
Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies

 , ,
, ,
Abstract
:1. Introduction
2. Triterpenes: Chemical Structures and Natural Occurrence
3. Bioavailability of Pentacyclic Triterpenes
3.1. Definition of Bioavailability and Challenges to Determine Pentacyclic Triterpene Bioavailability in a Complex Matrix
3.2. In Vitro Studies Carried out with Pentacyclic Triterpenes to Predict the In Vivo Bioavailability
3.3. Bioavailability of Bioactive Pentacyclic Triterpenes In Vivo
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Muffler, K.; Leipold, D.; Scheller, M.C.; Haas, C.; Steingroewer, J.; Bley, T.; Neuhaus, H.E.; Mirata, M.A.; Schrader, J.; Ulber, R. Biotransformation of triterpenes. Process Biochem. 2011, 46, 1–15. [Google Scholar] [CrossRef]
- Sheng, H.; Sun, H. Synthesis, biology and clinical significance of pentacyclic triterpenes: A multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat. Prod. Rep. 2011, 28, 543–593. [Google Scholar] [CrossRef] [PubMed]
- Siddique, H.R.; Saleem, M. Beneficial health effects of lupeol triterpene: A review of preclinical studies. Life Sci. 2011, 88, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Moreau, R.A.; Whitaker, B.D.; Hicks, K.B. Phytosterols, phytostanols, and their conjugates in foods: Structural diversity, quantitative analysis, and health-promoting uses. Prog. Lipid Res. 2002, 41, 457–500. [Google Scholar] [CrossRef]
- Szakiel, A.; Paczkowski, C.; Pensec, F.; Bertsch, C. Fruit cuticular waxes as a source of biologically active triterpenoids. Phytochem. Rev. 2012, 11, 263–284. [Google Scholar] [CrossRef] [PubMed]
- Pádua, T.A.; de Abreu, B.S.S.C.; Costa, T.E.M.M.; Nakamura, M.J.; Valente, L.M.M.; Henriques, M.G.; Siani, A.C.; Rosas, E.C. Anti-inflammatory effects of methyl ursolate obtained from a chemically derived crude extract of apple peels: Potential use in rheumatoid arthritis. Arch. Pharm. Res. 2014, 37, 1487–1495. [Google Scholar]
- Smina, T.P.; Mathew, J.; Janardhanan, K.K.; Devasagavam, T.P.A. Antioxidant activity and toxicity profile of total triterpenes isolated from Ganoderma lucidum (Fr.) P. Karst occurring in South India. Environ. Toxicol. Pharmacol. 2011, 32, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, R.H.; Kouzi, S.A. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 2004, 24, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Hamid, K.; Kam, A.; Wong, K.H.; Abdelhak, Z.; Razmovski-Naumovski, V.; Chan, K.; Li, K.M.; Groundwater, P.W.; Li, G.Q. The pentacyclic triterpenoids in herbal medicines and their pharmacological activities in diabetes and diabetic complications. Curr. Med. Chem. 2013, 20, 908–931. [Google Scholar] [CrossRef] [PubMed]
- Laszczyk, M.N. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med. 2009, 75, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Kalra, N.; Shukla, Y. Hepatoprotective effects of lupeol and mango pulp extract of carcinogen induced alteration in Swiss albino mice. Mol. Nutr. Food Res. 2007, 51, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.H.; Rasool, S.N.; Abdul Kareem, M.; Krushna, G.S.; Akhtar, P.M.; Devi, K.L. Maslinic acid protects againstisoproterenol-induce cardiotoxicity in albino Wistar rats. J. Med. Food. 2012, 15, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, R. Oleanolic acid and related triterpenoids from olives on vascular function: Molecular mechanisms and therapeutic perspectives. Curr. Med. Chem. 2015, 22, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Patlolla, J.M.R.; Rao, C.V. Triterpenoids for Cancer Prevention and Treatment: Current Status and Future Prospects. Curr. Pharm. Biotechnol. 2012, 13, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Theoduloz, C.; Schmeda-Hirschmann, G.; Razmilic, I.; Yáñez, T.; Rodríguez, J.A. Gastroprotective and ulcer-healing activity of oleanolic acid derivatives: In vitro-in vivo relationships. Life Sci. 2006, 29, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Tausch, L.; Henkel, A.; Siemoneit, U.; Poeckel, D.; Kather, N.; Franke, L.; Hofmann, B.; Schneider, G.; Angioni, C.; Geisslinger, G.; et al. Identification of human cathepsin G as a functional target of boswellic acids from the anti-inflammatory remedy frankincense. J. Immunol. 