Proline-Based Carbamates as Cholinesterase Inhibitors †

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results
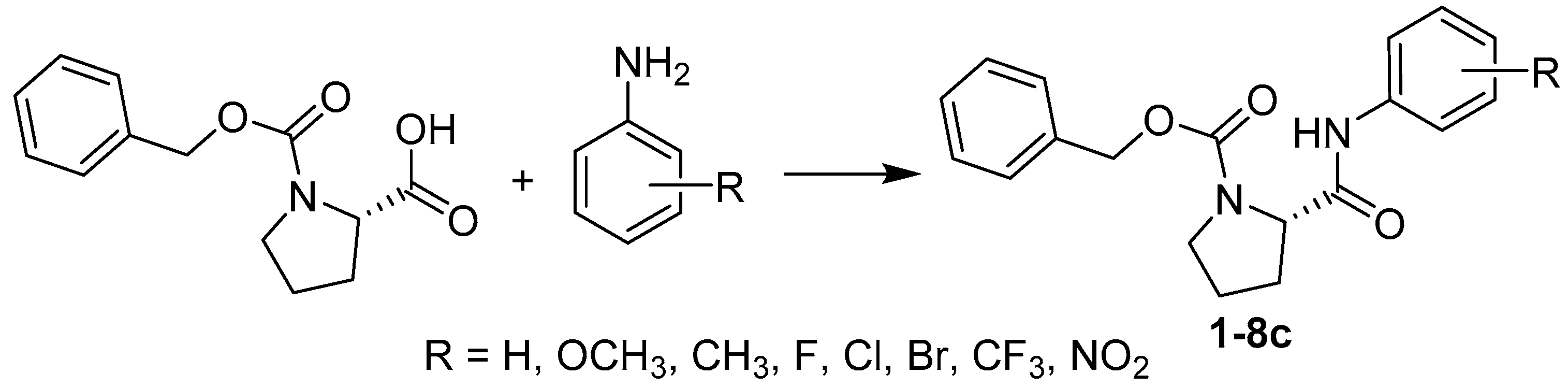
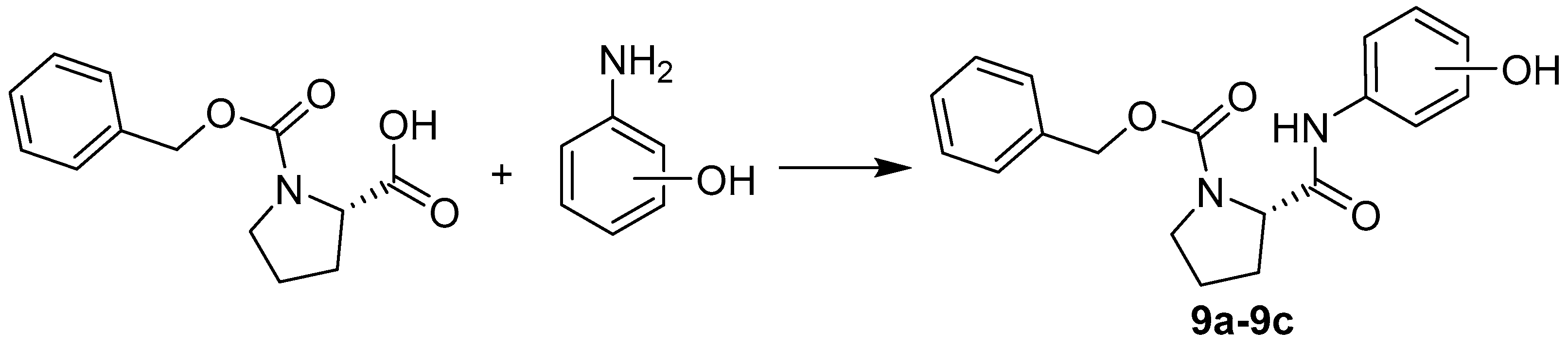
2.1. Chemistry
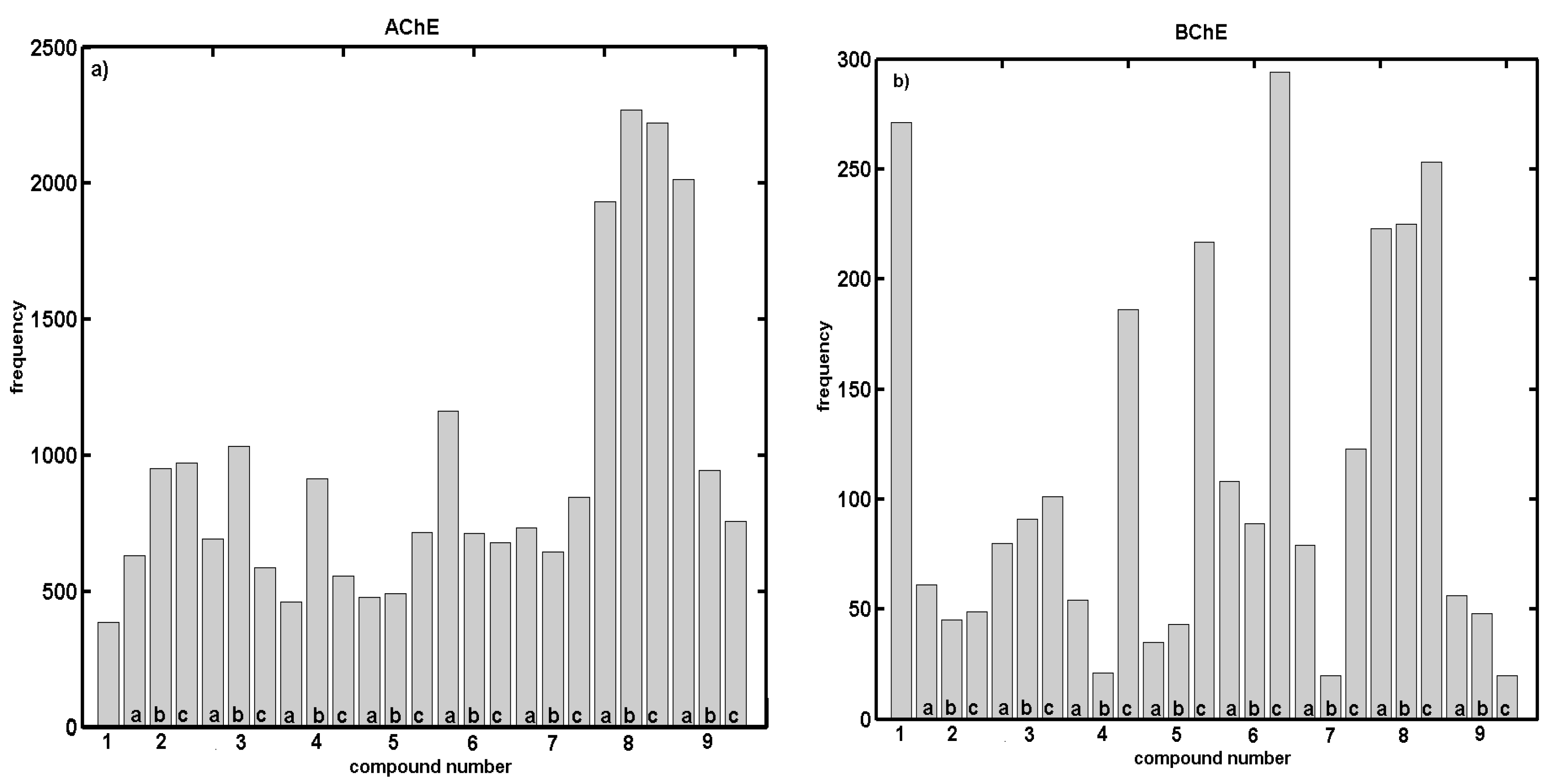
