Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions



Abstract
:
1. Introduction
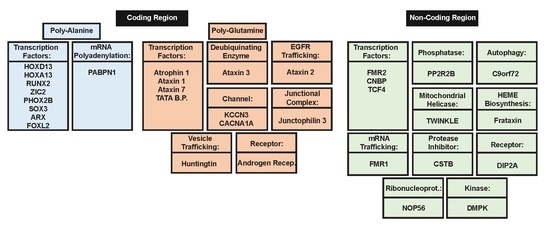
2. Polyalanine Repeat Expansions
2.1. Molecular Mechanisms of Poly-Ala Expansion Diseases
2.2. Genes Associated with Poly-Ala Expansion Diseases
3. Poly-Glutamine Repeat Expansions
3.1. Molecular Mechanisms of PolyQ Repeat Expansion Diseases
3.2. Genes Associated with PolyQ Expansion Diseases
4. Non-Coding Region Repeat Expansion
4.1. Molecular Mechanisms of Diseases Associated with Non-Coding Region Repeat Expansions
4.2. Genes Associated with Non-Coding Region Repeat Expansions
5. Intrinsic Disorder in Proteins Associated with Pathological Repeat Expansions
5.1. Disorder in Proteins Associated with Poly-Ala Expansion Diseases
5.2. Disorder in Proteins Associated with PolyQ Expansion Diseases
5.3. Disorder in Proteins Associated with Non-Coding Region Repeat Expansions
6. Summary
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 2010, 102, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Kissinger, C.R.; Villafranca, J.E.; Garner, E.; Guilliot, S.; Dunker, A.K. Thousands of proteins likely to have long disordered regions. Pac. Symp. Biocomput. 1998, 3, 437–448. [Google Scholar]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 161–171. [Google Scholar] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z. The protein trinity—Linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your id: Intrinsic disorder as an id for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Bondos, S.E.; Huang, F.; Oldfield, C.J. Intrinsically disordered proteins and multicellular organisms. Semin. Cell Dev. Biol. 2015, 37, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxreiter, M.; Toth-Petroczy, A.; Kraut, D.A.; Matouschek, A.; Lim, R.Y.; Xue, B.; Kurgan, L.; Uversky, V.N. Disordered proteinaceous machines. Chem. Rev. 2014, 114, 6806–6843. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Jakob, U.; Kriwacki, R.; Uversky, V.N. Conditionally and transiently disordered proteins: Awakening cryptic disorder to regulate protein function. Chem. Rev. 2014, 114, 6779–6805. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Oldfield, C.; Meng, J.; Huang, F.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Intrinsic protein disorder and protein-protein interactions. Pac. Symp. Biocomput. 2012, 116–127. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14–3-3 with their partners. BMC Genom. 2008, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.E. Nucleotide binding proteins. In Molecular Mechanism of Biological Recognition; Balaban, M., Ed.; Elsevier/North-Holland Biomedical Press: New York, NY, USA, 1979; pp. 79–94. [Google Scholar]
- Ng, K.P.; Potikyan, G.; Savene, R.O.; Denny, C.T.; Uversky, V.N.; Lee, K.A. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in scaffold proteins: Getting more from less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The triple power of D(3): Protein intrinsic disorder in degenerative diseases. Front. Biosci. (Landmark Ed.) 2014, 19, 181–258. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. (Landmark Ed.) 2009, 14, 5188–5238. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 89–100. [Google Scholar] [CrossRef]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Jorda, J.; Xue, B.; Uversky, V.N.; Kajava, A.V. Protein tandem repeats—The more perfect, the less structured. FEBS J. 2010, 277, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Hancock, J.M. Tandem and cryptic amino acid repeats accumulate in disordered regions of proteins. Genome Biol. 2009, 10, R59. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins evolve by repeat expansion. BioEssays 2003, 25, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.M.; Guigo, R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 2004, 14, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Kalita, M.K.; Ramasamy, G.; Duraisamy, S.; Chauhan, V.S.; Gupta, D. Protrepeatsdb: A database of amino acid repeats in genomes. BMC Bioinform. 2006, 7, 336. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Wang, J.; Malovannaya, A.; Xi, Y.; Li, W.; Guerra, R.; Hawke, D.H.; Qin, J.; Chen, J. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 2015, 11, 775. [Google Scholar] [CrossRef] [PubMed]
- Mier, P.; Alanis-Lobato, G.; Andrade-Navarro, M.A. Context characterization of amino acid homorepeats using evolution, position, and order. Proteins 2017, 85, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V. Tandem repeats in proteins: From sequence to structure. J. Struct. Biol. 2012, 179, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010, 11, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K. The biological effects of simple tandem repeats: Lessons from the repeat expansion diseases. Genome Res. 2008, 18, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Koshy, B.T.; Zoghbi, H.Y. The CAG/polyglutamine tract diseases: Gene products and molecular pathogenesis. Brain Pathol. 1997, 7, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.T., Jr.; Warren, S.T. Trinucleotide repeat expansion and human disease. Annu. Rev. Genet. 1995, 29, 703–728. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, N.J. Genetic anticipation. Expanding tandem repeats. Neurol. Clin. 1994, 12, 683–697. [Google Scholar] [PubMed]
- La Spada, A.R. Trinucleotide repeat instability: Genetic features and molecular mechanisms. Brain Pathol. 1997, 7, 943–963. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E. Repeat associated non-atg translation initiation: One DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 2011, 7, e1002018. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Xin, Q.; Li, L.; Li, J.; Qiu, R.; Guo, C.; Gong, Y.; Liu, Q. Eight-alanine duplication in homeobox d13 in a chinese family with synpolydactyly. Gene 2012, 499, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A.N.; Kornak, U.; Boddrich, A.; Suring, K.; Robinson, P.N.; Stiege, A.C.; Lurz, R.; Stricker, S.; Wanker, E.E.; Mundlos, S. A molecular pathogenesis for transcription factor associated poly-alanine tract expansions. Hum. Mol. Genet. 2004, 13, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Muragaki, Y.; Mundlos, S.; Upton, J.; Olsen, B.R. Altered growth and branching patterns in synpolydactyly caused by mutations in hoxd13. Science 1996, 272, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Utsch, B.; Becker, K.; Brock, D.; Lentze, M.J.; Bidlingmaier, F.; Ludwig, M. A novel stable polyalanine [poly(A)] expansion in the HOXA13 gene associated with hand-foot-genital syndrome: Proper function of poly(A)-harbouring transcription factors depends on a critical repeat length? Hum. Genet. 2002, 110, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Machida, J.; Yamaguchi, S.; Kimura, M.; Tatematsu, T.; Miyachi, H.; Matsushita, M.; Kitoh, H.; Ishiguro, N.; Nakayama, A.; et al. Characterisation of novel runx2 mutation with alanine tract expansion from japanese cleidocranial dysplasia patient. Mutagenesis 2016, 31, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, A.D.; Schrander-Stumpel, C.T.; Tserpelis, D.C.; Spee, M.K.; Stegmann, A.P.; Mancini, G.M.; Brooks, A.S.; Collee, M.; Maat-Kievit, A.; Simon, M.E.; et al. The unfolding clinical spectrum of holoprosencephaly due to mutations in shh, zic2, six3 and tgif genes. Eur. J. Hum. Genet. 2010, 18, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M., Jr. Holoprosencephaly: Clinical, anatomic, and molecular dimensions. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Klaskova, E.; Drabek, J.; Hobzova, M.; Smolka, V.; Seda, M.; Hyjanek, J.; Slavkovsky, R.; Stranska, J.; Prochazka, M. Significant phenotype variability of congenital central hypoventilation syndrome in a family with polyalanine expansion mutation of the PHOX2B gene. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2016, 160, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Di Lascio, S.; Belperio, D.; Benfante, R.; Fornasari, D. Alanine expansions associated with congenital central hypoventilation syndrome impair PHOX2B homeodomain-mediated dimerization and nuclear import. J. Biol. Chem. 2016, 291, 13375–13393. [Google Scholar] [CrossRef] [PubMed]
- Bachetti, T.; Di Duca, M.; Della Monica, M.; Grappone, L.; Scarano, G.; Ceccherini, I. Recurrence of CCHS associated PHOX2B poly-alanine expansion mutation due to maternal mosaicism. Pediatr. Pulmonol. 2014, 49, E45–E47. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Farlie, P.; Holbert, S.; Lockhart, P.; Thomas, P.Q. Polyalanine expansion mutations in the X-linked hypopituitarism gene SOX3 result in aggresome formation and impaired transactivation. Front. Biosci. 2007, 12, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A.; Mundlos, S. The other trinucleotide repeat: Polyalanine expansion disorders. Curr. Opin. Genet. Dev. 2005, 15, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Cossee, M.; Faivre, L.; Philippe, C.; Hichri, H.; de Saint-Martin, A.; Laugel, V.; Bahi-Buisson, N.; Lemaitre, J.F.; Leheup, B.; Delobel, B.; et al. Arx polyalanine expansions are highly implicated in familial cases of mental retardation with infantile epilepsy and/or hand dystonia. Am. J. Med. Genet. Part A 2011, 155, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Lisik, M.; Sieron, A.L. [Arx—One gene—Many phenotypes]. Neurologia i Neurochirurgia Polska 2008, 42, 338–344. [Google Scholar] [PubMed]
- Fan, J.; Zhou, Y.; Huang, X.; Zhang, L.; Yao, Y.; Song, X.; Chen, J.; Hu, J.; Ge, S.; Song, H.; et al. The combination of polyalanine expansion mutation and a novel missense substitution in transcription factor FOXL2 leads to different ovarian phenotypes in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) patients. Hum. Reprod. (Oxf. Engl.) 2012, 27, 3347–3357. [Google Scholar] [CrossRef] [PubMed]
- Beysen, D.; De Paepe, A.; De Baere, E. Foxl2 mutations and genomic rearrangements in BPES. Hum. Mutat. 2009, 30, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Schroder, R.; Zierz, S. Gcg repeats and phenotype in oculopharyngeal muscular dystrophy. Muscle Nerve 2001, 24, 120–122. [Google Scholar] [CrossRef]