2009, 183, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Skarke, C.; Kuczka, K.; Tausch, L.; Werz, O.; Rossmanith, T.; Barrett, J.S.; Harder, S.; Holtmeier, W.; Schwarz, J.A. Increased bioavailability of 11-keto-β-boswellic acid following single oral dose frankincense extract administration after a standardized meal in healthy male volunteers: Modeling and simulation considerations for evaluating drug exposures. J. Clin. Pharmacol. 2012, 52, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Sterk, V.; Büchele, B.; Simmet, T. Effect of food intake on the bioavailability of boswellic acids from a herbal preparation in healthy volunteers. Planta Med. 2004, 70, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Thawani, V.; Hingorani, L.; Shrivastava, M.; Bhate, V.R.; Khiyani, R. Pharmacokinetic study of 11-Keto beta-boswellic acid. Phytomedicine 2004, 11, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Kaeidi, A.; Esmaeili-Mahani, S.; Sheibani, V.; Abbasnejad, M.; Rasoulian, B.; Hajializadeh, Z.; Afrazi, S. Olive (Olea europaea L.) leaf extract attenuates early diabetic neuropathic pain through prevention of high glucose-induced apoptosis: In vitro and in vivo studies. J. Ethnopharmacol. 2011, 136, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.L.; Wu, H.; Liu, J.Z.; Hu, J.X.; Liao, N.; Peng, J.; Cao, P.P.; Liang, X.; Hai, C.X. Antidiabetic effect of oleanolic acid: A promising use of a traditional pharmacological agent. Phytother. Res. 2011, 25, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, X. Oral Bioavailability: Basic Principles, Advanced Concepts, and Applications, 1st ed.; John Wiley & Sons: New York, NY, USA, 2011; p. 568. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2009; p. 546. [Google Scholar]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants—Rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [PubMed]
- Andre, C.M.; Legay, S.; Deleruelle, A.; Nieuwenhuizen, N.; Punter, M.; Brendolise, C.; Cooney, J.M.; Lateur, M.; Hausman, J.-F.; Larondelle, Y.; et al. Multifunctional oxidosqualene cyclases and cytochrome P450 involved in the biosynthesis of apple fruit triterpenic acids. New Phytol. 2016, 211, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Andre, C.M.; Larsen, L.; Burgess, E.J.; Jensen, D.J.; Cooney, J.M.; Evers, D.; Laing, W.A. Unusual immuno-modulatory triterpene-caffeates in the skins of russeted varieties of apples and pears. J. Agric. Food Chem. 2013, 61, 2773–2779. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.P. Biological activity of defense-related plant secondary metabolites. In Plant-Derived Natural Products: Synthesis, Function and Application, 1st ed.; Osbourn, A., Lanzotti, V., Eds.; Springer: New York, NY, USA, 2009; pp. 283–299. [Google Scholar]
- Netala, V.R.; Ghosh, S.B.; Bobbu, P.; Anitha, D.; Tartte, V. Triterpenoid saponins: A review on biosynthesis, applications and mechanism of their action. Int. J. Pharm. Pharm. Sci. 2015, 7, 24–28. [Google Scholar]
- Andre, C.M.; Greenwood, J.M.; Walker, E.G.; Rassam, M.; Sullivan, M.; Evers, D.; Perry, N.B.; Laing, W.A. Anti-inflammatory procyanidins and triterpenes in 109 apple varieties. J. Agric. Food Chem. 2012, 60, 10546–10554. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Noshita, T.; Kidachi, Y.; Umetsu, H.; Hayashi, M.; Komiyama, K.; Funayama, S.; Ryoyama, K. Isolation of ursolic acid from apple peels and its specific efficacy as a potent antitumor agent. J. Health Sci. 2008, 54, 654–660. [Google Scholar] [CrossRef]
- Wang, W.H.; Chuang, H.Y.; Chen, C.H.; Chen, W.K.; Hwang, J.J. Lupeol acetate ameliorates collagen-induced arthritis and osteoclastogenesis of mice through improvement of microenvironment. Biomed. Pharmacother. 2016, 79, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.C.; Lin, M.C.; Mong, M.C.; Lin, C.Y. Bioavailability, distribution, and antioxidative effects of selected triterpenes in mice. J. Agric. Food Chem. 2012, 60, 7697–7701. [Google Scholar] [CrossRef] [PubMed]
- Juan, M.E.; Planas, J.M.; Ruiz-Gutierrez, V.; Daniel, H.; Wenzel, U. Antiproliferative and apoptosis-inducing effects of maslinic and oleanolic acids, two pentacyclic triterpenes from olives, on HT-29 colon cancer cells. Br. J. Nutr. 2008, 100, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, B.S.; Aslam, H.; Ali, S.T.; Begum, S.; Khatoon, N. Two new triterpenoids and a steroidal glycoside from the aerial parts of Ocimum basilicum. Chem. Pharm. Bull. 2007, 55, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Razboršek, M.I.; Vončina, D.B.; Doleček, V.; Vončina, E. Determination of oleanolic, betulinic and ursolic acid in lamiaceae and mass spectral fragmentation of their trimethylsilylated derivatives. Chromatographia 2008, 67, 433–440. [Google Scholar] [CrossRef]