2.2. In Vitro Evaluation of AChE- and BChE-Inhibiting Activity with CoMSA and SMV Procedure
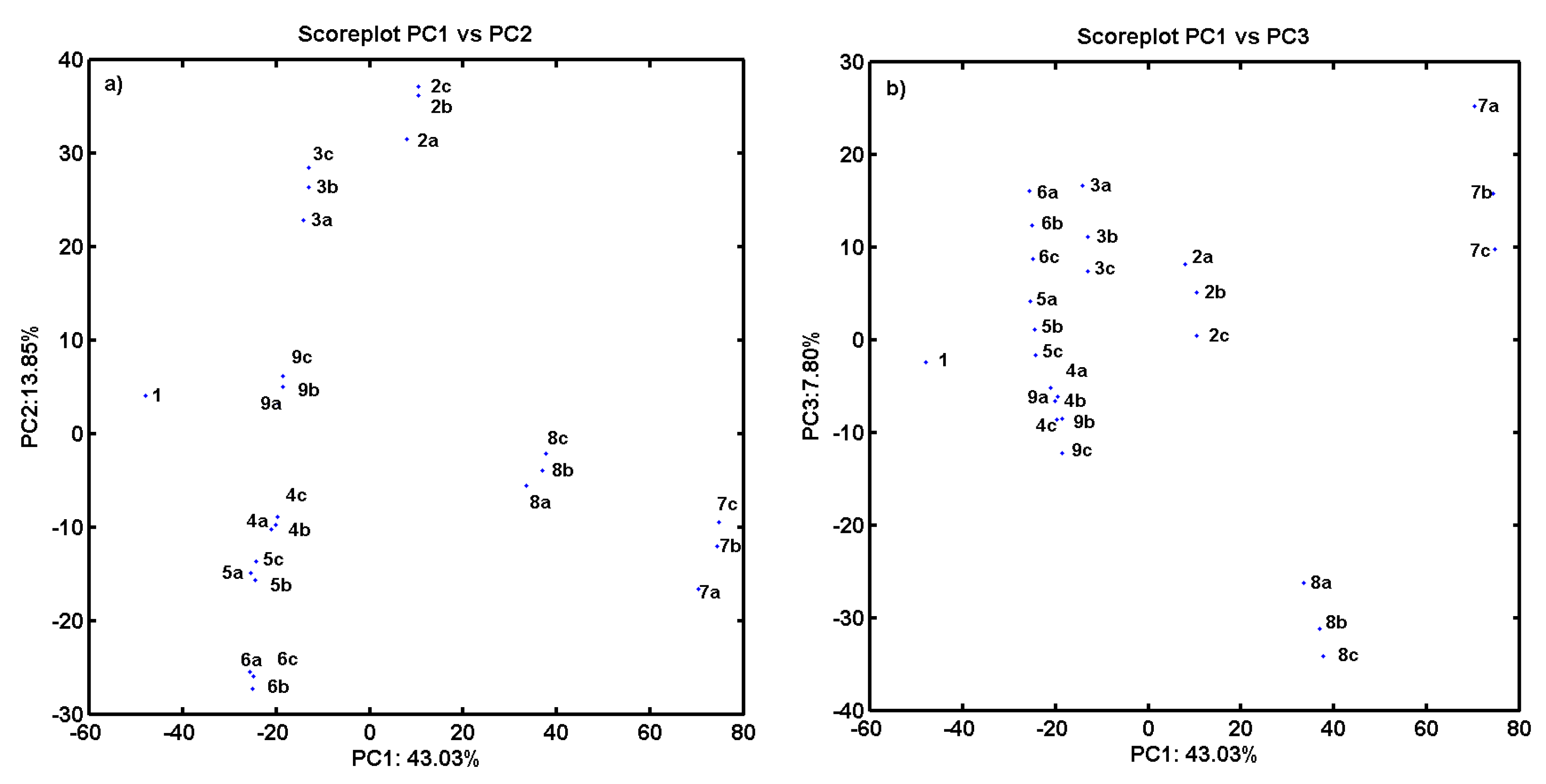
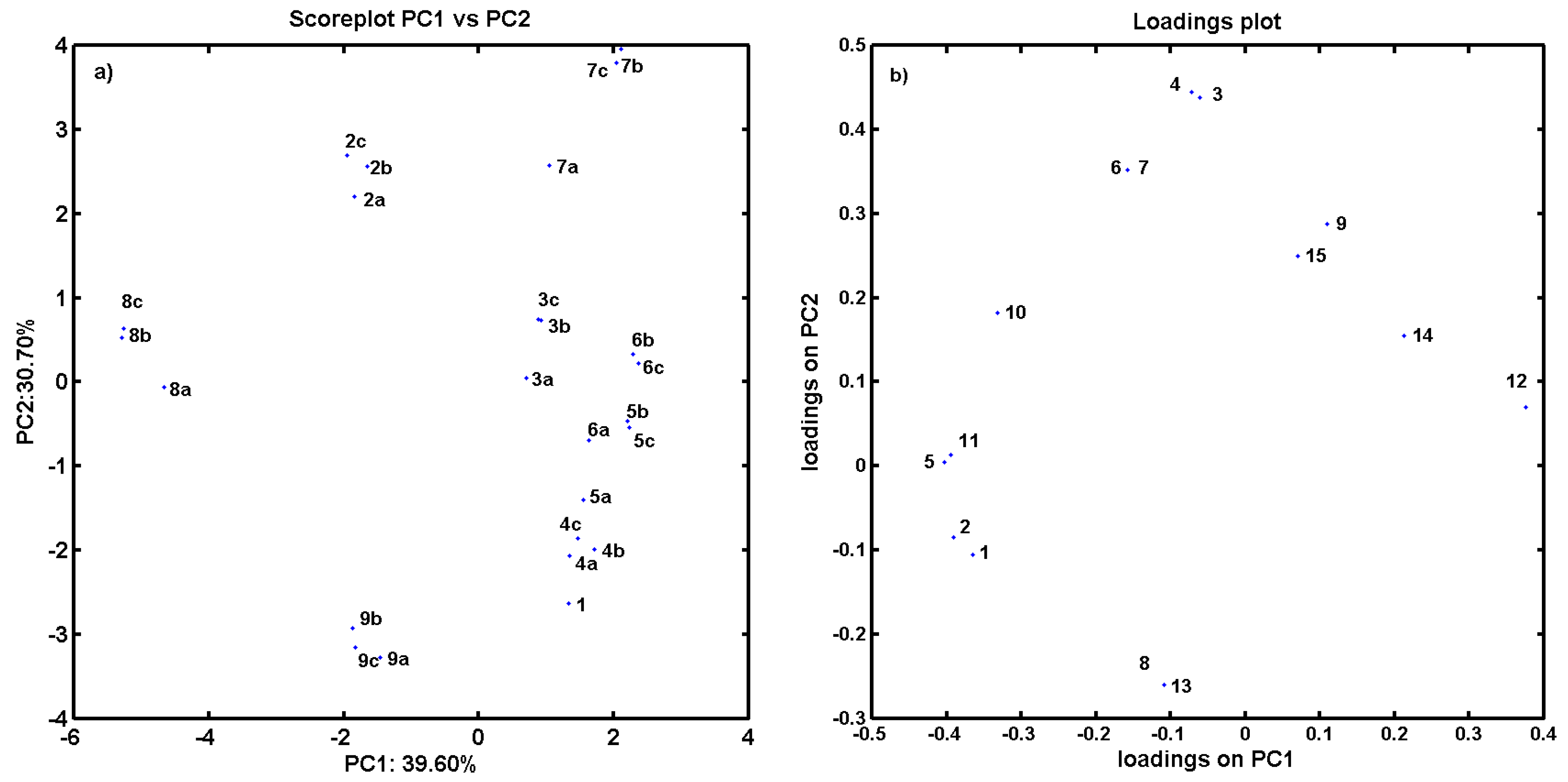
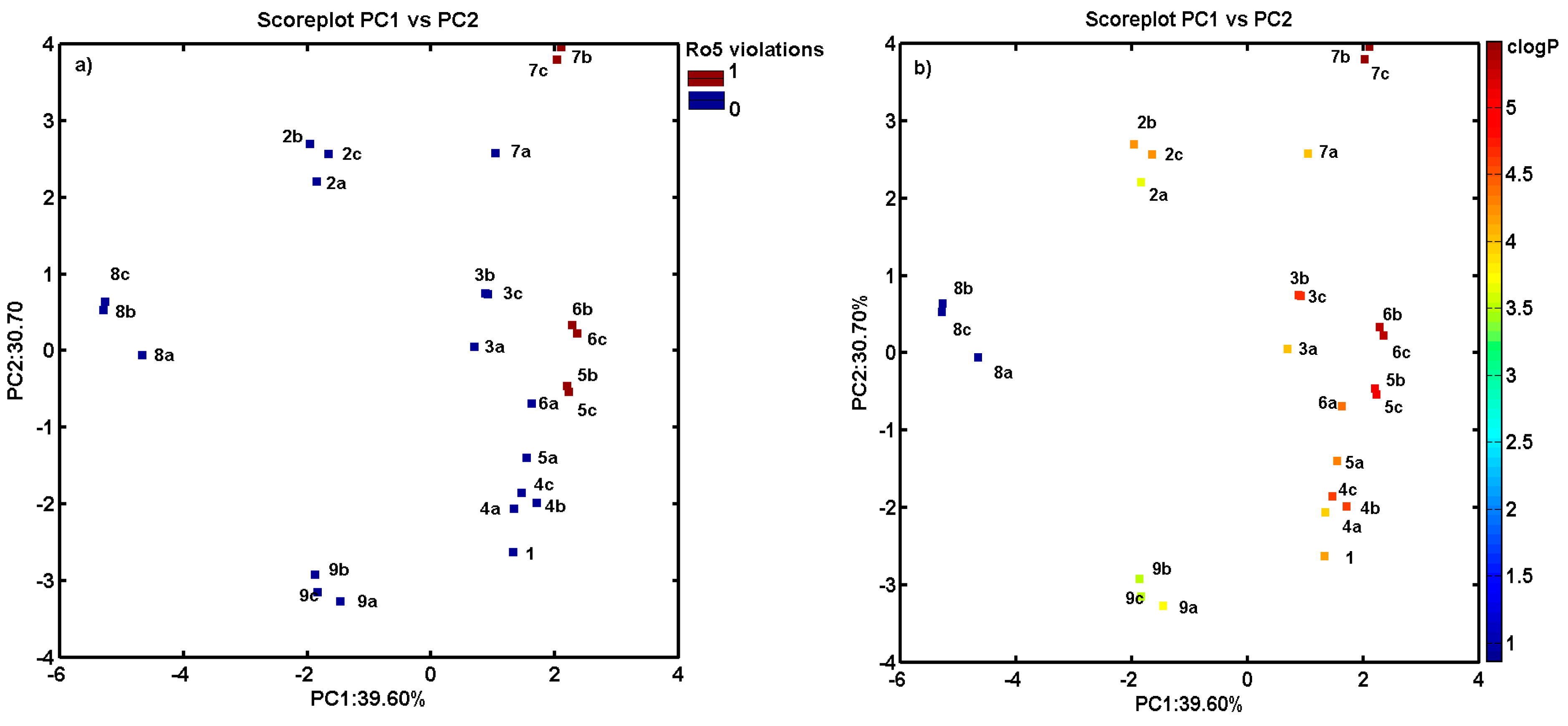
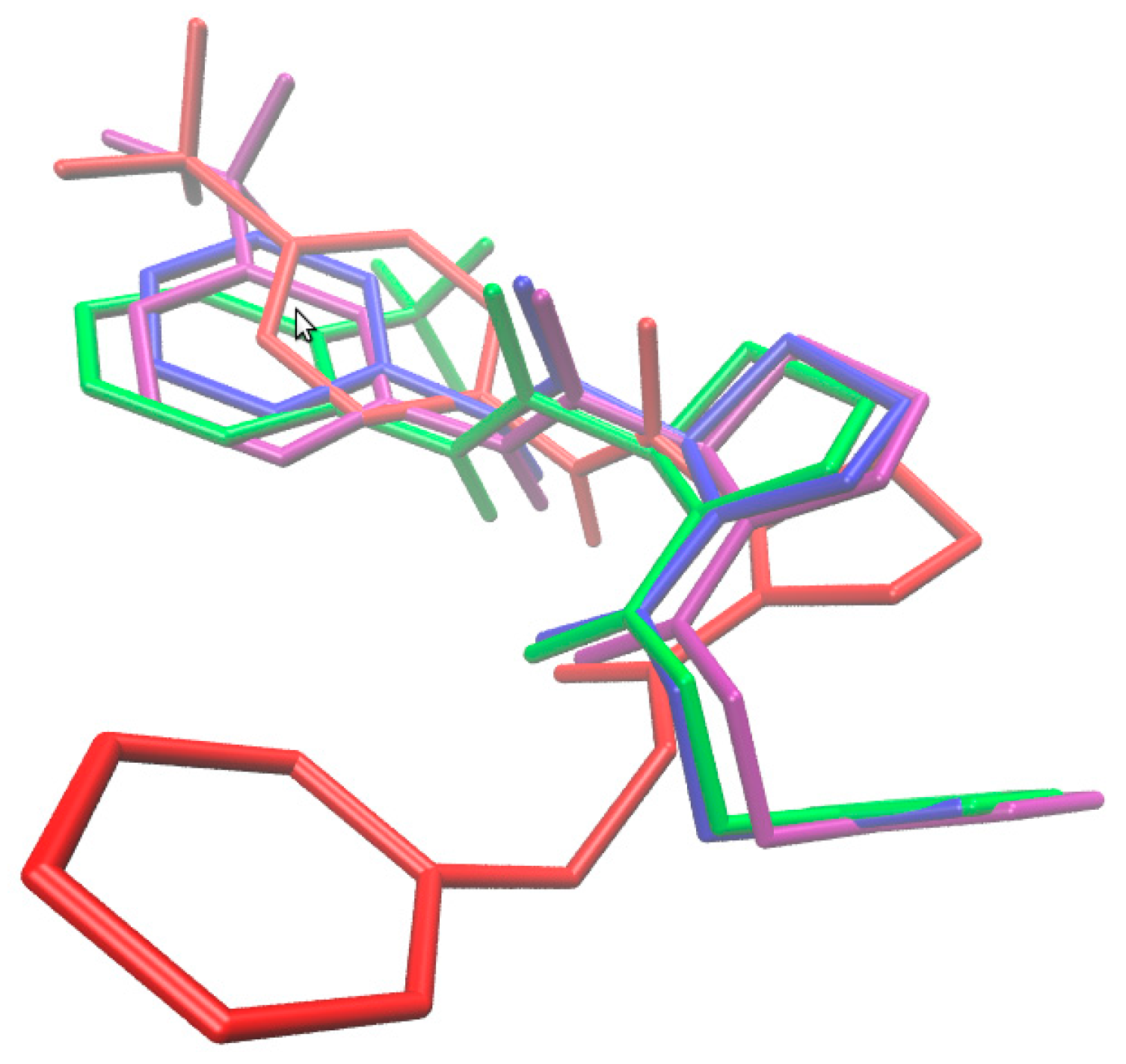
2.3. Consensus-Based 3D Pharmacophore Mapping
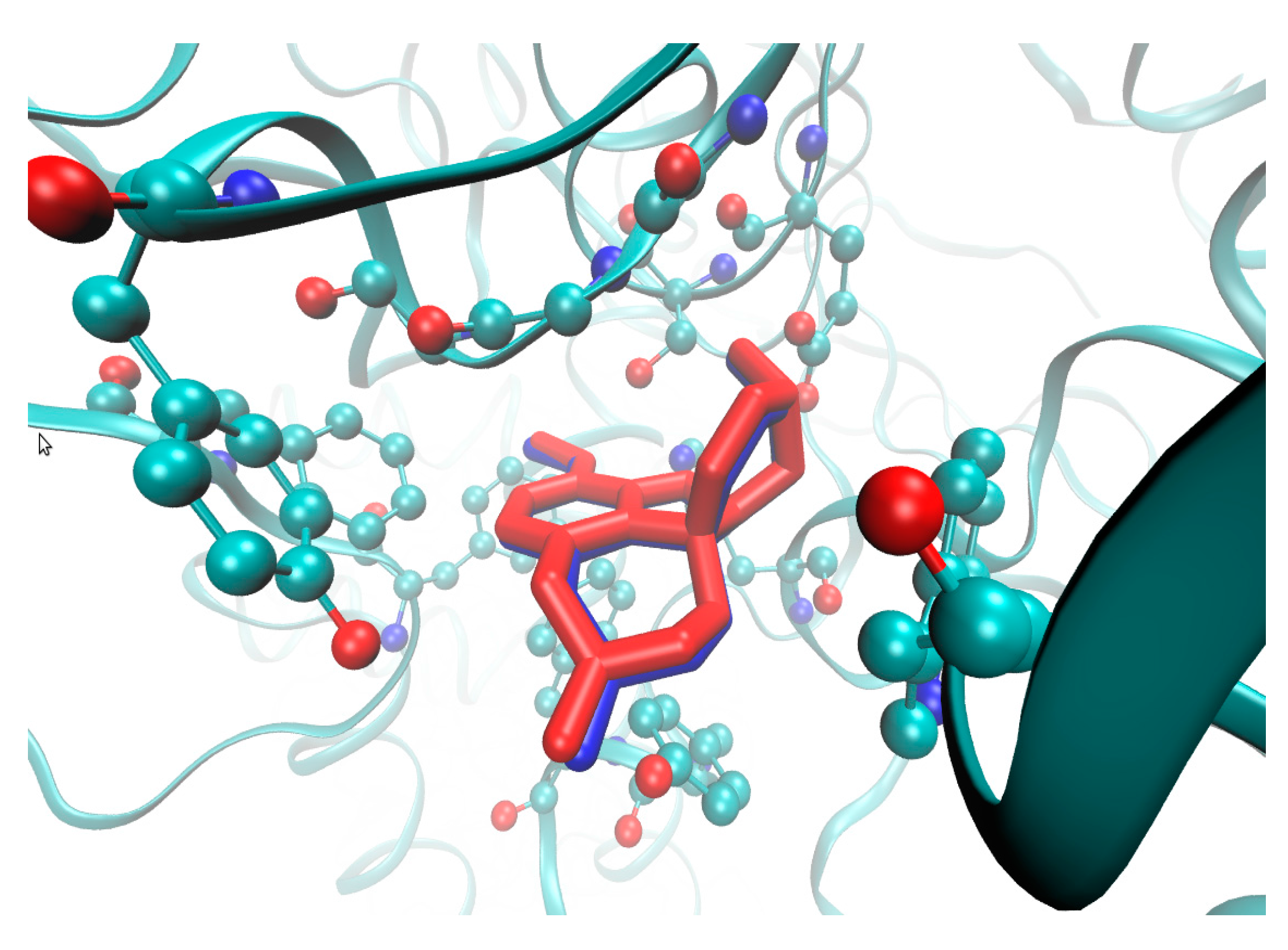