- Brais, B. Oculopharyngeal muscular dystrophy: A late-onset polyalanine disease. Cytogenet. Genome Res. 2003, 100, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, M.; Rankovic, V.; Tarasjev, A.; Orolicki, S.; Damjanovic, A.; Paunovic, V.R.; Romac, S. Schizophrenia and polymorphic CAG repeats array of calcium-activated potassium channel (KCNN3) gene in serbian population. Int. J. Neurosci. 2006, 116, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; O’Hearn, E.; Rosenblatt, A.; Callahan, C.; Hwang, H.S.; Ingersoll-Ashworth, R.G.; Fleisher, A.; Stevanin, G.; Brice, A.; Potter, N.T.; et al. A repeat expansion in the gene encoding junctophilin-3 is associated with huntington disease-like 2. Nat. Genet. 2001, 29, 377–378. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.L.; Holmes, S.E.; Rosenblatt, A.; Gourley, L.; O’Hearn, E.; Ross, C.A.; Seltzer, W.K.; Walker, R.H.; Ashizawa, T.; Rasmussen, A.; et al. Huntington’s disease-like 2 (HDL2) in North America and Japan. Ann. Neurol. 2004, 56, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Paulson, H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 2010, 67, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Bork, P. Heat repeats in the huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Goehler, H.; Lalowski, M.; Stelzl, U.; Waelter, S.; Stroedicke, M.; Worm, U.; Droege, A.; Lindenberg, K.S.; Knoblich, M.; Haenig, C.; et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to huntington’s disease. Mol. Cell 2004, 15, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, S.; Yanagisawa, H.; Ohsaki, E.; Shirayama, T.; Tadokoro, K.; Inoue, T.; Yamada, M. Structure and expression of the gene responsible for the triplet repeat disorder, dentatorubral and pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 8, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T.; et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.S.; McCall, A.E.; Cota, J.; Subramony, S.H.; Orr, H.T.; Hughes, M.R.; Zoghbi, H.Y. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1995, 10, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.M.; Weber, C.; Mandel, J.L.; Cancel, G.; Abbas, N.; et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, direct. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Durr, A.; Stevanin, G.; Cancel, G.; Duyckaerts, C.; Abbas, N.; Didierjean, O.; Chneiweiss, H.; Benomar, A.; Lyon-Caen, O.; Julien, J.; et al. Spinocerebellar ataxia 3 and machado-joseph disease: Clinical, molecular, and neuropathological features. Ann. Neurol. 1996, 39, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Riess, O.; Schols, L.; Bottger, H.; Nolte, D.; Vieira-Saecker, A.M.; Schimming, C.; Kreuz, F.; Macek, M., Jr.; Krebsova, A.; Macek, M.S.; et al. Sca6 is caused by moderate CAG expansion in the alpha1a-voltage-dependent calcium channel gene. Hum. Mol. Genet. 1997, 6, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Jodice, C.; Mantuano, E.; Veneziano, L.; Trettel, F.; Sabbadini, G.; Calandriello, L.; Francia, A.; Spadaro, M.; Pierelli, F.; Salvi, F.; et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum. Mol. Genet. 1997, 6, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Abbas, N.; Stevanin, G.; Durr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E.; et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Durr, A.; Stevanin, G.; Cancel, G.; Abbas, N.; Benomar, A.; Belal, S.; Lebre, A.S.; Abada-Bendib, M.; Grid, D.; et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum. Mol. Genet. 1998, 7, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Jeong, S.Y.; Uchihara, T.; Anno, M.; Nagashima, K.; Nagashima, T.; Ikeda, S.; Tsuji, S.; Kanazawa, I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in tata-binding protein. Hum. Mol. Genet. 2001, 10, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Maltecca, F.; Filla, A.; Castaldo, I.; Coppola, G.; Fragassi, N.A.; Carella, M.; Bruni, A.; Cocozza, S.; Casari, G.; Servadio, A.; et al. Intergenerational instability and marked anticipation in SCA-17. Neurology 2003, 61, 1441–1443. [Google Scholar] [CrossRef] [PubMed]
- Fujigasaki, H.; Verma, I.C.; Camuzat, A.; Margolis, R.L.; Zander, C.; Lebre, A.S.; Jamot, L.; Saxena, R.; Anand, I.; Holmes, S.E.; et al. SCA12 is a rare locus for autosomal dominant cerebellar ataxia: A study of an indian family. Ann. Neurol. 2001, 49, 117–121. [Google Scholar] [CrossRef]
- Holmes, S.E.; O’Hearn, E.E.; McInnis, M.G.; Gorelick-Feldman, D.A.; Kleiderlein, J.J.; Callahan, C.; Kwak, N.G.; Ingersoll-Ashworth, R.G.; Sherr, M.; Sumner, A.J.; et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat. Genet. 1999, 23, 391–392. [Google Scholar] [PubMed]
- Gray, S.J.; Gerhardt, J.; Doerfler, W.; Small, L.E.; Fanning, E. An origin of DNA replication in the promoter region of the human fragile X mental retardation (FMR1) gene. Mol. Cell. Biol. 2007, 27, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Winnepenninckx, B.; Debacker, K.; Ramsay, J.; Smeets, D.; Smits, A.; FitzPatrick, D.R.; Kooy, R.F. Cgg-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12Q13.1. Am. J. Hum. Genet. 2007, 80, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gecz, J.; Bielby, S.; Sutherland, G.R.; Mulley, J.C. Gene structure and subcellular localization of FMR2, a member of a new family of putative transcription activators. Genomics 1997, 44, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Gecz, J.; Gedeon, A.K.; Sutherland, G.R.; Mulley, J.C. Identification of the gene FMR2, associated with fraxe mental retardation. Nat. Genet. 1996, 13, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Shen, Y.; Gibbs, R.A.; Nelson, D.L. Identification of FMR2, a novel gene associated with the fraxe CCG repeat and CPG island. Nat. Genet. 1996, 13, 109–113. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Grabczyk, E.; Usdin, K. The GAA*TTC triplet repeat expanded in friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000, 28, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ricker, K.; Jacobsen, J.F.; Rasmussen, L.J.; Dick, K.A.; Kress, W.; Schneider, C.; Koch, M.C.; Beilman, G.J.; Harrison, A.R.; et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology 2003, 60, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Fang, P.; Pearson, C.E.; Jayakar, P.; Ashizawa, T.; Roa, B.B.; Nelson, D.L. Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: Repeat purity as a disease modifier? Am. J. Hum. Genet. 2006, 78, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Yamagata, T.; Burgess, D.L.; Rasmussen, A.; Grewal, R.P.; Watase, K.; Khajavi, M.; McCall, A.E.; Davis, C.F.; Zu, L.; et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000, 26, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, M.; Stevanin, G.; Synofzik, M.; Monin, M.L.; Duyckaerts, C.; Sato, N.; Streichenberger, N.; Vighetto, A.; Desestret, V.; Tesson, C.; et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J. Neurol. Neurosurg. Psychiatry 2015, 86, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.V.; Hussain, I.; Cunnusamy, K.; Graham, E.; Gong, X.; Neelam, S.; Xing, C.; Kittler, R.; Petroll, W.M. TCF4 triplet repeat expansion and nuclear RNA foci in fuchs’ endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Okumura, N.; Nakagawa, H.; Koizumi, N.; Ikeda, Y.; Ueno, M.; Yoshii, K.; Adachi, H.; Aleff, R.A.; Butz, M.L.; et al. Trinucleotide repeat expansion in the tcf4 gene in fuchs’ endothelial corneal dystrophy in japanese. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4865–4869. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G., Jr.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3’ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Tsilfidis, C.; MacKenzie, A.E.; Mettler, G.; Barcelo, J.; Korneluk, R.G. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat. Genet. 1992, 1, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Worth, P.F.; Houlden, H.; Giunti, P.; Davis, M.B.; Wood, N.W. Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat. Genet. 2000, 24, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Dalton, J.C.; Moseley, M.L.; Gardner, K.L.; Bird, T.D.; Ashizawa, T.; Seltzer, W.K.; Pandolfo, M.; Milunsky, A.; Potter, N.T.; et al. Spinocerebellar ataxia type 8: Molecular genetic comparisons and haplotype analysis of 37 families with ataxia. Am. J. Hum. Genet. 2004, 75, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Daughters, R.S.; Ranum, L.P. Bidirectional expression of the SCA8 expansion mutation: One mutation, two genes. Cerebellum 2008, 7, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E. Identification of mutations in cystatin b, the gene responsible for the unverricht-lundborg type of progressive myoclonus epilepsy (epm1). Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar] [PubMed]
- Lalioti, M.D.; Scott, H.S.; Genton, P.; Grid, D.; Ouazzani, R.; M’Rabet, A.; Ibrahim, S.; Gouider, R.; Dravet, C.; Chkili, T.; et al. A pcr amplification method reveals instability of the dodecamer repeat in progressive myoclonus epilepsy (epm1) and no correlation between the size of the repeat and age at onset. Am. J. Hum. Genet. 1998, 62, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, T.; Walsh, I.; Martin, A.J.; Tosatto, S.C. Mobidb: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics 2012, 28, 2080–2081. [Google Scholar] [CrossRef] [PubMed]
- Potenza, E.; Domenico, T.D.; Walsh, I.; Tosatto, S.C. Mobidb 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2014, 43, D315–D320. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Trochet, D.; Clement-Ziza, M.; Munnich, A.; Lyonnet, S. Polyalanine expansions in human. Hum. Mol. Genet. 2004, 13, R235–R243. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Debeane, F.; Trinh, Q.D.; Turcotte, J.F.; Corbeil-Girard, L.P.; Dicaire, M.J.; Saint-Denis, A.; Page, M.; Rouleau, G.A.; Brais, B. Polymorphism, shared functions and convergent evolution of genes with sequences coding for polyalanine domains. Hum. Mol. Genet. 2003, 12, 2967–2979. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Kanemura, Y. Molecular biology of pediatric hydrocephalus and hydrocephalus-related diseases. Neurol. Med. Chir. 2015, 55, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, J.P.; Murphy, R.M. Length-dependent aggregation of uninterrupted polyalanine peptides. Biochemistry 2011, 50, 9200–9211. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Pavone, P.; Scapagnini, G.; Romeo, L.; Lombardo, I.; Li Volti, G.; Corsello, G.; Pavone, L. The aristaless (arx) gene: One gene for many “interneuronopathies”. Front. Biosci. (Elite Ed.) 2010, 2, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Sherr, E.H. The arx story (epilepsy, mental retardation, autism, and cerebral malformations): One gene leads to many phenotypes. Curr. Opin. Pediatr. 2003, 15, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Beysen, D.; Moumne, L.; Veitia, R.; Peters, H.; Leroy, B.P.; De Paepe, A.; De Baere, E. Missense mutations in the forkhead domain of FOXL2 lead to subcellular mislocalization, protein aggregation and impaired transactivation. Hum. Mol. Genet. 2008, 17, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Zoghbi, H.Y. Trinucleotide repeats: Mechanisms and pathophysiology. Annu. Rev. Genom. Hum. Genet. 2000, 1, 281–328. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Zoghbi, H.Y. Fourteen and counting: Unraveling trinucleotide repeat diseases. Hum. Mol. Genet. 2000, 9, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Fischbeck, K.H.; Souders, D.; La Spada, A. A candidate gene for x-linked spinal muscular atrophy. Adv. Neurol. 1991, 56, 209–213. [Google Scholar] [PubMed]