- Ovesná, Z.; Vachálková, A.; Horváthová, K.; Tóthová, D. Pentacyclic triterpenoic acids: New chemoprotective compounds. Minireview. Neoplasma 2004, 51, 327–333. [Google Scholar] [PubMed]
- Raval, N.; Mistry, T.; Acharya, N.; Acharya, S. Development of glutathione-conjugated asiatic acid-loaded bovine serum albumin nanoparticles for brain-targeted drug delivery. J. Pharm. Pharmacol. 2015, 67, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Judy, W.V.; Hari, S.P.; Stogsdill, W.W.; Judy, J.S.; Naguib, Y.M.; Passwater, R. Antidiabetic activity of a standardized extract (glucosol) from Lagerstroemia speciosa leaves in type II diabetics: A dose-dependence study. J. Ethnopharmacol. 2003, 87, 115–117. [Google Scholar] [CrossRef]
- Ganbold, M.; Barker, J.; Ma, R.; Jones, L.; Carew, M. Cytotoxicity and bioavailability studies on a decoction of Oldenlandia diffusa and its fractions separated by HPLC. J. Ethnopharmacol. 2010, 131, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Rosemann, T.; Szecsenyi, J.; Hahn, E.G.; Willich, S.N.; Brinkhaus, B. Use of complementary and alternative medicine in Germany: A survey of patients with inflammatory bowel disease. BMC Complement. Altern. Med. 2006, 6, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Oleanolic acid and ursolic acid: Research perspectives. J. Ethnopharmacol. 2005, 100, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Avila, N.; Priego-Capote, F.; Ruiz-Jiménez, J.; de Castro, M.D.L. Fast and selective determination of triterpenic compounds in olive leaves by liquid chromatography–tandem mass spectrometry with multiple reaction monitoring after microwave-assisted extraction. Talanta 2009, 78, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Romero, C.; García, A.; Medina, E.; Ruiz-Méndez, M.V.; de Castro, A.; Brenes, M. Triterpenic acids in table olives. Food Chem. 2010, 118, 670–674. [Google Scholar] [CrossRef]
- Pérez-Camino, M.C.; Cert, A. Quantitative determination of hydroxy pentacyclic triterpene acids in vegetable oils. J. Agric. Food Chem. 1999, 47, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Dale, M.M.; Rang, H.P. Pharmacology, 6th ed.; Churchill Livingstone: London, UK, 2007; p. 829. [Google Scholar]
- Heaney, R.P. Factors influencing the measurement of bioavailability, taking calcium as a model. J. Nutr. 2001, 131, 1344S–1348S. [Google Scholar] [PubMed]
- Rein, M.J.; Renouf, M.; Cruz-Hernandez, C.; Actis-Goretta, L.; Thakkar, S.K.; da Silva Pinto, M. Bioavailability of bioactive food compounds: A challenging journey to bioefficacy. Br. J. Clin. Pharmacol. 2013, 75, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Van Willige, R.W.; Linssen, J.P.; Voragen, A.G. Influence of food matrix on absorption of flavour compounds by linear low-density polyethylene: Proteins and carbohydrates. J. Sci. Food Agric. 2000, 80, 1779–1789. [Google Scholar] [CrossRef]
- Riedl, J.; Linseisen, J.; Hoffmann, J.; Wolfram, G. Some dietary fibers reduce the absorption of carotenoids in women. J. Nutr. 1999, 129, 2170–2176. [Google Scholar] [PubMed]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Martin, Y.C. A practitioner’s perspective of the role of quantitative structure-activity analysis in medicinal chemistry. J. Med. Chem. 1981, 24, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.T.; Fischer, S.M.; Fricker, G.; Brandl, M. In vitro models to evaluate the permeability of poorly soluble drug entities: Challenges and perspectives. Eur. J. Pharm. Sci. 2012, 45, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Bérangère, C.; Caussarieu, N.; Morin, P.; Morin-Allory, L.; Lafosse, M. Rapid analysis of triterpenic acids by liquid chromatography using porous graphitic carbon and evaporative light scattering detection. J. Sep. Sci. 2004, 27, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Rafat, M.; Fong, K.W.; Goldsipe, A.; Stephenson, B.C.; Coradetti, S.T.; Sambandan, T.G.; Sinskey, A.J.; Rha, C. Association (micellization) and partitioning of aglycon triterpenoids. J. Colloid Interface Sci. 2008, 325, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Gareth, T. Medicinal Chemistry, An Introduction, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007; p. 621. [Google Scholar]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, C.Y.; Kong, A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Noh, K.; Han, C.Y.; Kim, S.G. Transactivation of genes encoding for phase II enzymes and phase III transporters by phytochemical antioxidants. Molecules 2010, 15, 6332–6348. [Google Scholar] [CrossRef] [PubMed]