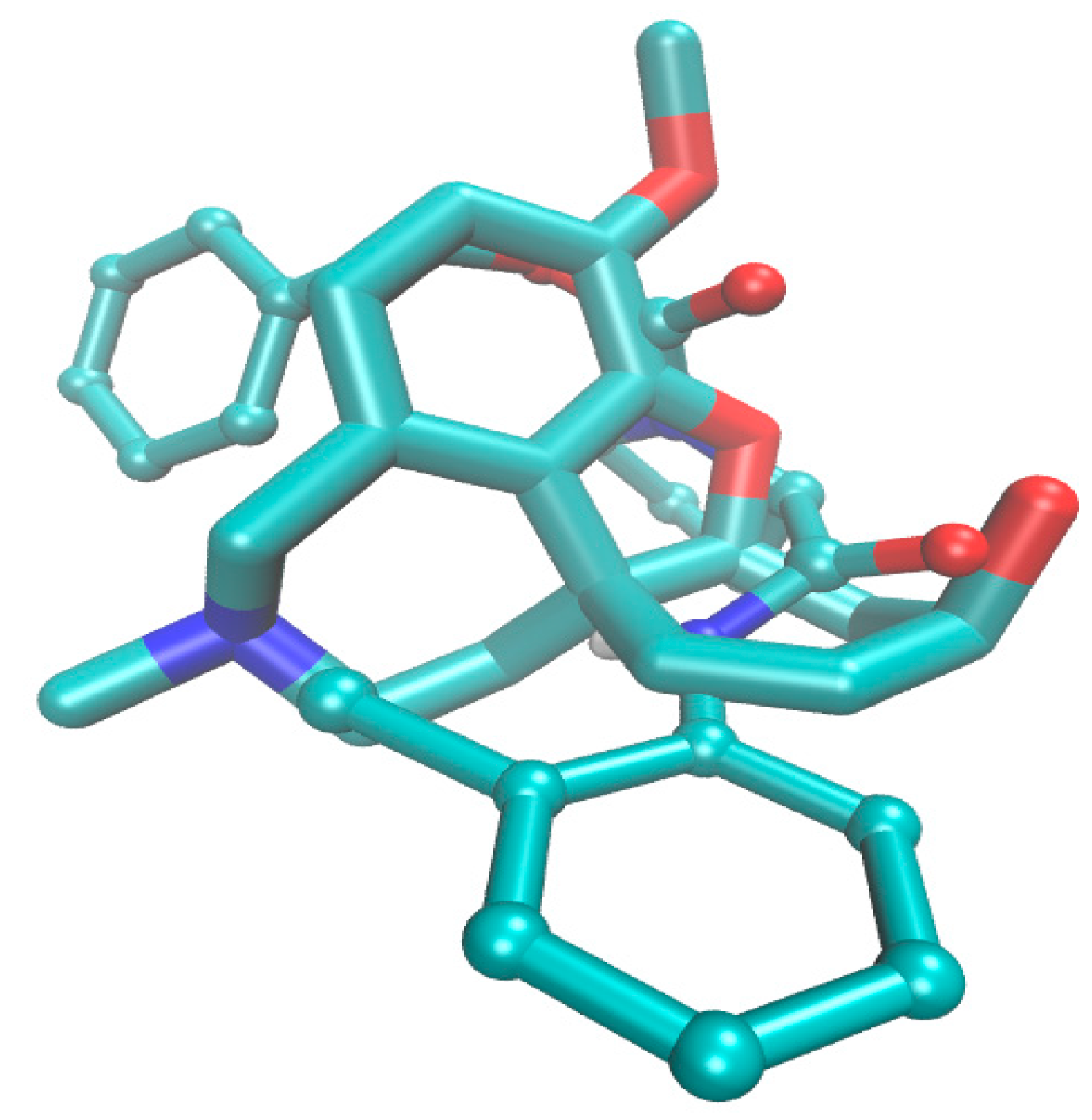
2.4. Molecular Docking Study
2.5. In Vitro Cytotoxicity Assay
3. Materials and Methods
3.1. General Methods
3.2. Synthesis
3.3. In Vitro Evaluation of AChE- and BChE-Inhibiting Activity
3.4. Comparative Molecular Surface Analysis
3.5. PLS Analysis
3.6. Iterative PLS-Based Variable Elimination
3.7. PCA Analysis
3.8. Model Builder
3.9. Molecular Modeling
3.10. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steinhilber, D.; Schubert-Zsilavecz, M.; Roth, H.J. Medizinische Chemie: Targets, Arzneistoffe, Chemische Biologie; Deutscher Apotheker Verlag: Stutgart, Germany, 2010. [Google Scholar]
- US Environmental Protection Agency—Pesticide Registration: Pesticide Data Submitters List (PDSL). 2016. Available online: https://www.epa.gov/sites/production/files/2016-04/documents/dslchem_0.pdf (accessed on 4 September 2017).
