Characterization of the Ornithine Hydroxylation Step in Albachelin Biosynthesis


Abstract
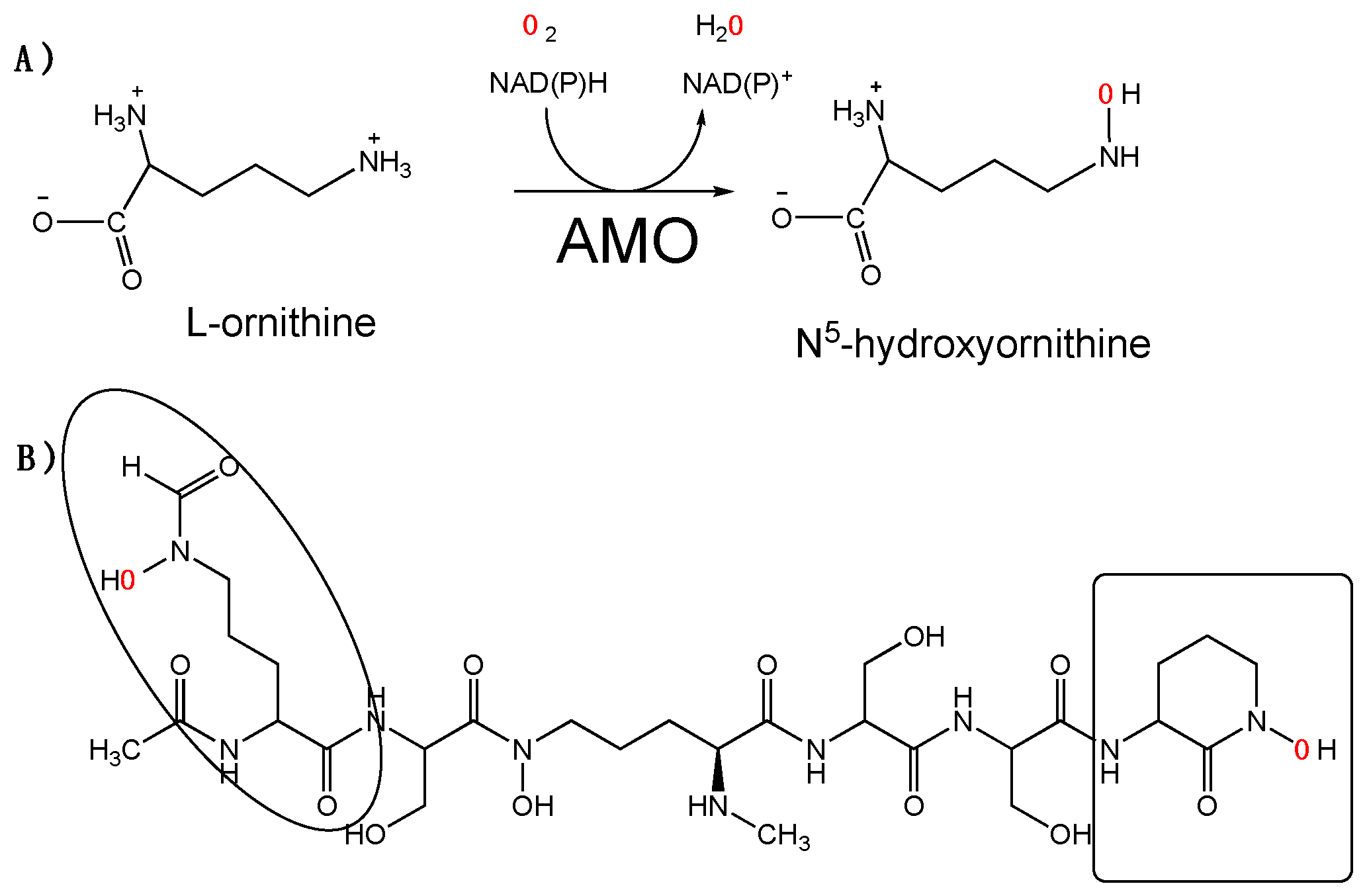
:1. Introduction
2. Results
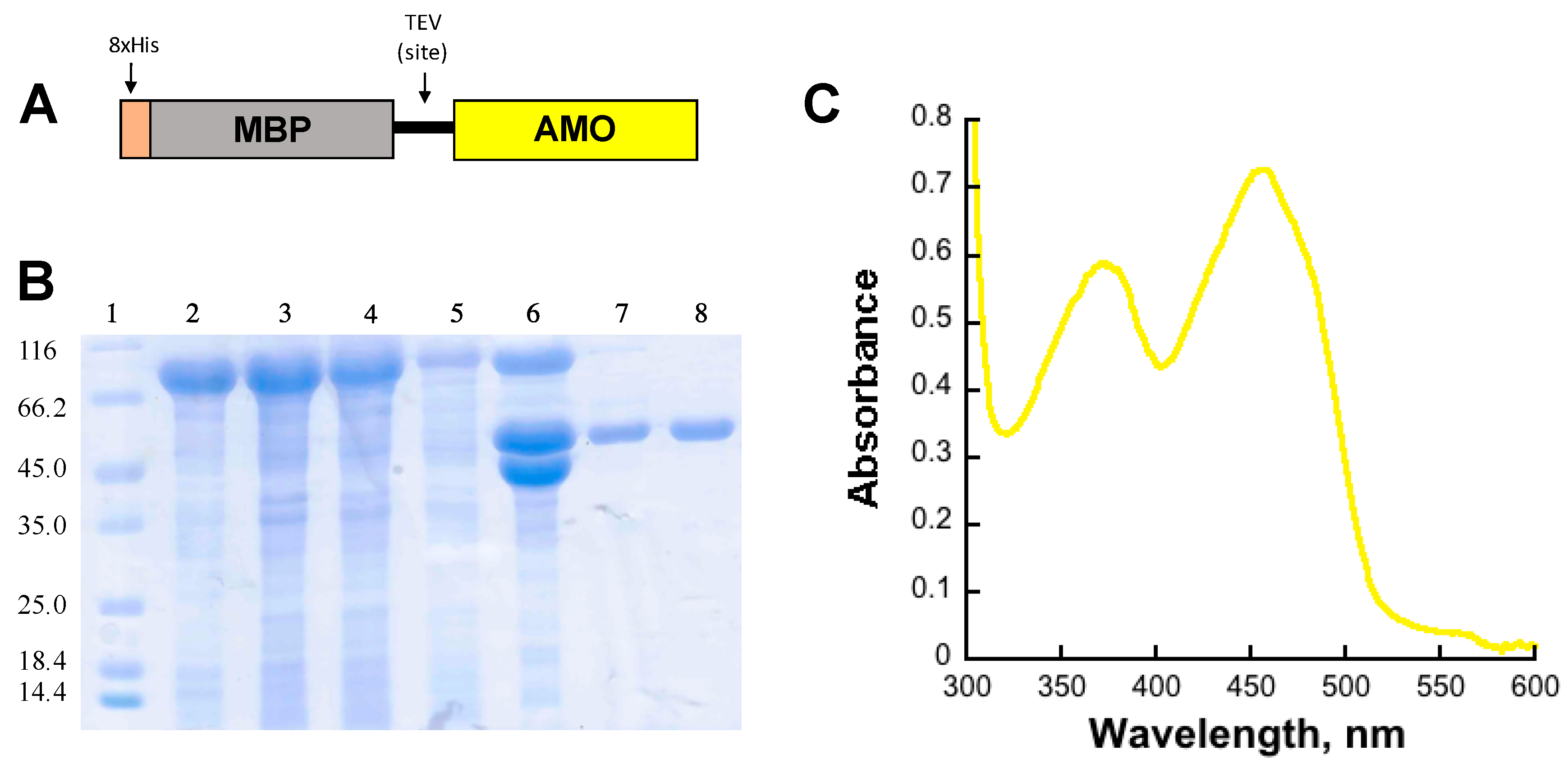
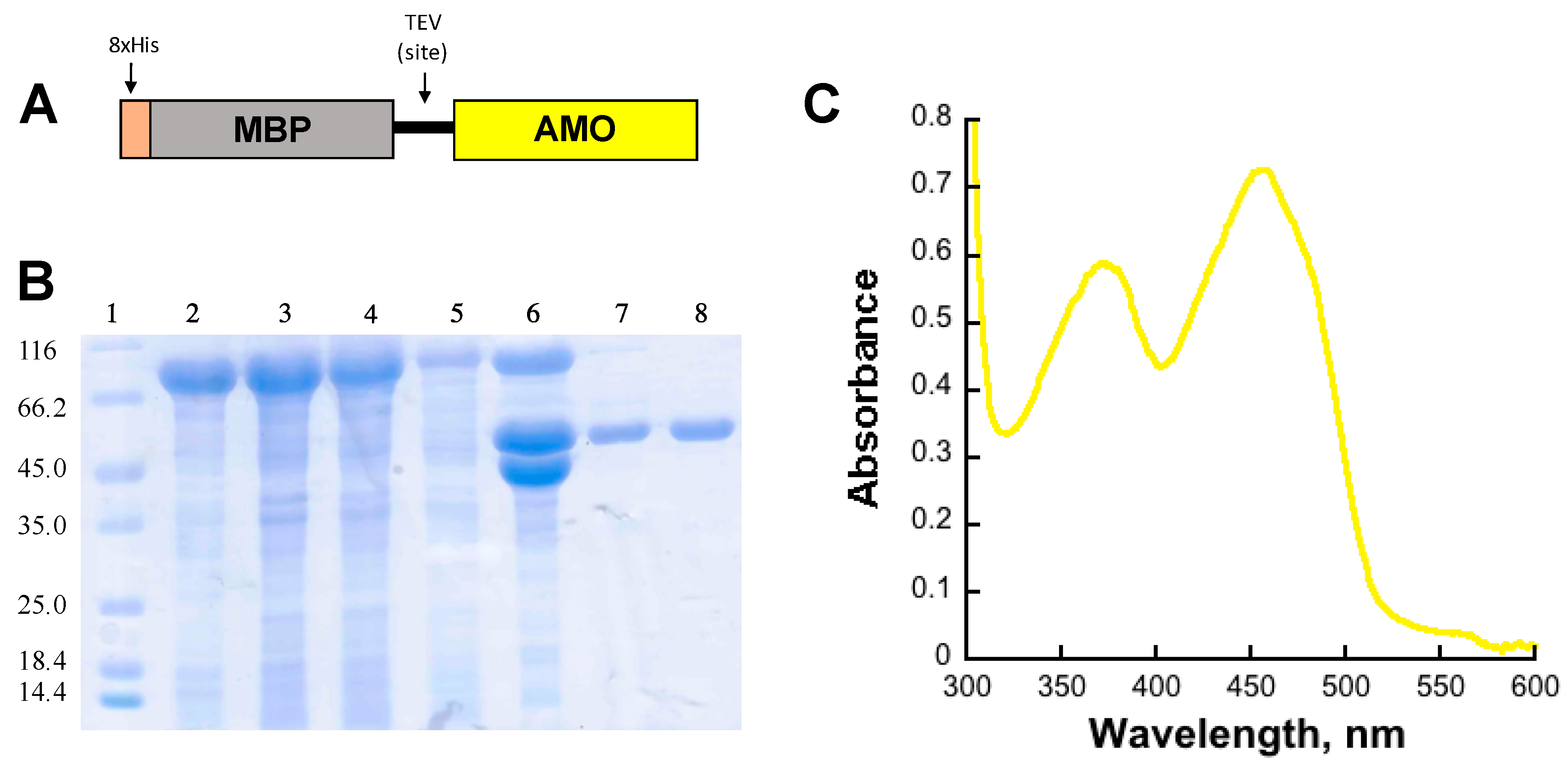
2.1. Expression and Purification of AMO
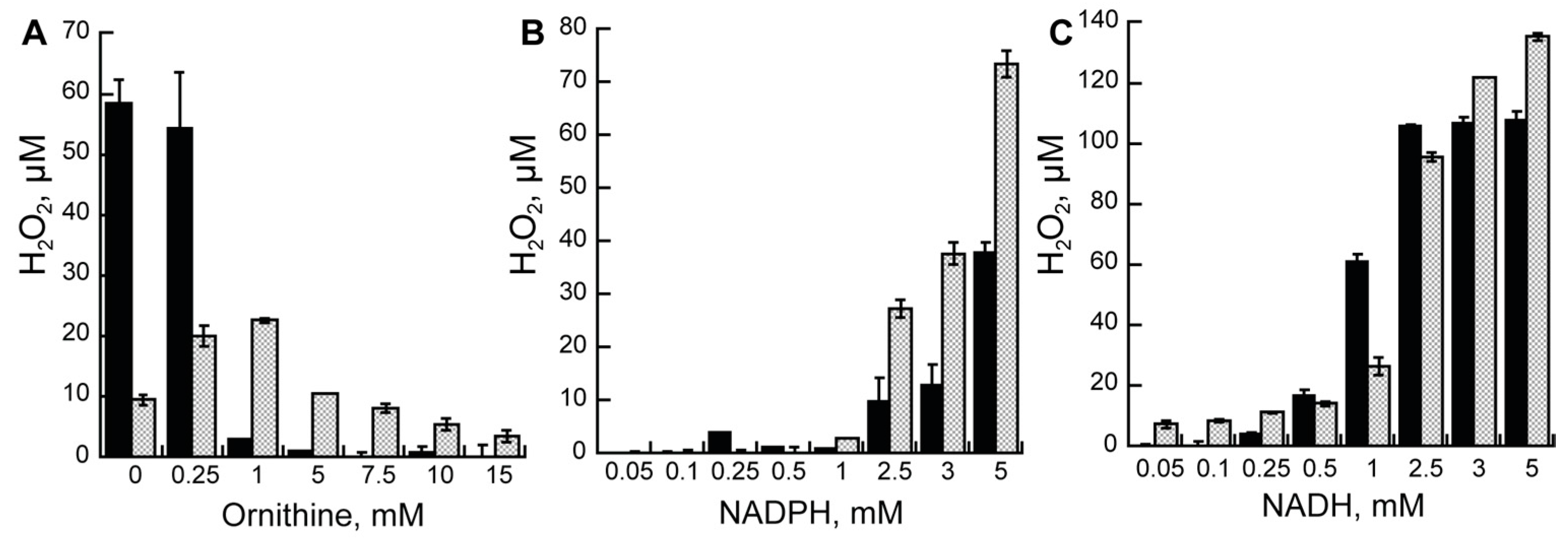
2.2. Steady-State Kinetics Varying Ornithine
2.3. Steady-State Kinetics Varying Reduced Nicotinamide Concentrations
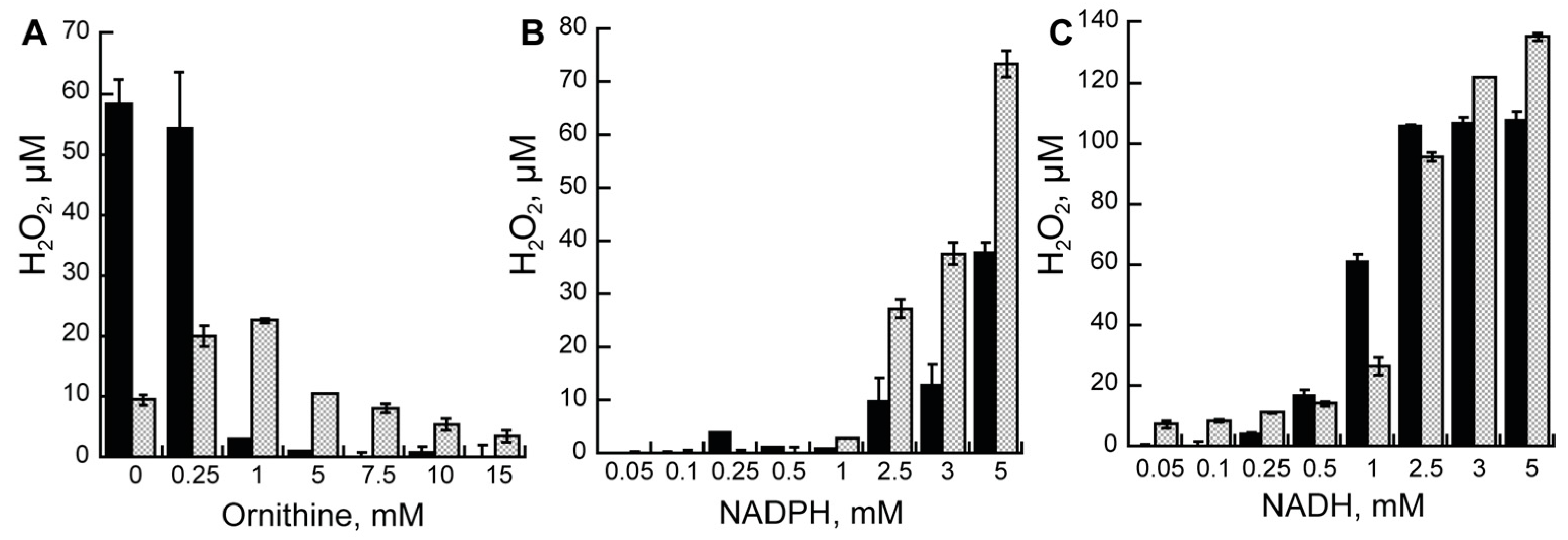
2.4. Hydrogen Peroxide Formation
3. Discussion
4. Materials and Methods
AMO Expression and Purification
5. Determination of the Extinction Coefficient of Bound Flavin to Holo-AMO
6. Oxygen Consumption Assay
7. Product Formation Assay: Determination of N-hydroxylation
8. Detection of Hydrogen Peroxide Formation
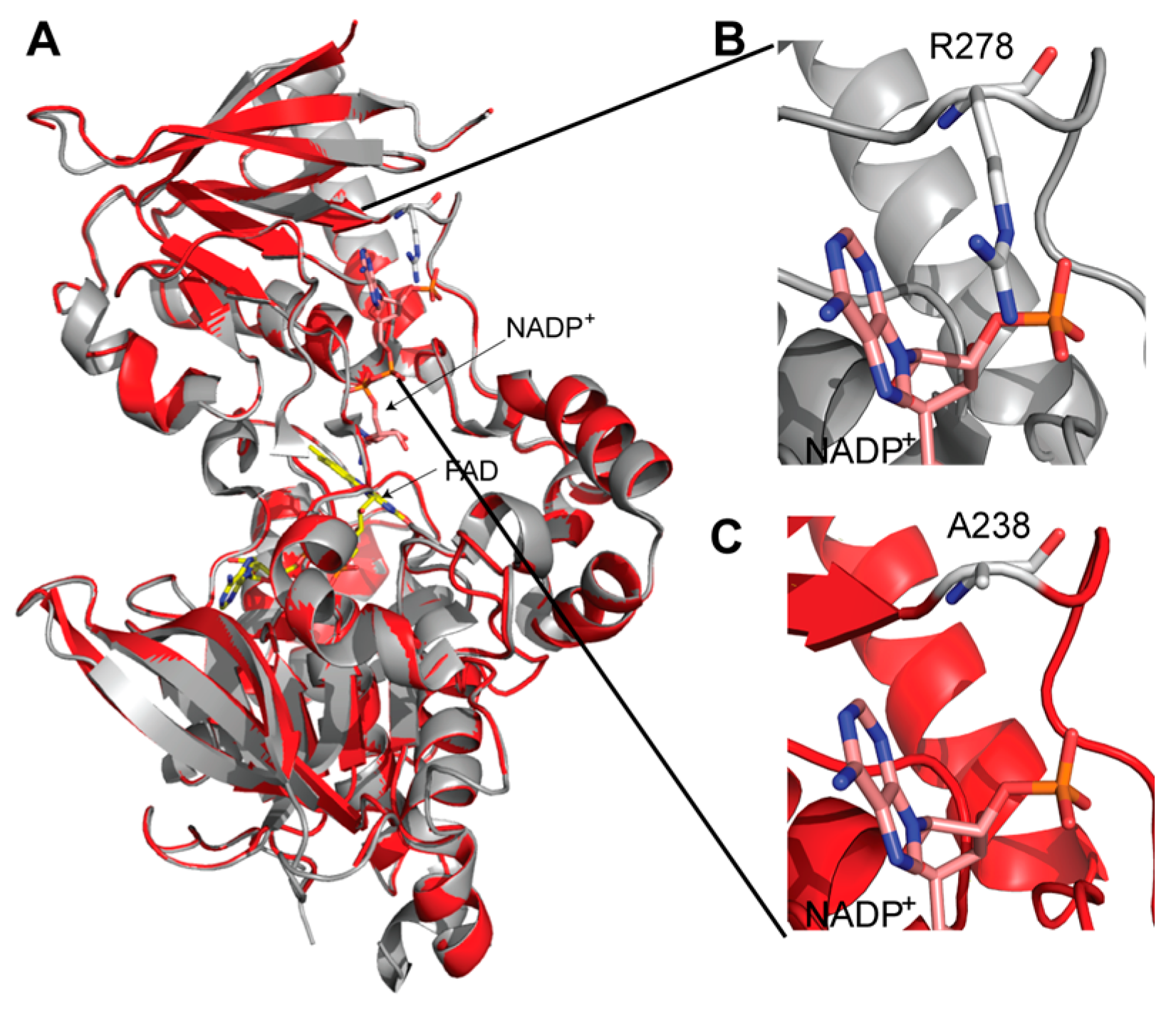
9. Three-Dimensional Model
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kodani, S.; Komaki, H.; Suzuki, M.; Hemmi, H.; Ohnishi-Kameyama, M. Isolation and structure determination of new siderophore albachelin from Amycolatopsis alba. Biometals 2015, 28, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, W.J.