- Ferrigno, P.; Silver, P.A. Polyglutamine expansions: Proteolysis, chaperones, and the dangers of promiscuity. Neuron 2000, 26, 9–12. [Google Scholar] [CrossRef]
- La Spada, A.R.; Weydt, P.; Pineda, V.V. Frontiers in neuroscience huntington’s disease pathogenesis: Mechanisms and pathways. In Neurobiology of Huntington’s Disease: Applications to Drug Discovery; Lo, D.C., Hughes, R.E., Eds.; CRC Press/Taylor & Francis LLC.: Boca Raton, FL, USA, 2011. [Google Scholar]
- Brouwer, J.R.; Willemsen, R.; Oostra, B.A. Microsatellite repeat instability and neurological disease. BioEssays 2009, 31, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Li, X.J. Aggregation of n-terminal huntingtin is dependent on the length of its glutamine repeats. Hum. Mol. Genet. 1998, 7, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.; Preisinger, E.; Dranovsky, A.; Goldgaber, D.; Housman, D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11404–11409. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.K.; Paulson, H.L.; Pendse, S.J.; Saionz, S.J.; Bonini, N.M.; Pittman, R.N. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell Biol. 1998, 143, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hu, H.Y. Sequestration of cellular interacting partners by protein aggregates: Implication in a loss-of-function pathology. FEBS J. 2016, 283, 3705–3717. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.A.; Jiang, H.; Ross, C.A. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: Evidence for a compact beta-sheet structure. Hum. Mol. Genet. 2005, 14, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.J.; Cyr, D.M. Amyloid in neurodegenerative diseases: Friend or foe? Semin. Cell Dev. Biol. 2011, 22, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Orr, H.T. Polyglutamine diseases: Protein cleavage and aggregation. Curr. Opin. Neurobiol. 1999, 9, 566–570. [Google Scholar] [CrossRef]
- Ross, C.A.; Wood, J.D.; Schilling, G.; Peters, M.F.; Nucifora, F.C., Jr.; Cooper, J.K.; Sharp, A.H.; Margolis, R.L.; Borchelt, D.R. Polyglutamine pathogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Preisinger, E.; Jordan, B.M.; Kazantsev, A.; Housman, D. Evidence for a recruitment and sequestration mechanism in huntington’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E. Protein aggregation and pathogenesis of huntington’s disease: Mechanisms and correlations. Biol. Chem. 2000, 381, 937–942. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Taylor, J.P.; Taye, A.A.; Robitschek, J.; Li, M.; Walcott, J.; Merry, D.; Chai, Y.; Paulson, H.; Sobue, G.; et al. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 2000, 9, 2197–2202. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Fischbeck, K.H. Polyglutamine and CBP: Fatal attraction? Nat. Med. 2001, 7, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Berthelier, V.; Yang, W.; Wetzel, R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 2001, 311, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Berthelier, V.; Hamilton, J.B.; O’Nuallain, B.; Wetzel, R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 2002, 41, 7391–7399. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Pope, B.J.; Owen, D.; Wanker, E.E.; Scherzinger, E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of sup35 and of the amyloid beta-peptide of amyloid plaques. Proc. Natl. Acad. Sci. USA 2002, 99, 5596–5600. [Google Scholar] [CrossRef] [PubMed]
- Butland, S.L.; Devon, R.S.; Huang, Y.; Mead, C.L.; Meynert, A.M.; Neal, S.J.; Lee, S.S.; Wilkinson, A.; Yang, G.S.; Yuen, M.M.; et al. CAG-encoded polyglutamine length polymorphism in the human genome. BMC Genom. 2007, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Mitchell, C.; Essop, F.; Tager, S.; Temlett, J.; Stevanin, G.; Ross, C.; Rudnicki, D.; Margolis, R. Junctophilin 3 (JPH3) expansion mutations causing huntington disease like 2 (HDL2) are common in south african patients with african ancestry and a huntington disease phenotype. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Seixas, A.I.; Holmes, S.E.; Takeshima, H.; Pavlovich, A.; Sachs, N.; Pruitt, J.L.; Silveira, I.; Ross, C.A.; Margolis, R.L.; Rudnicki, D.D. Loss of junctophilin-3 contributes to huntington disease-like 2 pathogenesis. Ann. Neurol. 2012, 71, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.L.; Li, S.H.; Young, W.S.; Wagster, M.V.; Stine, O.C.; Kidwai, A.S.; Ashworth, R.G.; Ross, C.A. Drpla gene (atrophin-1) sequence and mRNA expression in human brain. Brain Res. Mol. Brain Res. 1996, 36, 219–226. [Google Scholar] [CrossRef]
- Giorgetti, E.; Lieberman, A.P. Polyglutamine androgen receptor-mediated neuromuscular disease. Cell. Mol. Life Sci. 2016, 73, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A. Spinal and bulbar muscular atrophy. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; Gene Reviews is a registered trademark of the University of Washington; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sun, Y.M.; Lu, C.; Wu, Z.Y. Spinocerebellar ataxia: Relationship between phenotype and genotype—A review. Clin. Genet. 2016, 90, 305–314. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J. Structural and functional alterations in the androgen receptor in spinal bulbar muscular atrophy. Biochem. Soc. Trans. 2001, 29, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Duenas, A.M.; Goold, R.; Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain 2006, 129, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, K.; Krzyzosiak, W.J. Patterns of CAG repeat interruptions in SCA1 and SCA2 genes in relation to repeat instability. Hum. Mutat. 2004, 24, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Tagawa, K.; Oka, T.; Sasabe, T.; Ito, H.; Shiwaku, H.; La Spada, A.R.; Okazawa, H. Ataxin-7 associates with microtubules and stabilizes the cytoskeletal network. Hum. Mol. Genet. 2012, 21, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Brunt, E.R.; de Vos, R.A.; Dijk, F.; van der Want, H.J.; Rub, U.; den Dunnen, W.F. The p62 antibody reveals various cytoplasmic protein aggregates in spinocerebellar ataxia type 6. Clin. Neuropathol. 2009, 28, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Chandy, K.G.; Fantino, E.; Wittekindt, O.; Kalman, K.; Tong, L.L.; Ho, T.H.; Gutman, G.A.; Crocq, M.A.; Ganguli, R.; Nimgaonkar, V.; et al. Isolation of a novel potassium channel gene HSKCA3 containing a polymorphic CAG repeat: A candidate for schizophrenia and bipolar disorder? Mol. Psychiatry 1998, 3, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.; Modai, I.; Ziv, H.; Amir, S.; Halperin, T.; Weizman, A.; Navon, R. An association of CAG repeats at the KCNN3 locus with symptom dimensions of schizophrenia. Biol. Psychiatry 2002, 51, 788–794. [Google Scholar] [CrossRef]
- Dror, V.; Shamir, E.; Ghanshani, S.; Kimhi, R.; Swartz, M.; Barak, Y.; Weizman, R.; Avivi, L.; Litmanovitch, T.; Fantino, E.; et al. HKCA3/KCNN3 potassium channel gene: Association of longer CAG repeats with schizophrenia in israeli ashkenazi jews, expression in human tissues and localization to chromosome 1q21. Mol. Psychiatry 1999, 4, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Gargus, J.J.; Fantino, E.; Gutman, G.A. A piece in the puzzle: An ion channel candidate gene for schizophrenia. Mol. Med. Today 1998, 4, 518–524. [Google Scholar] [CrossRef]
- Perutz, M.F. Glutamine repeats and inherited neurodegenerative diseases: Molecular aspects. Curr. Opin. Struct. Biol. 1996, 6, 848–858. [Google Scholar] [CrossRef]
- Ross, C.A. Polyglutamine pathogenesis: Emergence of unifying mechanisms for huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar] [CrossRef]
- Bates, G. Huntingtin aggregation and toxicity in huntington’s disease. Lancet 2003, 361, 1642–1644. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Serra, H.G.; Zoghbi, H.Y.; Orr, H.T. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum. Mol. Genet. 2001, 10, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef]
- Gusella, J.; MacDonald, M. No post-genetics era in human disease research. Nat. Rev. Genet. 2002, 3, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Tsuiji, H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016, 1647, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Pritchard, M.; Kremer, E.; Lynch, M.; Nancarrow, J.; Baker, E.; Holman, K.; Mulley, J.C.; Warren, S.T.; Schlessinger, D.; et al. Fragile x genotype characterized by an unstable region of DNA. Science 1991, 252, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Saul, R.A.; Tarleton, J.C. FMR1-related disorders. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; Gene Reviews is a registered trademark of the University of Washington; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Eberhart, D.E.; Malter, H.E.; Feng, Y.; Warren, S.T. The fragile x mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 1996, 5, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a cgg repeat coincident with a breakpoint cluster region exhibiting length variation in fragile x syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Knight, S.J.; Hirst, M.C.; Roche, A.; Christodoulou, Z.; Huson, S.M.; Winter, R.; Fitchett, M.; McKinley, M.J.; Lindenbaum, R.H.; Nakahori, Y.; et al. Molecular studies of the fragile x syndrome. Am. J. Med. Genet. 1992, 43, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J.; Hagerman, R.J. Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.; Webb, J.; Grimley, S.; Conway, G.; Jacobs, P. Studies of fraxa and fraxe in women with premature ovarian failure. J. Med. Genet. 1998, 35, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.K.; Marcus, M.; Epstein, M.P.; Allen, E.G.; Anido, A.E.; Paquin, J.J.; Yadav-Shah, M.; Sherman, S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. (Oxf. Engl.) 2005, 20, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Hayward, B.; Nakamura, A.J.; Bonner, W.M.; Usdin, K. Evidence for chromosome fragility at the frataxin locus in friedreich ataxia. Mutat. Res. 2015, 781, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9orf72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., 3rd; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to C9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9ORF72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9ORF72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic prn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–e1117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 2016, 167, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.; Day, J.W. Myotonic dystrophy: Clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr. Neurol. Neurosci. Rep. 2002, 2, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Abe, K.; Matsuura, T.; Ikeda, Y.; Hitomi, T.; Akechi, Y.; Habu, T.; Liu, W.; Okuda, H.; Koizumi, A. Expansion of intronic GGCCTG hexanucleotide repeat in nop56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011, 89, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalioti, M.D.; Scott, H.S.; Antonarakis, S.E. Altered spacing of promoter elements due to the dodecamer repeat expansion contributes to reduced expression of the cystatin B gene in EPM1. Hum. Mol. Genet. 1999, 8, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Virtaneva, K.; D’Amato, E.; Miao, J.; Koskiniemi, M.; Norio, R.; Avanzini, G.; Franceschetti, S.; Michelucci, R.; Tassinari, C.A.; Omer, S.; et al. Unstable minisatellite expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat. Genet. 1997, 15, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Paulson, H.L. Polyglutamine neurodegeneration: Protein misfolding revisited. Trends Neurosci. 2008, 31, 521–528. [Google Scholar] [CrossRef] [PubMed]