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]-benzamides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef] [PubMed]
- Fajkusova, D.; Pesko, M.; Keltosova, S.; Guo, J.; Oktabec, Z.; Vejsova, M.; Kollar, P.; Coffey, A.; Csollei, J.; Kralova, K.; et al. Anti-infective and herbicidal activity of N-substituted 2-aminobenzothiazoles. Bioorg. Med. Chem. 2012, 20, 7059–7068. [Google Scholar] [CrossRef] [PubMed]
- Pauk, K.; Zadrazilova, I.; Imramovsky, A.; Vinsova, J.; Pokorna, M.; Masarikova, M.; Cizek, A.; Jampilek, J. New derivatives of salicylamides: Preparation and antimicrobial activity against various bacterial species. Bioorg. Med. Chem. 2013, 21, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Nevin, E.; Soral, M.; Kushkevych, I.; Gonec, T.; Bobal, P.; Kollar, P.; Coffey, A.; O´Mahony, J.; Liptaj, T.; et al. Synthesis and antimycobacterial properties of ring-substituted 6-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2015, 23, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Zadrazilova, I.; Nevin, E.; Soral, M.; Gonec, T.; Kollar, P.; Oravec, M.; Coffey, A.; O’Mahony, J.; Liptaj, T.; et al. Ring-Substituted 8-hydroxyquinoline-2-carboxanilides as potential antimycobacterial agents. Bioorg. Med. Chem. 2015, 23, 4188–4196. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Pospisilova, S.; Kauerova, T.; Kos, J.; Dohanosova, J.; Oravec, M.; Kollar, P.; Coffey, A.; Liptaj, T.; Cizek, A.; Jampilek, J. N-Alkoxyphenylhydroxynaphthalenecarboxamides and their antimycobacterial activity. Molecules 2016, 21, 1068. [Google Scholar] [CrossRef] [PubMed]
- Kauerova, T.; Kos, J.; Gonec, T.; Jampilek, J.; Kollar, P. Antiproliferative and pro-apoptotic effect of novel nitro-substituted hydroxynaphthanilides on human cancer cell lines. Int. J. Mol. Sci. 2016, 17, 1219. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, M.; Angelina, E.; Lima, S.; Gonec, T.; Otevrel, J.; Marvanova, P.; Padrtova, T.; Mokry, P.; Bobal, P.; Acosta, L.M.; et al. Search of new structural scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017, 139, 461–481. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Pesko, M.; Monreal-Ferriz, J.; Kralova, K.; Vinsova, J.; Jampilek, J. Photosynthesis-inhibiting efficiency of 4-chloro-2-(chlorophenylcarbamoyl)phenyl alkyl-carbamates. Bioorg. Med. Chem. Lett. 2011, 21, 4564–4567. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Stranik, J.; Pesko, M.; Kos, J.; Oravec, M.; Kralova, K.; Jampilek, J. Photosynthesis-inhibiting activity of 1-[(2-chlorophenyl)carbamoyl]- and 1-[(2-nitrophenyl)carbamoyl]naphthalen-2-yl alkylcarbamates. Molecules 2017, 22, 1199. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J.; Brychtova, K. Azone analogues: Classification, design, and transdermal penetration principles. Med. Res. Rev. 2012, 32, 907–947. [Google Scholar] [CrossRef] [PubMed]
- Pejchal, V.; Stepankova, S.; Padelkova, Z.; Imramovsky, A.; Jampilek, J. 1,3-Substituted imidazolidine-2,4,5-triones: Synthesis and inhibition of cholinergic enzymes. Molecules 2011, 16, 7565–7582. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Stepankova, S.; Vanco, J.; Pauk, K.; Monreal-Ferriz, J.; Vinsova, J.; Jampilek, J. Acetylcholinesterase-inhibiting activity of salicylanilide n-alkylcarbamates and their molecular docking. Molecules 2012, 17, 10142–10158. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Pejchal, V.; Stepankova, S.; Vorcakova, K.; Jampilek, J.; Vanco, J.; Simunek, P.; Kralovec, K.; Bruckova, L.; Mandikova, J.; et al. Synthesis and in vitro evaluation of new derivatives of 2-substituted-6-fluorobenzo[d]thiazoles as cholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 1735–1748. [Google Scholar] [CrossRef] [PubMed]
- Cygler, M.; Schrag, J.D.; Sussman, J.L.; Harel, M.; Silman, I.; Gentry, M.K.; Doctor, B.P. Relationship between sequence conservation and 3-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993, 2, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.E.; Perez, R.G.; Kobayashi, H. Cholinesterase inhibitor therapy in Alzheimer’s disease: The limits and tolerability of irreversible CNS-selective acetylcholinesterase inhibition in primates. J. Alzheimers Dis. 2017, 55, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15, PCC.12r01412. [Google Scholar] [CrossRef] [PubMed]
- Cholinesterase Inhibitors. Available online: https://www.drugs.com/drug-class/cholinesterase-inhibitors.html (accessed on 4 September 2017).
- NIH—National Institute on Aging: Alzheimer’s Disease Fact Sheet. Available online: https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet (accessed on 4 September 2017).
- Alzheimers.net. 2016 Alzheimer’s Statistics. Available online: http://www.alzheimers.net/resources/alzheimers-statistics (accessed on 4 September 2017).
- Bajic, V.; Milovanovic, E.S.; Spremo-Potparevic, B.; Zivkovic, L.; Milicevic, Z.; Stanimirovic, J.; Bogdanovic, N.; Isenovic, E.R. Treatment of Alzheimer’s disease: Classical therapeutic approach. Curr. Pharm. Anal. 2016, 12, 82–90. [Google Scholar] [CrossRef]
- Brus, B.; Kosak, U.; Turk, S.; Pislar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.P.; Gobec, S. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef] [PubMed]