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, H.; Del Campo, J.S.M.; Dai, Y.; Adly, C.; El-Sohaimy, S.; Sobrado, P. Mechanism of Rifampicin Inactivation in Nocardia farcinica. PLoS ONE 2016, 11, e0162578. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.K.; Abdelwahab, H.; Martin Del Campo, J.S.; Mehra-Chaudhary, R.; Sobrado, P.; Tanner, J.J. The Structure of the Antibiotic Deactivating, N-hydroxylating Rifampicin Monooxygenase. J. Biol. Chem. 2016, 291, 21553–21562. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, Q.; Li, S.; Lin, Q.; Fu, P.; Zhang, G.; Zhang, H.; Shi, R.; Zhu, W.; Zhang, C. Insights into caerulomycin A biosynthesis: A two-component monooxygenase CrmH-catalyzed oxime formation. J. Am. Chem. Soc. 2013, 135, 18750–18753. [Google Scholar] [CrossRef] [PubMed]
- Chocklett, S.W.; Sobrado, P. Aspergillus fumigatus SidA is a highly specific ornithine hydroxylase with bound flavin cofactor. Biochemistry 2010, 49, 6777–6783. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Fedkenheuer, M.; Chocklett, S.W.; Qi, J.; Oppenheimer, M.; Sobrado, P. Dual role of NADP(H) in the reaction of a flavin dependent N-hydroxylating monooxygenase. Biochim. Biophys. Acta 2012, 