- O’Hearn, E.; Holmes, S.E.; Margolis, R.L. Spinocerebellar ataxia type 12. Handb. Clin. Neurol. 2012, 103, 535–547. [Google Scholar] [PubMed]
- Dong, Y.; Wu, J.J.; Wu, Z.Y. Identification of 46 CAG repeats within PPP2R2B as probably the shortest pathogenic allele for SCA12. Parkinsonism Relat. Disord. 2015, 21, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61 (Suppl. 7), 176–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)p(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. Iupred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. Prdos: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. Espritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. Anchor: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. Ronn: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. Globplot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. Disprot: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [PubMed]
- Toth-Petroczy, A.; Oldfield, C.J.; Simon, I.; Takagi, Y.; Dunker, A.K.; Uversky, V.N.; Fuxreiter, M. Malleable machines in transcription regulation: The mediator complex. PLoS Comput. Biol. 2008, 4, e1000243. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Uversky, V.N. Drugs for ‘protein clouds’: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Raynes, R.; Powell, C.; Xue, B.; Uversky, V.N. HSF transcription factor family, heat shock response, and protein intrinsic disorder. Curr. Protein Pept. Sci. 2012, 13, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006, 359, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Grzeschik, K.H. Synpolydactyly: Clinical and molecular advances. Clin. Genet. 2008, 73, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Cantile, M.; Franco, R.; Tschan, A.; Baumhoer, D.; Zlobec, I.; Schiavo, G.; Forte, I.; Bihl, M.; Liguori, G.; Botti, G.; et al. HOX D13 expression across 79 tumor tissue types. Int. J. Cancer 2009, 125, 1532–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantile, M.; Franco, R.; Forte, I.; Cerrone, M.; Anniciello, A.; Liguori, G.; Manna, A.; Corrado, A.; Aquino, G.; Terracciano, L.; et al. HOX D13 expression across 79 tumor tissue types and its prognostic role in pancreatic cancer. Virchows Arch. 2009, 455, 391. [Google Scholar]
- Lappin, T.R.; Grier, D.G.; Thompson, A.; Halliday, H.L. HOX genes: Seductive science, mysterious mechanisms. Ulster Med. J. 2006, 75, 23–31. [Google Scholar] [PubMed]
- Cantile, M.; Scognamiglio, G.; La Sala, L.; La Mantia, E.; Scaramuzza, V.; Valentino, E.; Tatangelo, F.; Losito, S.; Pezzullo, L.; Chiofalo, M.G.; et al. Aberrant expression of posterior HOX genes in well differentiated histotypes of thyroid cancers. Int. J. Mol. Sci. 2013, 14, 21727–21740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Liu, J.S.; Guo, S.C.; He, S.C.; Qiu, G.L.; Lu, J.; Wang, J.; Fan, L.; Zhao, W.; Che, X.M. Hottip and HOXA13 are oncogenes associated with gastric cancer progression. Oncol. Rep. 2016, 35, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Williams, M.E.; Innis, J.W. Range of HOX/TALE superclass associations and proteins domain requirements for HOXA13 : MEIS interaction. Dev. Biol. 2005, 277, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Tsai, C.J.; Nussinov, R. Analysis of ordered and disordered protein complexes reveals structural features discriminating between stable and unstable monomers. J. Mol. Biol. 2004, 341, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.H.; Panchenko, A.R. Intrinsic disorder and protein multibinding in domain, terminal, and linker regions. Mol. Biosyst. 2010, 6, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Hu, G.; Yang, J.Y.; Peng, Z.L.; Uversky, V.N.; Kurgan, L. In various protein complexes, disordered protomers have large per-residue surface areas and area of protein-, DNA- and RNA-binding interfaces. FEBS Lett. 2015, 589, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Morrison, N.A.; Stephens, A.S.; Osato, M.; Pasco, J.A.; Fozzard, N.; Stein, G.S.; Polly, P.; Griffiths, L.R.; Nicholson, G.C. Polyalanine repeat polymorphism in runx2 is associated with site-specific fracture in post-menopausal females. PLoS ONE 2013, 8, e72740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.P.; Wang, Y.Y.; He, L.; Huang, G.L.; Du, Y.; Zhang, G.; Yan, X.L.; Xia, P.Y.; Ye, B.Q.; Wang, S.; et al. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J. Clin. Investig. 2015, 125, 3795–3808. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Kanai, M.; Kijima, K.; Akaba, K.; Hashimoto, M.; Hasegawa, H.; Otaki, S.; Koizumi, T.; Kusuda, S.; Ogawa, Y.; et al. Molecular analysis of congenital central hypoventilation syndrome. Hum. Genet. 2003, 114, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Laudier, B.; Attie-Bitach, T.; Trang, H.; de Pontual, L.; Gener, B.; Trochet, D.; Etchevers, H.; Ray, P.; Simonneau, M.; et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 2003, 33, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; O’Brien, L.M.; Gozal, D.; Trang, H.; Nordenskjold, A.; Laudier, B.; Svensson, P.J.; Uhrig, S.; Cole, T.; Munnich, A.; et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am. J. Hum. Genet. 2005, 76, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Bourdeaut, F.; Trochet, D.; Janoueix-Lerosey, I.; Ribeiro, A.; Deville, A.; Coz, C.; Michiels, J.F.; Lyonnet, S.; Amiel, J.; Delattre, O. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Cancer Lett. 2005, 228, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Ronce, N.; Hamel, B.C.; Thomas, P.; Lespinasse, J.; Raynaud, M.; Paringaux, C.; Van Bokhoven, H.; Kalscheuer, V.; Fryns, J.P.; et al. Transcription factor SOX3 is involved in x-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet. 2002, 71, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.S.; Cundall, M.; Turton, J.; Rizotti, K.; Mehta, A.; Palmer, R.; Wong, J.; Chong, W.K.; Al-Zyoud, M.; El-Ali, M.; et al. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am. J. Hum. Genet. 2005, 76, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J. Clin. Investig. 2011, 121, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, R.W.; Jiang, Y.G.; Zou, Y.B.; Guo, W. Overexpression of SOX3 is associated with diminished prognosis in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2013, 20 (Suppl. 3), S459–S466. [Google Scholar] [CrossRef] [PubMed]
- Vural, B.; Chen, L.C.; Saip, P.; Chen, Y.T.; Ustuner, Z.; Gonen, M.; Simpson, A.J.; Old, L.J.; Ozbek, U.; Gure, A.O. Frequency of SOX group b (SOX1, 2, 3) and zic2 antibodies in turkish patients with small cell lung carcinoma and their correlation with clinical parameters. Cancer 2005, 103, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Trubetskoy, A.; Suzuki, T.; Jenkins, N.A.; Copeland, N.G.; Lenz, J. Genome-based identification of cancer genes by proviral tagging in mouse retrovirus-induced T-cell lymphomas. J. Virol. 2003, 77, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Papalopulu, N.; Vogt, P.K.; Li, J. The oncogenic potential of the high mobility group box protein SOX3. Cancer Res. 2000, 60, 6303–6306. [Google Scholar] [PubMed]
- Gure, A.O.; Stockert, E.; Scanlan, M.J.; Keresztes, R.S.; Jager, D.; Altorki, N.K.; Old, L.J.; Chen, Y.T. Serological identification of embryonic neural proteins as highly immunogenic tumor antigens in small cell lung cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 4198–4203. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, T.; Poirier, K.; Friocourt, G.; Bahi, N.; Beaumont, D.; Fauchereau, F.; Ben Jeema, L.; Zemni, R.; Vinet, M.C.; Francis, F.; et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in x-linked mental retardation. Hum. Mol. Genet. 2002, 11, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Das, S.; Petras, K.; Kitamura, K.; Morohashi, K.; Abuelo, D.N.; Barr, M.; Bonneau, D.; Brady, A.F.; Carpenter, N.J.; et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 2004, 23, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Partington, M.; Kerr, B.; Mangelsdorf, M.; Gecz, J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am. J. Med. Genet. 2002, 112, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Stromme, P.; Mangelsdorf, M.E.; Shaw, M.A.; Lower, K.M.; Lewis, S.M.; Bruyere, H.; Lutcherath, V.; Gedeon, A.K.; Wallace, R.H.; Scheffer, I.E.; et al. Mutations in the human ortholog of aristaless cause x-linked mental retardation and epilepsy. Nat. Genet. 2002, 30, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Yanazawa, M.; Sugiyama, N.; Miura, H.; Iizuka-Kogo, A.; Kusaka, M.; Omichi, K.; Suzuki, R.; Kato-Fukui, Y.; Kamiirisa, K.; et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and x-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 2002, 32, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Stromme, P.; Bakke, S.J.; Dahl, A.; Gecz, J. Brain cysts associated with mutation in the aristaless related homeobox gene, ARX. J. Neurol. Neurosurg. Psychiatry 2003, 74, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Verdin, H.; De Baere, E. Blepharophimosis, ptosis, and epicanthus inversus. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Harris, S.E.; Chand, A.L.; Winship, I.M.; Gersak, K.; Aittomaki, K.; Shelling, A.N. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol. Hum. Reprod. 2002, 8, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Laissue, P.; Lakhal, B.; Benayoun, B.A.; Dipietromaria, A.; Braham, R.; Elghezal, H.; Philibert, P.; Saad, A.; Sultan, C.; Fellous, M.; et al. Functional evidence implicating FOXL2 in non-syndromic premature ovarian failure and in the regulation of the transcription factor osr2. J. Med. Genet. 2009, 46, 455–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, P.J.; Leung, D.; Chu, S. Genetics and genomics of ovarian sex cord-stromal tumors. Clin. Genet. 2017, 91, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zhou, D.; Tong, S.; Gao, Y.; Teng, M.; Niu, L. Crystal structure and possible dimerization of the single RRM of human PABPN1. Proteins 2008, 71, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluijs, B.M.; van Engelen, B.G.; Hoefsloot, L.H. Oculopharyngeal muscular dystrophy (OPMD) due to a small duplication in the PABPN1 gene. Hum. Mutat. 2003, 21, 553. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Kanto, K.; Hatakeyama, K.; Ide, T.; Wakabayashi-Nakao, K.; Watanabe, Y.; Sakura, N.; Terashima, M.; Yamaguchi, K.; Mochizuki, T. Exosome-mediated extracellular release of polyadenylate-binding protein 1 in human metastatic duodenal cancer cells. Proteomics 2014, 14, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, J.; Watanabe, K.; Sano, A.; Nagase, T.; Nakajima, J.; Fukayama, M.; Yatomi, Y.; Ohishi, N.; Takai, D. Alternative polyadenylation is associated with lower expression of PABPN1 and poor prognosis in non-small cell lung cancer. Cancer Sci. 2014, 105, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Grube, S.; Gerchen, M.F.; Adamcio, B.; Pardo, L.A.; Martin, S.; Malzahn, D.; Papiol, S.; Begemann, M.; Ribbe, K.; Friedrichs, H.; et al. A CAG repeat polymorphism of KCNN3 predicts SK3 channel function and cognitive performance in schizophrenia. EMBO Mol. Med. 2011, 3, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mab action by the alkyl-lipid ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed]
