390/molecules22101609
APA StyleGodshaw, J., Hopfer, H., Nelson, J., & Ebeler, S. E. (2017). Comparison of Dilution, Filtration, and Microwave Digestion Sample Pretreatments in Elemental Profiling of Wine by ICP-MS. Molecules, 22(10), 1609. https://doi.org/10.3390/molecules22101609