1824, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Robinson, R.; Sobrado, P. Monitoring the reductive and oxidative half-reactions of a flavin-dependent monooxygenase using stopped-flow spectrophotometry. J. Vis. Exp. 2012, 3803. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Badieyan, S.; Sobrado, P. C4a-hydroperoxyflavin formation in N-hydroxylating flavin monooxygenases is mediated by the 2’-OH of the nicotinamide ribose of NADP(+). Biochemistry 2013, 52, 9089–9091. [Google Scholar] [CrossRef] [PubMed]
- Olucha, J.; Lamb, A.L. Mechanistic and structural studies of the N-hydroxylating flavoprotein monooxygenases. Bioorg. Chem. 2011, 39, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, S.; Fedkenheuer, M.; Vogelaar, N.J.; Robinson, H.H.; Sobrado, P.; Mattevi, A. Structural insight into the mechanism of oxygen activation and substrate selectivity of flavin-dependent N-hydroxylating monooxygenases. Biochemistry 2012, 51, 7043–7045. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.; Avila, D.; Sobrado, P. Effect of pH on the Reductive and Oxidative Half-reactions of Aspergillus fumigatus Siderophore A. In Flavins and Flavoproteins; Miller, S., Hille, R., Palfey, B., Eds.; Lulu: Raleigh, NC, USA, 2013; pp. 289–294. [Google Scholar]