- Steinestel, K.; Eder, S.; Ehinger, K.; Schneider, J.; Genze, F.; Winkler, E.; Wardelmann, E.; Schrader, A.J.; Steinestel, J. The small conductance calcium-activated potassium channel 3 (SK3) is a molecular target for Edelfosine to reduce the invasive potential of urothelial carcinoma cells. Tumour Biol. 2016, 37, 6275–6283. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, L.; Gueguinou, M.; Potier-Cartereau, M.; Vandecasteele, G.; Bougnoux, P.; Chevalier, S.; Chantome, A.; Vandier, C. cAMP-PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3-Orai1 complex. Pflugers Arch. 2014, 466, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [PubMed]
- Stevanin, G.; Camuzat, A.; Holmes, S.E.; Julien, C.; Sahloul, R.; Dode, C.; Hahn-Barma, V.; Ross, C.A.; Margolis, R.L.; Durr, A.; et al. CAG/CTG repeat expansions at the huntington’s disease-like 2 locus are rare in huntington’s disease patients. Neurology 2002, 58, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.H.; Rasmussen, A.; Rudnicki, D.; Holmes, S.E.; Alonso, E.; Matsuura, T.; Ashizawa, T.; Davidoff-Feldman, B.; Margolis, R.L. Huntington’s disease—Like 2 can present as chorea-acanthocytosis. Neurology 2003, 61, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Faber, P.W.; Barnes, G.T.; Srinidhi, J.; Chen, J.; Gusella, J.F.; MacDonald, M.E. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Boutell, J.M.; Thomas, P.; Neal, J.W.; Weston, V.J.; Duce, J.; Harper, P.S.; Jones, A.L. Aberrant interactions of transcriptional repressor proteins with the huntington’s disease gene product, huntingtin. Hum. Mol. Genet. 1999, 8, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Schaeper, U.; Boyd, J.M.; Verma, S.; Uhlmann, E.; Subramanian, T.; Chinnadurai, G. Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in negative modulation of oncogenic transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 10467–10471. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Majumder, P.; Sarkar, S.; Giri, K.; Mukhopadhyay, D.; Bhattacharyya, N.P. Huntingtin interacting protein HYPK is intrinsically unstructured. Proteins 2008, 71, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Nopoulos, P.C. Huntington disease: A single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar] [PubMed]
- Onodera, O.; Oyake, M.; Takano, H.; Ikeuchi, T.; Igarashi, S.; Tsuji, S. Molecular cloning of a full-length cDNA for dentatorubral-pallidoluysian atrophy and regional expressions of the expanded alleles in the CNS. Am. J. Hum. Genet. 1995, 57, 1050–1060. [Google Scholar] [PubMed]
- Yazawa, I.; Nukina, N.; Hashida, H.; Goto, J.; Yamada, M.; Kanazawa, I. Abnormal gene product identified in hereditary dentatorubral-pallidoluysian atrophy (DRPLA) brain. Nat. Genet. 1995, 10, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Sibinga, N.E. Atrophin proteins interact with the fat1 cadherin and regulate migration and orientation in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 6955–6965. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Yazawa, I. Pathological accumulation of Atrophin-1 in dentatorubralpallidoluysian atrophy. Int. J. Clin. Exp. Pathol. 2011, 4, 378–384. [Google Scholar] [PubMed]
- Chen, H.; Fang, Y.; Zhu, H.; Li, S.; Wang, T.; Gu, P.; Fang, X.; Wu, Y.; Liang, J.; Zeng, Y.; et al. Protein-protein interaction analysis of distinct molecular pathways in two subtypes of colorectal carcinoma. Mol. Med. Rep. 2014, 10, 2868–2874. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Denayer, S.; Van Tilborgh, N.; Kerkhofs, S.; Helsen, C.; Haelens, A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl. Recept. Signal. 2008, 6, e008. [Google Scholar] [CrossRef] [PubMed]
- Lavery, D.N.; McEwan, I.J. Structural characterization of the native NH2-terminal transactivation domain of the human androgen receptor: A collapsed disordered conformation underlies structural plasticity and protein-induced folding. Biochemistry 2008, 47, 3360–3369. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J.; Lavery, D.; Fischer, K.; Watt, K. Natural disordered sequences in the amino terminal domain of nuclear receptors: Lessons from the androgen and glucocorticoid receptors. Nucl. Recept. Signal. 2007, 5, e001. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Rousso, E.; Anheim, M.; Cossee, M.; Tranchant, C. A family with early-onset and rapidly progressive x-linked spinal and bulbar muscular atrophy. Neurology 2005, 64, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.C.; Plaga, A.R.; Shankar, E.; Gupta, S. Androgen receptor-related diseases: What do we know? Andrology 2016, 4, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Gui, H.; Jin, F.; Heck, B.W.; Lin, P.; Ma, J.; Fondell, J.D.; Tsai, C.C. Ataxin-1 and brother of ataxin-1 are components of the notch signalling pathway. EMBO Rep. 2011, 12, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Kim, S.J.; Ka, S.; Choi, I.; Kang, S. USP7, a ubiquitin-specific protease, interacts with ataxin-1, the SCA1 gene product. Mol. Cell. Neurosci. 2002, 20, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Allen, M.D.; Veprintsev, D.B.; Lowe, J.; Bycroft, M. The structure of the AXH domain of spinocerebellar ataxin-1. J. Biol. Chem. 2004, 279, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Servadio, A.; Koshy, B.; Armstrong, D.; Antalffy, B.; Orr, H.T.; Zoghbi, H.Y. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat. Genet. 1995, 10, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.R.; An, H.T.; Ko, J.; Kang, S. Ataxin-1 regulates epithelial-mesenchymal transition of cervical cancer cells. Oncotarget 2017, 8, 18248–18259. [Google Scholar] [CrossRef] [PubMed]
- Nonis, D.; Schmidt, M.H.; van de Loo, S.; Eich, F.; Dikic, I.; Nowock, J.; Auburger, G. Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell. Signal. 2008, 20, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Golatta, M.; Wullner, U.; Lengauer, T. Structural and functional analysis of ataxin-2 and ataxin-3. Eur. J. Biochem. 2004, 271, 3155–3170. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Nkiliza, A.; Chartier-Harlin, M.C. ATXN2 a culprit with multiple facets. Oncotarget 2017, 8, 34028. [Google Scholar] [CrossRef] [PubMed]
- Nkiliza, A.; Mutez, E.; Simonin, C.; Lepretre, F.; Duflot, A.; Figeac, M.; Villenet, C.; Semaille, P.; Comptdaer, T.; Genet, A.; et al. RNA-binding disturbances as a continuum from spinocerebellar ataxia type 2 to parkinson disease. Neurobiol. Dis. 2016, 96, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.N.; Loomis, S.J.; Kang, J.H.; Allingham, R.R.; Gharahkhani, P.; Khor, C.C.; Burdon, K.P.; Aschard, H.; Chasman, D.I.; Igo, R.P., Jr.; et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat. Genet. 2016, 48, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, R.; Westermann, F.; Wittke, I.; Nowock, J.; Schwab, M. Ataxin-2 promotes apoptosis of human neuroblastoma cells. Oncogene 2003, 22, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Senic-Matuglia, F.; Di Fiore, P.P.; Polo, S.; Hodsdon, M.E.; De Camilli, P. Deubiquitinating function of ataxin-3: Insights from the solution structure of the Josephin Domain. Proc. Natl. Acad. Sci. USA 2005, 102, 12700–12705. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Gong, L.; Williams, A.J.; Sakai, N.; Todi, S.V.; Paulson, H.L. JOSD1, a membrane-targeted deubiquitinating enzyme, is activated by ubiquitination and regulates membrane dynamics, cell motility, and endocytosis. J. Biol. Chem. 2013, 288, 17145–17155. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.; Breuer, P. Josephin Domain-containing proteins from a variety of species are active de-ubiquitination enzymes. Biol. Chem. 2007, 388, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Macfarlan, T.; Pittman, R.N.; Chakravarti, D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J. Biol. Chem. 2002, 277, 45004–45012. [Google Scholar] [CrossRef] [PubMed]
- Masino, L.; Musi, V.; Menon, R.P.; Fusi, P.; Kelly, G.; Frenkiel, T.A.; Trottier, Y.; Pastore, A. Domain architecture of the polyglutamine protein ataxin-3: A globular domain followed by a flexible tail. FEBS Lett. 2003, 549, 21–25. [Google Scholar] [CrossRef]
- Albrecht, M.; Hoffmann, D.; Evert, B.O.; Schmitt, I.; Wullner, U.; Lengauer, T. Structural modeling of ataxin-3 reveals distant homology to adaptins. Proteins 2003, 50, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.X.; Tang, Y.; Ma, Y. Ataxin-3 expression correlates with the clinicopathologic features of gastric cancer. Int. J. Clin. Exp. Med. 2014, 7, 973–981. [Google Scholar] [PubMed]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1a-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Terwindt, G.; Kors, E.; Haan, J.; Vermeulen, F.; Van den Maagdenberg, A.; Frants, R.; Ferrari, M. Mutation analysis of the CACNA1A calcium channel subunit gene in 27 patients with sporadic hemiplegic migraine. Arch. Neurol. 2002, 59, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene cacnl1a4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- Carrera, P.; Piatti, M.; Stenirri, S.; Grimaldi, L.M.; Marchioni, E.; Curcio, M.; Righetti, P.G.; Ferrari, M.; Gelfi, C. Genetic heterogeneity in italian families with familial hemiplegic migraine. Neurology 1999, 53, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillon, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.G.; Tournier-Lasserve, E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Stam, A.H.; Vanmolkot, K.R.; Kremer, H.P.; Gartner, J.; Brown, J.; Leshinsky-Silver, E.; Gilad, R.; Kors, E.E.; Frankhuizen, W.S.; Ginjaar, H.B.; et al. CACNA1A R1347Q: A frequent recurrent mutation in hemiplegic migraine. Clin. Genet. 2008, 74, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 2016, 99, 287–298.