- Kosak, U.; Brus, B.; Knez, D.; Sink, R.; Zakelj, S.; Trontelj, J.; Pislar, A.; Slenc, J.; Gobec, M.; Zivin, M.; et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.A.; Gutierrez, M.; Ramirez, D.; Alzate-Morales, J.; Bernal, C.C.; Guiza, F.M.; Romero Bohorquez, A.R. Novel N-allyl/propargyl tetrahydroquinolines: Synthesis via three-component cationic imino Diels-Alder reaction, binding prediction, and evaluation as cholinesterase inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Brus, B.; Coquelle, N.; Sosic, I.; Sink, R.; Brazzolotto, X.; Mravljak, J.; Colletier, J.P.; Gobec, S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2015, 23, 4442–4452. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Meena, P.; Singh, A.; Jameel, E.; Maqbool, M.; Mobashir, M.; Shandilya, A.; Tiwari, M.; Hoda, N.; Jayaram, B. Synthesis and screening of triazolopyrimidine scaffold as multi-functional agents for Alzheimer’s disease therapies. Eur. J. Med. Chem. 2016, 119, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Shevtsova, E.F.; Makhaeva, G.F.; Grigoriev, V.V.; Boltneva, N.P.; Kovaleva, N.V.; Lushchekina, S.V.; Shevtsov, P.N.; Neganova, M.E.; Redkozubova, O.M.; et al. Novel conjugates of aminoadamantanes with carbazole derivatives as potential multitarget agents for AD treatment. Sci. Rep. 2017, 7, 45627. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Rao, P.P. 2,4-Disubstituted quinazolines as amyloid-β aggregation inhibitors with dual cholinesterase inhibition and antioxidant properties: Development and structure-activity relationship (SAR) studies. Eur. J. Med. Chem. 2017, 126, 823–843. [Google Scholar] [CrossRef] [PubMed]
- Kozakiewicz, D.; Polanski, J.; Jampilek, J.; Imramovsky, A.; Stepankova, S. New Carbamate Derivatives and Their Application. U.S. Patent 420626, 23 February 2017. [Google Scholar]
- Stanton, D.T. QSAR and QSPR model interpretation using partial least squares (PLS) analysis. Curr. Comput. Aided Drug Des. 2012, 8, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Magdziarz, T.; Polanski, J. Pharmacophore-based database mining for probing fragmental drug-likeness of diketo acid analogues. SAR&QSAR Environ. Res. 2012, 23, 185–204. [Google Scholar]
- Albericio, F. Developments in peptide and amide synthesis. Curr. Opin. Chem. Biol. 2004, 8, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Lanigan, R.M.; Sheppard, T.D. Recent developments in amide synthesis: Direct amidation of carboxylic acids and transamidation reactions. Eur. J. Org. Chem. 2013, 7453–7465. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- De Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical routes for amide bond formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Bobal, P.; Sujan, J.; Pesko, M.; Guo, J.; Kralova, K.; Pavlacka, L.; Vesely, L.; Kreckova, E.; Kos, J.; et al. Investigating the spectrum of biological activity of substituted quinoline-2-carboxamides and their isosteres. Molecules 2012, 17, 613–644. [Google Scholar] [CrossRef] [PubMed]
- Bobal, P.; Sujan, J.; Otevrel, J.; Imramovsky, A.; Padelkova, Z.; Jampilek, J. Microwave-assisted synthesis of new substituted anilides of quinaldic acid. Molecules 2012, 17, 1292–1306. [Google Scholar] [CrossRef] [PubMed]
- Pizova, H.; Bobal, P. An optimized and scalable synthesis of propylphosphonic anhydride for general use. Tetrahedron Lett. 2015, 56, 2014–2017. [Google Scholar] [CrossRef]
- Mukayama, T. Asymmetric synthesis based on chiral diamines having pyrrolidine ring. Tetrahedron 1981, 37, 4111–4119. [Google Scholar] [CrossRef]
- Rhyoo, H.Y.; Yoon, Y.A.; Park, H.J.; Chung, Y.K. Use of amino amides derived from proline as chiral ligands in the ruthenium(II)-catalyzed transfer hydrogenation reaction of ketones. Tetrahedron Lett. 2001, 42, 5045–5048. [Google Scholar] [CrossRef]
- Devillers, J. Methods for building QSARs. Methods Mol. Biol. 2013, 930, 3–27. [Google Scholar] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Salvino, J.M.; Mallamo, J.P. Knowledge-based chemoinformatic approaches to drug discovery. Drug Discov. Today 2006, 11, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. Hansch Analysis and Related Approaches; Wiley-VCH Verlag: Weinheim, Germany, 1993. [Google Scholar]
- Bak, A.; Polanski, J. The 4D-QSAR study on anti-HIV HEPT analogues. Bioorg. Med. Chem. 2006, 14, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.; Magdziarz, T.; Bak, A.; Chilmonczyk, Z.; Kasprzycka-Guttman, T.; Misiewicz-Krzeminska, I.; Skupinska, K.; Polanski, J. Does molecular docking reveal alternative chemopreventive mechanism of activation of oxidoreductase by sulforaphane isothiocyanates? J. Mol. Model. 2010, 16, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Zentgraf, M.; Steuber, H.; Koch, C.; La Motta, C.; Sartini, S.; Sotriffer, C.A.; Klebe, G. How reliable are current docking approaches for structure-based drug design? Lessons from aldose reductase. Angew. Chem. Int. Ed. Engl. 2007, 46, 3575–3580. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Heigh, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of docking. Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Suffness, M.; Douros, J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Warren, S.; O’Brien, P. Synthesis of (R)- or (S)-diphenylphosphinoyl hydroxy aldehydes and 1,2-diols using Mukaiyama’s bicyclic aminal methodology and Sharpless asymmetric dihydroxylation. J. Chem. Soc. Perkin Trans. 1996, 1, 2129–2138. [Google Scholar]
- Wang, Z.; Zhou, W. Synthesis and in vitro antibacterial activity of (2S)-N-(substitutedphenyl)-1-[(2R)-2-[(formylhydroxyamino)methyl]-1-oxohexyl]-2-pyrrolidinecarboxamides as potential peptide deformylase inhibitors. Chem. Biol. Drug Des. 2009, 73, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Evindar, G.; Batey, R.A. Parallel synthesis of a library of benzoxazoles and benzothiazoles using ligand-accelerated copper-catalyzed cyclizations of ortho-halobenzanilides. J. Org. Chem. 2006, 71, 1802–1808. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.U.; Roberts, C.D.; Abadi, A.D.M.; Griffith, R.C.; Leivers, M.R. Preparation of Proline Amides for Treating Flaviviridae Family Virus Infection. U.S. Patent WO/2007/070556, 21 June 2007. [Google Scholar]