- Robinson, R.; Franceschini, S.; Fedkenheuer, M.; Rodriguez, P.J.; Ellerbrock, J.; Romero, E.; Echandi, M.P.; Martin Del Campo, J.S.; Sobrado, P. Arg279 is the key regulator of coenzyme selectivity in the flavin-dependent ornithine monooxygenase SidA. Biochim. Biophys. Acta 2014, 1844, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Meneely, K.M.; Lamb, A.L. Biochemical characterization of a flavin adenine dinucleotide-dependent monooxygenase, ornithine hydroxylase from Pseudomonas aeruginosa, suggests a novel reaction mechanism. Biochemistry 2007, 46, 11930–11937. [Google Scholar] [CrossRef] [PubMed]
- Meneely, K.M.; Barr, E.W.; Bollinger, J.M., Jr.; Lamb, A.L. Kinetic mechanism of ornithine hydroxylase (PvdA) from Pseudomonas aeruginosa: Substrate triggering of O2 addition but not flavin reduction. Biochemistry 2009, 48, 4371–4376. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Sobrado, P. Substrate binding modulates the activity of Mycobacterium smegmatis G, a flavin-dependent monooxygenase involved in the biosynthesis of hydroxamate-containing siderophores. Biochemistry 2011, 50, 8489–8496. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.M.; Rodriguez, P.J.; Sobrado, P. Mechanistic studies on the flavin-dependent N(6)-lysine monooxygenase MbsG reveal an unusual control for catalysis. Arch. Biochem. Biophys. 2014, 550–551, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Robinson, R.M.; Martin Del Campo, J.S.; Keul, N.D.; Rodriguez, P.J.; Robinson, H.H.; Mattevi, A.; Sobrado, P. An Unprecedented NADPH Domain Conformation in Lysine Monooxygenase NbtG Provides Insights into Uncoupling of Oxygen Consumption from Substrate Hydroxylation. J. Biol. Chem. 2015, 290, 12676–12688. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, H.; Robinson, R.; Rodriguez, P.; Adly, C.; El-Sohaimy, S.; Sobrado, P. Identification of structural determinants of NAD(P)H selectivity and lysine binding in lysine N(6)-monooxygenase. Arch. Biochem. Biophys. 2016, 606, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Blommel, P.G.; Martin, P.A.; Wrobel, R.L.; Steffen, E.; Fox, B.G. High efficiency single step production of expression plasmids from cDNA clones using the Flexi Vector cloning system. Protein Expr. Purif. 2006, 47, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Grabski, A.; Mehler, M.; Drott, D. The Overnight Express Autoinduction System: High-density cell growth and protein expression while you sleep. Nat. Methods 2005, 2, 233–235. [Google Scholar] [CrossRef]
- Studier, F.W. Stable Expression Clones and Auto-Induction for Protein Production in E. coli. In Structural Genomics: General Applications; Chen, Y.W., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 17–32. [Google Scholar]
- Macheroux, P. UV-Visible Spectroscopy as a Tool to Study Flavoproteins. In Flavoprotein Protocols; Chapman, S.K., Reid, G.A., Eds.; Humana Press: Totowa, NJ, USA, 1999; pp. 1–7. [Google Scholar]
- Csaky, T.Z. On the estimation of bound hydroxylamine in biological materials. Acta Chem. Scand. 1948, 450–454. [Google Scholar] [CrossRef]
- Tomlinson, G.; Cruickshank, W.H.; Viswanatha, T. Sensitivity of substituted hydroxylamines to determination by iodine oxidation. Anal. Biochem. 1971, 44, 670–679. [Google Scholar] [CrossRef]
- Gillam, A.H.; Lewis, A.G.; Andersen, R.J. Quantitative determination of hydroxamic acids. Anal. Chem. 1981, 53, 841–844. [Google Scholar] [CrossRef]