- Reinson, K.; Oiglane-Shlik, E.; Talvik, I.; Vaher, U.; Ounapuu, A.; Ennok, M.; Teek, R.; Pajusalu, S.; Murumets, U.; Tomberg, T.; et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am. J. Med. Genet. A 2016, 170, 2173–2176. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.; Ozaki, M.; Mizoguchi, T.; Allingham, R.R.; Li, Z.; Haripriya, A.; Nakano, S.; Uebe, S.; Harder, J.M.; Chan, A.S.; et al. A common variant mapping to CACNA1A is associated with susceptibility to exfoliation syndrome. Nat. Genet. 2015, 47, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Lai, M.D.; Phan, N.N.; Sun, Z.; Lin, Y.C. Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [PubMed]
- Palhan, V.B.; Chen, S.; Peng, G.H.; Tjernberg, A.; Gamper, A.M.; Fan, Y.; Chait, B.T.; La Spada, A.R.; Roeder, R.G. Polyglutamine-expanded ataxin-7 inhibits staga histone acetyltransferase activity to produce retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 8472–8477. [Google Scholar] [CrossRef] [PubMed]
- Stevanin, G.; Durr, A.; Brice, A. Clinical and molecular advances in autosomal dominant cerebellar ataxias: From genotype to phenotype and physiopathology. Eur. J. Hum. Genet. 2000, 8, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Burwinkel, B.; Michailidou, K.; Arias-Perez, J.I.; Zamora, M.P.; Menendez-Rodriguez, P.; Hardisson, D.; Mendiola, M.; Gonzalez-Neira, A.; Pita, G.; et al. Common non-synonymous SNPs associated with breast cancer susceptibility: Findings from the breast cancer association consortium. Hum. Mol. Genet. 2014, 23, 6096–6111. [Google Scholar] [CrossRef] [PubMed]
- Kalvala, A.; Gao, L.; Aguila, B.; Dotts, K.; Rahman, M.; Nana-Sinkam, S.P.; Zhou, X.; Wang, Q.E.; Amann, J.; Otterson, G.A.; et al. Rad51C-ATXN7 fusion gene expression in colorectal tumors. Mol. Cancer 2016, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Bharti, B.; Mishra, R. Spleen-specific isoforms of Pax5 and ataxin-7 as potential proteomic markers of lymphoma-affected spleen. Mol. Cell. Biochem. 2015, 402, 181–191. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Orphanides, G.; Lane, W.S.; Reinberg, D. Requirement of RSF and fact for transcription of chromatin templates in vitro. Science 1998, 282, 1900–1904. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.; Sinn, E.; Yamamoto, T.; Wang, J.; Roy, A.; Horikoshi, M.; Roeder, R.G. Highly conserved core domain and unique N terminus with presumptive regulatory motifs in a human tata factor (TFIID). Nature 1990, 346, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.G.; Tanese, N.; Pugh, B.F.; Tjian, R. Functional domains and upstream activation properties of cloned human TATA binding protein. Science 1990, 248, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.C.; Lieberman, P.M.; Schmidt, M.C.; Zhou, Q.; Pei, R.; Berk, A.J. Cloning of a transcriptionally active human TATA binding factor. Science 1990, 248, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Gouge, J.; Satia, K.; Guthertz, N.; Widya, M.; Thompson, A.J.; Cousin, P.; Dergai, O.; Hernandez, N.; Vannini, A. Redox signaling by the RNA polymerase III TFIIB-related factor Brf2. Cell 2015, 163, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.K.; Panov, K.I.; Cabart, P.; Russell, J.; Zomerdijk, J.C. TBP-TAF complex SL1 directs Rna polymerase i pre-initiation complex formation and stabilizes upstream binding factor at the rDNA promoter. J. Biol. Chem. 2005, 280, 29551–29558. [Google Scholar] [CrossRef] [PubMed]
- Hochheimer, A.; Tjian, R. Diversified transcription initiation complexes expand promoter selectivity and tissue-specific gene expression. Genes Dev. 2003, 17, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.J.; Shah, A.G.; Fang, Z.H.; Ward, E.G.; Warren, S.T.; Li, S.; Li, X.J. Polyglutamine domain modulates the TBP-TFIIB interaction: Implications for its normal function and neurodegeneration. Nat. Neurosci. 2007, 10, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K. The TATA box binding protein. Curr. Opin. Struct. Biol. 1996, 6, 69–75. [Google Scholar] [CrossRef]
- Lescure, A.; Lutz, Y.; Eberhard, D.; Jacq, X.; Krol, A.; Grummt, I.; Davidson, I.; Chambon, P.; Tora, L. The N-terminal domain of the human TATA-binding protein plays a role in transcription from TATA-containing RNA polymerase II and III promoters. EMBO J. 1994, 13, 1166–1175. [Google Scholar] [PubMed]
- Kenny, P.J.; Zhou, H.; Kim, M.; Skariah, G.; Khetani, R.S.; Drnevich, J.; Arcila, M.L.; Kosik, K.S.; Ceman, S. MOV10 and fmrp regulate AGO2 association with microRNA recognition elements. Cell Rep. 2014, 9, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Bechara, E.G.; Didiot, M.C.; Melko, M.; Davidovic, L.; Bensaid, M.; Martin, P.; Castets, M.; Pognonec, P.; Khandjian, E.W.; Moine, H.; et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009, 7, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didiot, M.C.; Tian, Z.; Schaeffer, C.; Subramanian, M.; Mandel, J.L.; Moine, H. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 2008, 36, 4902–4912. [Google Scholar] [CrossRef] [PubMed]