- Zeror, S.; Collin, J.; Fiaud, J.; Zouioueche, L.A. Evaluation of ligands for ketone reduction by asymmetric hydride transfer in water by multi-substrate screening. Adv. Synth. Catal. 2008, 350, 197–204. [Google Scholar] [CrossRef]
- Cox, C.; Lectka, T. Intramolecular catalysis of amide isomerization: Kinetic consequences of the 5-NH- -Na hydrogen bond in prolyl peptides. J. Am. Chem. Soc. 1998, 120, 10660–10668. [Google Scholar] [CrossRef]
- Jacobs, J.W.; Patel, D.; Lewis, J.; Ni, Z. Preparation of Novel Heterocyclic Urea Compounds, Particularly N-hydroxy-2-[N-substituted-N-[(2-substituted-pyrrolidin-1-yl)carbonyl]amino]acetamides, with Activity as Peptide Deformylase Inhibitors, Their Compositions and Methods of Use as Antimicrobials. U.S. Patent 20020119962, 29 August 2002. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ou, S.; Kwok, K.C.; Wang, Y.; Bao, H. An improved method to determine SH and –S–S– group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Naturforsch. 2004, 59, 293–296. [Google Scholar]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2006, 46, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Zupan, J.; Gasteiger, J. Neural Networks and Drug Design for Chemists, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Centner, V.; Massart, D.L.; de Noord, O.E.; de Jong, S.; Vandeginste, B.M.V.; Sterna, C. Elimination of uninformative variables for multivariate calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, A.; Drobek, L.; Dombek, V.; Bak, A. Modeling of experimental data on trace elements and organic compounds content in industrial waste dumps. Chemosphere 2016, 162, 189–198. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds 1–9c are available from author H. Pizova. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R | clogP | PSA (Å) | PV | AChE IC50 (μM) | BChE IC50 (μM) | SI * | tox. IC50 (μM) |
| 1 | H | 4.1632 | 49.446 | 148.212 | 105.88 ± 2.20 | 55.57 ± 1.01 | 1.91 | >30 |
| 2a | 2-OCH3 | 3.6486 | 51.491 | 160.579 | 130.99 ± 1.29 | 51.53 ± 0.75 | 2.54 | >30 |
| 2b | 3-OCH3 | 4.2386 | 61.506 | 165.704 | 126.13 ± 2.79 | 58.47 ± 0.42 | 2.16 | >30 |
| 2c | 4-OCH3 | 4.2386 | 61.876 | 148.051 | 109.36 ± 0.22 | 117.40 ± 0.45 | 0.93 | >30 |
| 3a | 2-CH3 | 4.0122 | 45.255 | 141.705 | 61.94 ± 1.35 | 98.27 ± 5.88 | 0.63 | >30 |
| 3b | 3-CH3 | 4.6622 | 49.460 | 129.946 | 81.06 ± 0.03 | 114.84 ± 4.83 | 0.71 | >30 |
| 3c | 4-CH3 | 4.6622 | 49.466 | 132.090 | 123.41 ± 0.44 | 153.11 ± 2.29 | 0.81 | >30 |
| 4a | 2-F | 3.9638 | 48.435 | 144.032 | 51.87 ± 0.76 | 59.97 ± 0.00 | 0.86 | >30 |
| 4b | 3-F | 4.5638 | 49.474 | 131.146 | 69.88 ± 0.21 | 34.61 ± 0.11 | 2.02 | >30 |
| 4c | 4-F | 4.5638 | 49.441 | 144.623 | 87.23 ± 1.00 | 58.14 ± 1.39 | 1.50 | >30 |
| 5a | 2-Cl | 4.2838 | 48.725 | 135.310 | 46.35 ± 1.05 | 53.47 ± 0.52 | 0.87 | >30 |
| 5b | 3-Cl | 5.1338 | 49.486 | 134.686 | 48.27 ± 0.11 | 64.20 ± 7.14 | 0.75 | >30 |
| 5c | 4-Cl | 5.1338 | 49.435 | 133.841 | 77.34 ± 3.80 | 147.34 ± 1.18 | 0.52 | >30 |
| 6a | 2-Br | 4.4038 | 49.183 | 137.829 | 84.14 ± 4.39 | 27.38 ± 0.85 | 3.07 | >30 |
| 6b | 3-Br | 5.2838 | 49.440 | 137.166 | 74.16 ± 0.20 | 47.68 ± 0.08 | 1.56 | >30 |
| 6c | 4-Br | 5.2838 | 49.462 | 133.509 | 81.32 ± 0.67 | 28.21 ± 0.50 | 2.88 | >30 |
| 7a | 2-CF3 | 4.0470 | 42.920 | 142.558 | 73.24 ± 2.88 | 44.89 ± 1.66 | 1.63 | >30 |
| 7b | 3-CF3 | 5.4970 | 49.450 | 118.742 | 68.91 ± 0.06 | 39.46 ± 1.40 | 1.75 | >30 |
| 7c | 4-CF3 | 5.4970 | 49.438 | 124.821 | 88.65 ± 1.60 | 75.48 ± 2.94 | 1.17 | >30 |
| 8a | 2-NO2 | 0.8712 | 110.799 | 195.483 | 152.21 ± 0.01 | 48.69 ± 0.80 | 3.13 | >30 |
| 8b | 3-NO2 | 0.8712 | 134.854 | 207.158 | 64.99 ± 0.97 | 55.93 ± 0.63 | 1.16 | >30 |
| 8c | 4-NO2 | 0.8712 | 134.702 | 209.563 | 84.52 ± 0.06 | 58.75 ± 0.27 | 1.44 | >30 |
| 9a | 2-OH | 3.7262 | 93.856 | 164.611 | 153.40 ± 9.97 | 42.01 ± 1.26 | 3.65 | >30 |
| 9b | 3-OH | 3.4962 | 105.477 | 172.921 | 122.00 ± 1.38 | 86.22 ± 0.40 | 1.41 | >30 |
| 9c | 4-OH | 3.4962 | 105.171 | 173.231 | 91.08 ± 0.27 | 132.97 ± 0.39 | 0.68 | >30 |
| RIV | – | – | – | – | 50.10 ± 3.08 | 19.95 ± 0.31 | 25.11 | – |
| GLT | – | – | – | – | 4.0 ± 0.13 | 7.96 ± 0.59 | 0.50 | – |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizova, H.; Havelkova, M.; Bobal, P.; Stepankova, S.; Kauerova, T.; Bak, A.; Kollar, P.; Kozik, V.; Oravec, M.; Imramovsky, A.; et al. Proline-Based Carbamates as Cholinesterase Inhibitors. Molecules 2017, 22, 1969. https://doi.org/10.3390/molecules22111969
Pizova H, Havelkova M, Bobal P, Stepankova S, Kauerova T, Bak A, Kollar P, Kozik V, Oravec M, Imramovsky A, et al. Proline-Based Carbamates as Cholinesterase Inhibitors. Molecules. 2017; 22(11):1969. https://doi.org/10.3390/molecules22111969
Chicago/Turabian StylePizova, Hana, Marketa Havelkova, Pavel Bobal, Sarka Stepankova, Tereza Kauerova, Andrzej Bak, Peter Kollar, Violetta Kozik, Michal Oravec, Ales Imramovsky, and et al. 2017. "Proline-Based Carbamates as Cholinesterase Inhibitors" Molecules 22, no. 11: 1969. https://doi.org/10.3390/molecules22111969
APA StylePizova, H., Havelkova, M., Bobal, P., Stepankova, S., Kauerova, T., Bak, A., Kollar, P., Kozik, V., Oravec, M., Imramovsky, A., & Jampilek, J. (2017). Proline-Based Carbamates as Cholinesterase Inhibitors. Molecules, 22(11), 1969. https://doi.org/10.3390/molecules22111969