- Kelly, L.; Sternberg, M.J.E. Protein structure prediction on the Wed: A case study using Phyre server. Nat. Protocols 2009, 4, 363–371. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 1.3r1; Schrödinger, LLC: New York, NY, USA, 2010. [Google Scholar]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Parameter | Holo-AMO | Recon-AMO |
|---|---|---|---|
| Oxygraph | kcat (s−1) | 0.340 ± 0.030 | 0.200 ± 0.010 |
| (Orn) | KM(Orn) (mM) | 0.305 ± 0.108 | 0.107 ± 0.052 |
| kcat/KM(Orn) (mM−1 s−1) | 1.12 ± 0.410 | 1.70 ± 0.810 | |
| Oxygraph | kcat (s−1) | 0.340 ± 0.020 | 0.170 ± 0.010 |
| (Lys) | KM(Lys) (mM) | 0.560 ± 0.110 | 0.180 ± 0.070 |
| kcat/KM(Lys) (mM−1 s−1) | 0.630 ± 0.120 | 0.950 ± 0.040 | |
| Product formation | kcat (s−1) | 0.200 ± 0.006 | 0.010 ± 0.003 |
| (Orn) | KM(Orn) (mM) | 0.234 ± 0.048 | 0.240 ± 0.035 |
| kcat/KM(Orn) (mM−1 s−1) | 0.860 ± 0.200 | 0.040 ± 0.010 | |
| %Coupling (Orn) | 59% | 5% |
| Assay * | Parameters | Holo-AMO | Recon-AMO | ||
|---|---|---|---|---|---|
| NADPH | NADH | NADPH | NADH | ||
| Oxygraph | kcat, (s−1) | 0.25 ± 0.01 | 0.15 ± 0.01 | 0.17 ± 0.01 | 0.18 ± 0.01 |
| KM(NAD(P)H, (mM) | 1.40 ± 0.16 | 0.32 ± 0.10 | 2.70 ± 0.35 | 1.60 ± 0.20 | |