- Antar, L.N.; Li, C.; Zhang, H.; Carroll, R.C.; Bassell, G.J. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 2006, 32, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Lombaert, I.M.; Hauser, B.R.; Patel, V.N.; Hoffman, M.P. Exosomal microRNA transport from salivary mesenchyme regulates epithelial progenitor expansion during organogenesis. Dev. Cell 2017, 40, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Bensaid, M.; Melko, M.; Bechara, E.G.; Davidovic, L.; Berretta, A.; Catania, M.V.; Gecz, J.; Lalli, E.; Bardoni, B. FRAXE-associated mental retardation protein (FMR2) is an RNA-binding protein with high affinity for G-quartet RNA forming structure. Nucleic Acids Res. 2009, 37, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Chio, A.; Borghero, G.; Restagno, G.; Mora, G.; Drepper, C.; Traynor, B.J.; Sendtner, M.; Brunetti, M.; Ossola, I.; Calvo, A.; et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9orf72. Brain 2012, 135, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Daoud, H.; Suhail, H.; Sabbagh, M.; Belzil, V.; Szuto, A.; Dionne-Laporte, A.; Khoris, J.; Camu, W.; Salachas, F.; Meininger, V.; et al. C9orf72 hexanucleotide repeat expansions as the causative mutation for chromosome 9p21-linked amyotrophic lateral sclerosis and frontotemporal dementia. Arch. Neurol. 2012, 69, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Redondo, A.; Dols-Icardo, O.; Rojas-Garcia, R.; Esteban-Perez, J.; Cordero-Vazquez, P.; Munoz-Blanco, J.L.; Catalina, I.; Gonzalez-Munoz, M.; Varona, L.; Sarasola, E.; et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in spain and different populations worldwide. Hum. Mutat. 2013, 34, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J.; et al. C9orf72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Liu, Y.; Banez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA FOCI from antisense transcripts in C9orf72 als and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA FOCI. Acta Neuropathol. 2013, 126, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9orf72 hexanucleotide repeat form nuclear RNA FOCI and undergo repeat-associated non-ATG translation in C9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobagyi, T.; Shaw, C.E. P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked ftld and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Arzberger, T.; Grasser, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, N.; Alhothali, M.; Alfonso, M.H.; Breydo, L.; Uversky, V.N. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2017, 74, 1297–1318. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, R.A.; Napoli, E.; Wong, A.; Zhan, S.; Reutenauer, L.; Morin, D.; Buckpitt, A.R.; Taroni, F.; Lonnerdal, B.; Ristow, M.; et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005, 14, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.A.; Gakh, O.; Park, S.; Cui, J.; Mooney, S.M.; Sampson, M.; Ferreira, G.C.; Isaya, G. Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry 2005, 44, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Watkins, N.J.; Lemm, I.; Ingelfinger, D.; Schneider, C.; Hossbach, M.; Urlaub, H.; Luhrmann, R. Assembly and maturation of the U3 snoRNP in the nucleoplasm in a large dynamic multiprotein complex. Mol. Cell 2004, 16, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Hayano, T.; Yanagida, M.; Yamauchi, Y.; Shinkawa, T.; Isobe, T.; Takahashi, N. Proteomic analysis of human Nop56p-associated pre-ribosomal ribonucleoprotein complexes. Possible link between Nop56p and the nucleolar protein treacle responsible for treacher collins syndrome. J. Biol. Chem. 2003, 278, 34309–34319. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.; Schulz, B.; Blume, A.; Wahle, E.; Rudolph, R.; Schwarz, E. Trinucleotide expansions leading to an extended poly-l-alanine segment in the poly (A) binding protein PABPN1 cause fibril formation. Protein Sci. 2003, 12, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, R. Physical chemistry of polyglutamine: Intriguing tales of a monotonous sequence. J. Mol. Biol. 2012, 421, 466–490. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, B.; Kerenyi, A.; Bogar, F.; Paragi, G.; Penke, B.; Rakhely, G. Studying the structural properties of polyalanine and polyglutamine peptides. J. Mol. Model. 2007, 13, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Pelassa, I.; Cora, D.; Cesano, F.; Monje, F.J.; Montarolo, P.G.; Fiumara, F. Association of polyalanine and polyglutamine coiled coils mediates expansion disease-related protein aggregation and dysfunction. Hum. Mol. Genet. 2014, 23, 3402–3420. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Lee, J.M.; Pinto, R.M.; Gillis, T.; St Claire, J.C.; Wheeler, V.C. Quantification of age-dependent somatic CAG repeat instability in HDH CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS ONE 2011, 6, e23647. [Google Scholar] [CrossRef] [PubMed]
- Swami, M.; Hendricks, A.E.; Gillis, T.; Massood, T.; Mysore, J.; Myers, R.H.; Wheeler, V.C. Somatic expansion of the huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet. 2009, 18, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Dragileva, E.; Hendricks, A.; Teed, A.; Gillis, T.; Lopez, E.T.; Friedberg, E.C.; Kucherlapati, R.; Edelmann, W.; Lunetta, K.L.; MacDonald, M.E.; et al. Intergenerational and striatal CAG repeat instability in huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 2009, 33, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Gonitel, R.; Moffitt, H.; Sathasivam, K.; Woodman, B.; Detloff, P.J.; Faull, R.L.; Bates, G.P. DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 3467–3472. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Lebel, L.A.; Vrbanac, V.; Teed, A.; te Riele, H.; MacDonald, M.E. Mismatch repair gene MSH2 modifies the timing of early disease in Hdh (Q111) striatum. Hum. Mol. Genet. 2003, 12, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed]