| kcat/KM(NAD(P)H, (mM−1 s−1) | 0.160 ± 0.015 | 0.50 ± 10 | 0.060 ± 0.005 | 0.11 ± 0.01 | |
| Product formation | kcat, (s−1) | 0.16 ± 0.05 | N/A | 0.08 ± 0.03 | 0.040 ± 0.010 |
| KM(NAD(P)H, (mM) | 0.60 ± 0.20 | N/A | 2.60 ± 2.0 | 0.60 ± 0.30 | |
| kcat/KM(NAD(P)H, (mM−1 s−1) | 0.260 ± 0.020 | N/A | 0.03 ± 0.01 | 0.08 ± 0.02 | |
| %Coupling | 64% | 47% | 22% | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bufkin, K.; Sobrado, P. Characterization of the Ornithine Hydroxylation Step in Albachelin Biosynthesis. Molecules 2017, 22, 1652. https://doi.org/10.3390/molecules22101652
Bufkin K, Sobrado P. Characterization of the Ornithine Hydroxylation Step in Albachelin Biosynthesis. Molecules. 2017; 22(10):1652. https://doi.org/10.3390/molecules22101652
Chicago/Turabian StyleBufkin, Kendra, and Pablo Sobrado. 2017. "Characterization of the Ornithine Hydroxylation Step in Albachelin Biosynthesis" Molecules 22, no. 10: 1652. https://doi.org/10.3390/molecules22101652
APA StyleBufkin, K., & Sobrado, P. (2017). Characterization of the Ornithine Hydroxylation Step in Albachelin Biosynthesis. Molecules, 22(10), 1652. https://doi.org/10.3390/molecules22101652