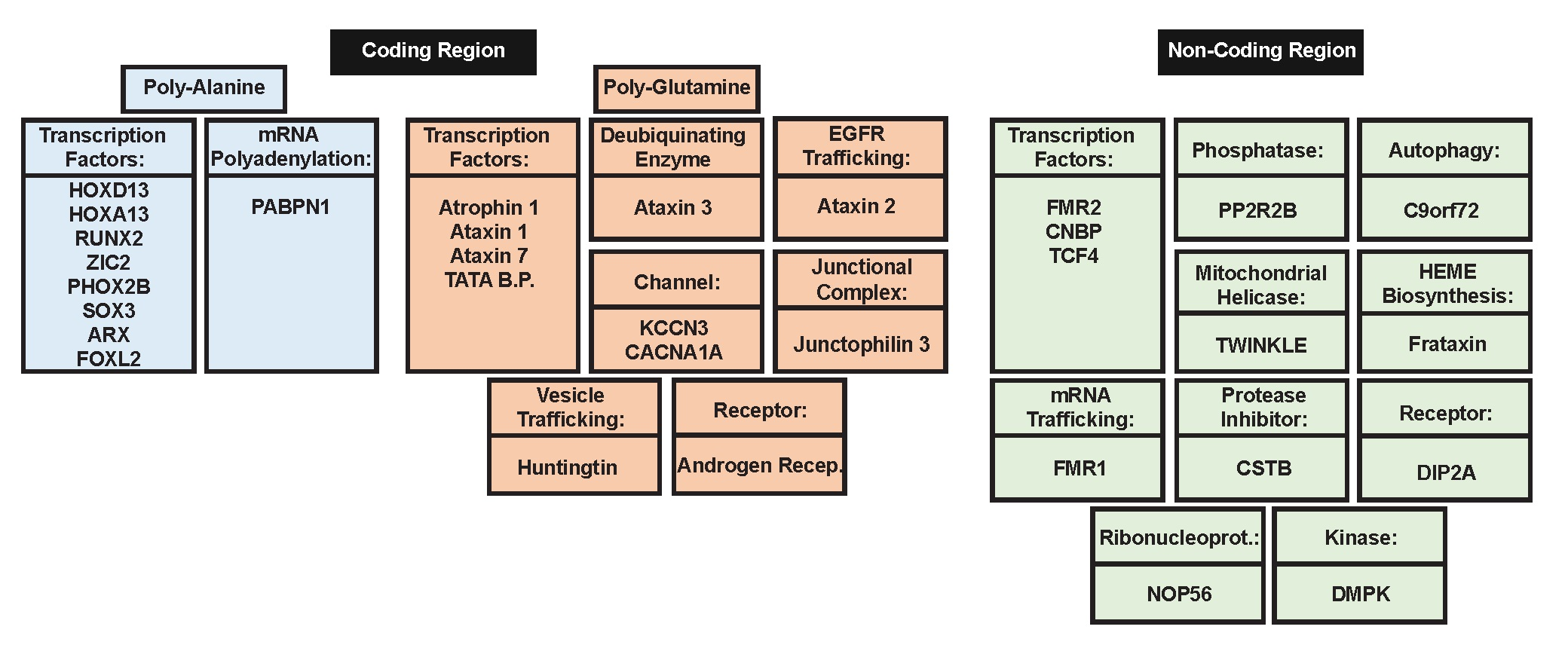{kind=link}
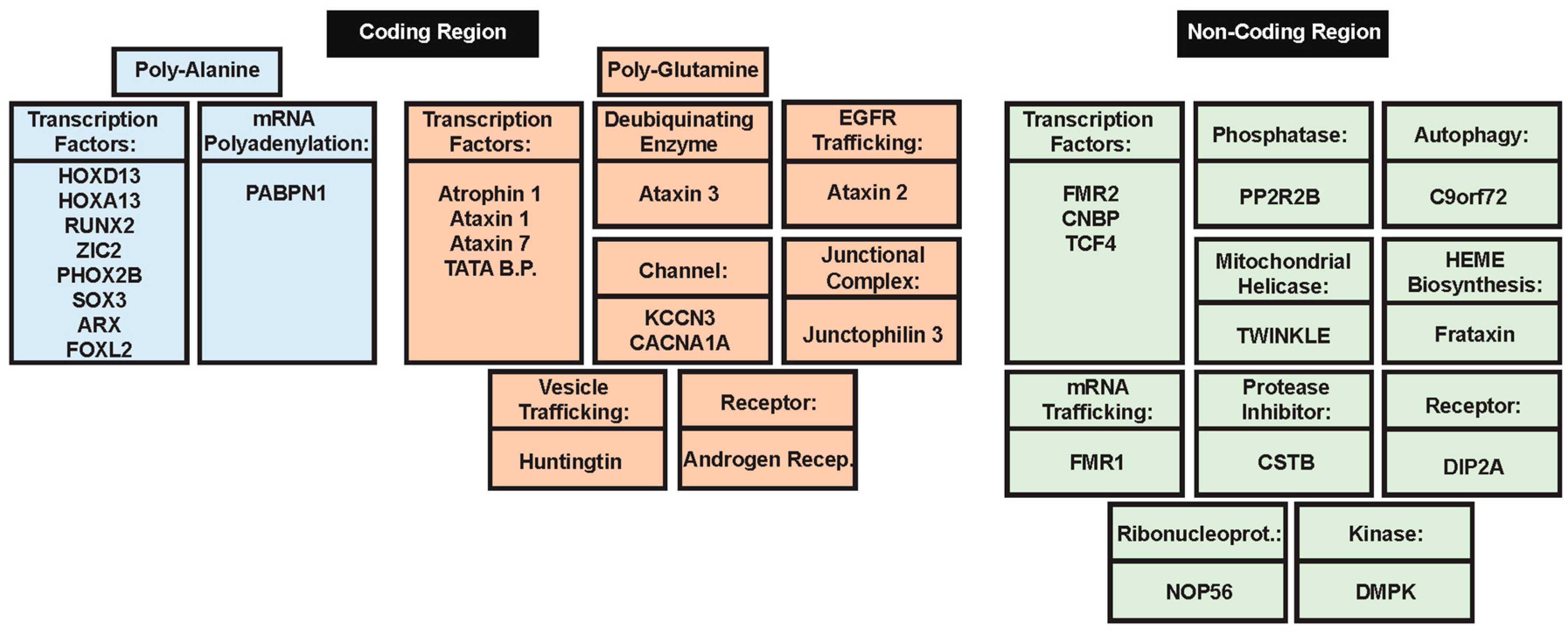{kind=link}
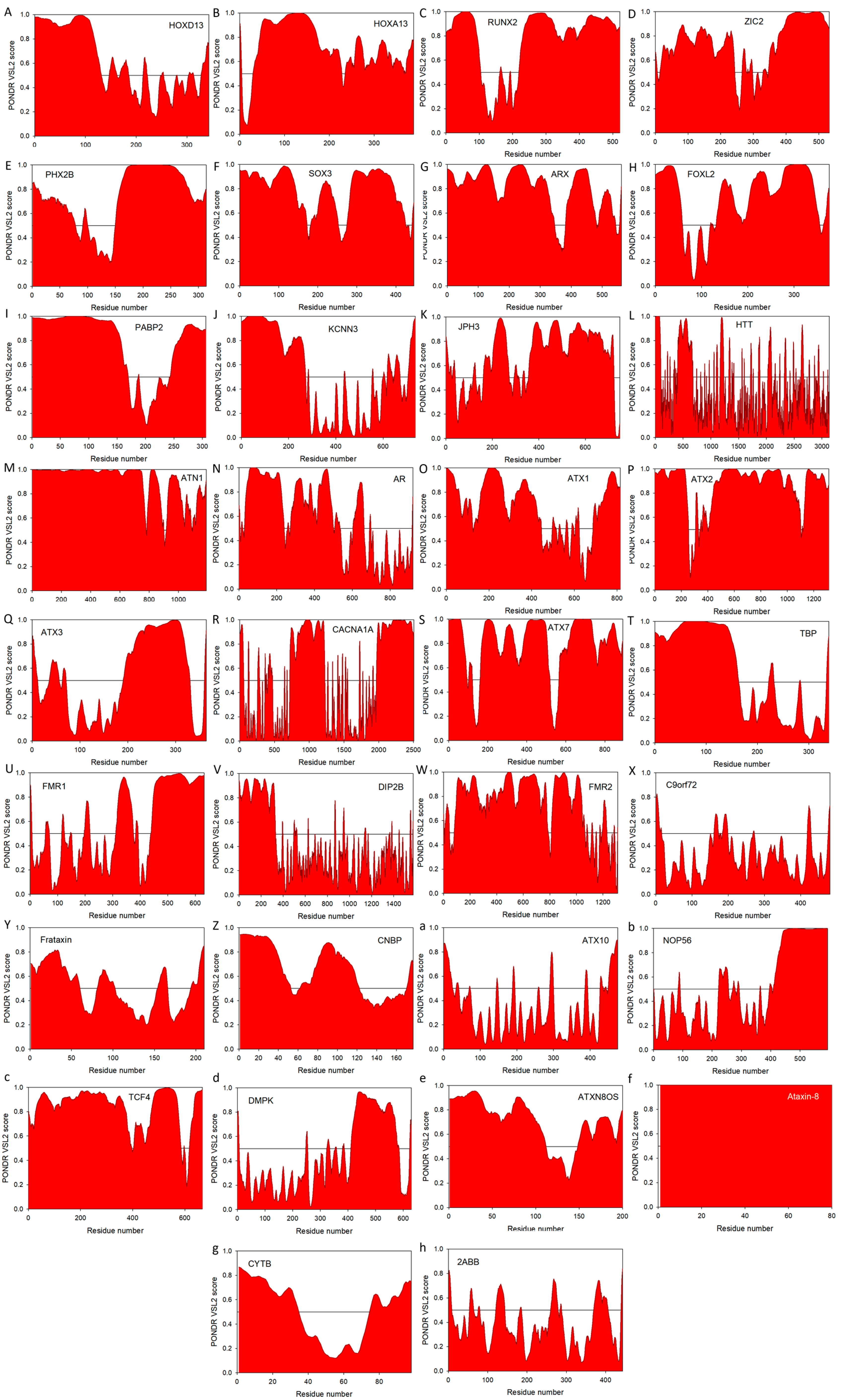{kind=link}
| Repeat Location | Gene | Disease a | Repeat Sequence | WT Length | Pathogenic Length b | % Disorder c | References | |
|---|---|---|---|---|---|---|---|---|
| Poly-alanine | Exon | HOXD13 | SPD II | GCG | 15 | >21 | 33.82 | [64,65,66] |
| Exon | HOXA13 | HFGS | GCG | 12 | >17 | 34.28 | [67] | |
| Exon | RUNX2 | CCD | GCG | 17 | >26 | 62.96 | [65,68] | |
| Exon | ZIC2 | HPE | GCG | 9 | -- | 54.89 | [69,70] | |
| Exon | PHOX2B | CCHS | GCG | 20 | -- | 54.15 | [71,72,73] | |
| Exon-X chrom. | SOX3 | XLMR + GHD | GCG | 15 | >25 | 51.12 | [74,75] | |
| Exon-X chrom. | ARX | XLMR | GCG | 16 | >17, >22 | 59.07 | [76,77] | |
| Exon | FOXL2 | BPEIS | GCG | 14 | >21, >24 | 47.34 | [78,79] | |
| Exon | PABPN1 | OPMD | GCG | 10 | >11, >16 | 59.80 | [80,81] | |
| Poly-glutamine | Exon | KCNN3 | Schizo. d | CAG | -- | -- | 37.50 | [82] |
| Exon | JPH3 | HDL2 | CAG/CTG | 6 to 28 | >41 | 51.07 | [83,84,85] | |
| Exon | HTT | HD | CAG | 6 to 35 | >35 | 19.10 | [86,87] | |
| Exon | ATN1 | DRPLA | CAG | 3 to 36 | >48 | 86.05 | [88,89] | |
| Exon | AR | SBMA | CAG | 9 to 36 | >37 | 54.13 | [90] | |
| Exon | ATXN1 | SCA1 | CAG | 6 to 39 | >39 | 54.97 | [91] | |
| Exon | ATXN2 | SCA2 | CAG | 14 to 32 | >33 | 79.13 | [92,93] | |
| Exon | ATXN3 | SCA3 | CAG | 12 to 40 | >54 | 42.03 | [82,94] | |
| Exon | CACNA1A | SCA6 | CAG | 4 to 18 | >20 | 42.08 | [95,96] | |
| Exon | ATXN7 | SCA7 | CAG | 7 to 17 | >33 | 71.30 | [97,98] | |
| Exon | TBP | SCA17 | CAG | 25 to 42 | >44 | 46.31 | [99,100] | |
| Non-coding | 5′ UTR | PPP2R2B | SCA12 | CAG | 7 to 32 | >54 | 7.67 | [101,102] |
| 5′ UTR-X chrom. | FMR1 | FXMR, FXTAS | CGG | 6 to 55 | >200, >55 | 38.29 | [103] | |
| 5′ UTR | DIP2B | FRA12A MR | CGG | 6 to 23 | >200 | 19.73 | [104] | |
| 5′ UTR-X chrom. | FMR2 | FRAXE MR | GCC | -- | >200 | 58.12 | [105,106,107] | |
| 5′ UTR | C9orf72 | C9ALS/FTD | GGGGCC | -- | Unknown | 2.70 | [108,109] | |
| Intron | FXN | FRDA | GAA | 7 to 22 | >66 | 40.00 | [110,111] | |
| Intron | CNBP/zfn9 | DM2 | CCTG | <27 | >75 | 19.77 | [112,113] | |
| Intron | ATXN10 | SCA10 | ATTCT | 10 to 29 | >279 | 5.26 | [114,115] | |
| Intron | NOP56 | SCA36 | GGCCTG | 3 to 8 | >1500 | 26.26 | [116] | |
| Intron | TCF4 | FECD | CTG | -- | >50 | 88.01 | [117,118] | |
| 3′ UTR | DMPK | DM1 | CTG | 5 to 37 | >50 | 14.63 | [119,120] | |
| 3′ UTR | ATXN8OS | SCA8 e | CTG | 6 to 37 | >74 | 68.00 f | [121,122,123] | |
| Exon | ATXN8 | SCA8 e | CAG | 15 to 50 | 71 to 1300 | 100.00 f | [124] | |
| Promoter | CSTB | EPM1 | CCCCGCCCCGCG | 2 to 3 | >14 | 40.82 | [125,126] |
| Gene | Repeat Sequence | Sense Translation | Antisense Translation |
|---|---|---|---|
| FXS | CGG-CGG-CGG-CGG | R-R-R-R | A-A-A-A |
| GGC-GGC-GGC-GGC | G-G-G-G | P-P-P-P | |
| GCG-GCG-GCG-GCG | A-A-A-A | R-R-R-R | |
| DIP2B | CGG-CGG-CGG-CGG | R-R-R-R | A-A-A-A |
| GGC-GGC-GGC-GGC | G-G-G-G | P-P-P-P | |
| GCG-GCG-GCG-GCG | A-A-A-A | R-R-R-R | |
| FMR2 | GCC-GCC-GCC-GCC | A-A-A-A | R-R-R-R |
| CCG-CCG-CCG-CCG | P-P-P-P | G-G-G-G | |
| CGC-CGC-CGC-CGC | R-R-R-R | A-A-A-A | |
| C9orf72 | GGG-GCC-GGG-GCC | G-A-G-A | P-R-P-R |
| GGG-CCG-GGG-CCG | G-P-G-P | P-G-P-G | |
| GGC-CGG-GGC-CGG | G-R-G-R | P-A-P-A | |
| FXN | GAA-GAA-GAA-GAA | E-E-E-E | L-L-L-L |
| AAG-AAG-AAG-AAG | K-K-K-K | F-F-F-F | |
| AGA-AGA-AGA-AGA | R-R-R-R | S-S-S-S | |
| CNBP | CCT-GCC-TGC-CTG | P-A-C-L | G-R-T-G-G |
| CTG-CCT-GCC-TGC | L-P-A-C | G-G-R-T-G | |
| TGC-CTG-CCT-GCC | C-L-P-A | T-G-G-G-R | |
| ATXN10 | ATT-CTA-TTC-TAT-TCT | I-L-F-F-T | STOP-D-K-I-R |
| TTC-TAT-TCT-ATT-CTA | F-F-S-I-L | K-I-R-STOP-D | |
| TCT-ATT-CTA-TTC-TAT | S-I-L-F-F | R-STOP-D-K-I | |
| TCF4 | CTG-CTG-CTG-CTG | L-L-L-L | D-D-D-D |
| TGC-TGC-TGC-TGC | C-C-C-C | P-P-P-P | |
| GCT-GCT-GCT-GCT | A-A-A-A | R-R-R-R | |
| DMPK | CTG-CTG-CTG-CTG | L-L-L-L | D-D-D-D |
| TGC-TGC-TGC-TGC | C-C-C-C | P-P-P-P | |
| GCT-GCT-GCT-GCT | A-A-A-A | R-R-R-R | |
| JPH3 | CTG-CTG-CTG-CTG | L-L-L-L | D-D-D-D |
| TGC-TGC-TGC-TGC | C-C-C-C | P-P-P-P | |
| GCT-GCT-GCT-GCT | A-A-A-A | R-R-R-R | |
| ATXN8 | CTG-CTG-CTG-CTG | L-L-L-L | D-D-D-D |
| TGC-TGC-TGC-TGC | C-C-C-C | P-P-P-P | |
| GCT-GCT-GCT-GCT | A-A-A-A | R-R-R-R | |
| CSTB | CCC-CGC-CCC-GCG | P-R-P-A | G-A-G-R |
| CCC-GCC-CCG-CGC | P-A-P-R | G-R-G-A | |
| CCG-CCC-CGC-GCC | P-P-R-A | G-G-A-R |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darling, A.L.; Uversky, V.N. Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions. Molecules 2017, 22, 2027. https://doi.org/10.3390/molecules22122027
Darling AL, Uversky VN. Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions. Molecules. 2017; 22(12):2027. https://doi.org/10.3390/molecules22122027
Chicago/Turabian StyleDarling, April L., and Vladimir N. Uversky. 2017. "Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions" Molecules 22, no. 12: 2027. https://doi.org/10.3390/molecules22122027