- Berger, V.; Gabriel, A.F.; Sergent, T.; Trouet, A.; Larondelle, Y.; Schneider, Y.J. Interaction of ochratoxin A with human intestinal Caco-2 cells: Possible implication of a multidrug resistance-associated protein (MRP2). Toxicol. Lett. 2003, 140–141, 465–476. [Google Scholar] [CrossRef]
- Cháirez-Ramírez, M.H.; Sánchez-Burgos, J.A.; Gomes, C.; Moreno-Jiménez, M.R.; González-Laredo, R.F.; Bernad-Bernad, M.J.; Medina-Torres, L.; Ramírez-Mares, M.V.; Gallegos-Infante, J.A.; Rocha-Guzmán, N.E. Morphological and release characterization of nanoparticles formulated with poly (dl-lactide-co-glycolide) (PLGA) and lupeol: In vitro permeability and modulator effect on NF-κB in Caco-2 cell system stimulated with TNF-α. Food Chem. Toxicol. 2015, 85, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man-fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Jaggi, M.; Singh, M.K.; Mukherjee, R.; Burman, A.C. Pharmacological evaluation of C-3 modified betulinic acid derivatives with potent anticancer activity. Investig. New Drugs 2008, 26, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.W.; Kim, Y.H.; Kim, H.H.; Ji, H.Y.; Yoo, S.D.; Choi, W.R.; Lee, S.M.; Han, C.K.; Lee, H.S. Dose-linear pharmacokinetics of oleanolic acid after intravenous and oral administration in rats. Biopharm. Drug Dispos. 2007, 2, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.H.; Du, Z.; Wang, G.N.; Chan, H.M.; Chang, Q.; Lai, L.C.; Chow, A.H.; Zheng, Y. Spray freeze drying with polyvinylpyrrolidone and sodium caprate for improved dissolution and oral bioavailability of oleanolic acid, a BCS Class IV compound. Int. J. Pharm. 2011, 404, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Gao, Y.; Wang, M.; Fang, L.; Ping, Q. Propylene glycol-linked amino acid/dipeptide diester prodrugs of oleanolic acid for PepT1-mediated transport: Synthesis, intestinal permeability, and pharmacokinetics. Mol. Pharm. 2013, 10, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Z.; Ye, Z.; Hauck, C.; Murphy, P.A.; McCoy, J.A.; Widrlechner, M.P.; Reddy, M.B.; Hendrich, S. Permeability of rosmarinic acid in Prunella vulgaris and ursolic acid in Salvia officinalis extracts across Caco-2 cell monolayers. J. Ethnopharmacol. 2011, 137, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, H.; Sun, F.; Sun, S.; Zhu, Z.; Chai, Y. Biopharmaceutical and pharmacokinetic characterization of asiatic acid in Centella asiatica as determined by a sensitive and robust HPLC-MS method. J. Ethnopharmacol. 2015, 163, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Krüger, P.; Kanzer, J.; Hummel, J.; Fricker, G.; Schubert-Zsilavecz, M.; Abdel-Tawab, M. Permeation of boswellia extract in the Caco-2 model and possible interactions of its constituents KBA and AKBA with OATP1B3 and MRP2. Eur. J. Pharm. Sci. 2009, 36, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hüsch, J.; Gerbeth, K.; Fricker, G.; Setzer, C.; Zirkel, J.; Rebmann, H.; Schubert-Zsilavecz, M.; Abdel-Tawab, M. Effect of phospholipid-based formulations of boswellia serrata extract on the solubility, permeability, and absorption of the individual boswellic acid constituents present. J. Nat. Prod. 2012, 75, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Gerbeth, K.; Hüsch, J.; Fricker, G.; Werz, O.; Schubert-Zsilavecz, M.; Abdel-Tawab, M. In vitro metabolism, permeation, and brain availability of six major boswellic acids from Boswellia serrata gum resins. Fitoterapia 2013, 84, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Udeani, G.O.; Zhao, G.M.; Geun Shin, Y.; Cooke, B.P.; Graham, J.; Beecher, C.W.; Kinghorn, A.D.; Pezzuto, J.M. Pharmacokinetics and tissue distribution of betulinic acid in CD-1 mice. Biopharm. Drug Dispos. 1999, 20, 379–383. [Google Scholar] [CrossRef]
- Godugu, C.; Patel, A.R.; Doddapaneni, R.; Somagoni, J.; Singh, M. Approaches to improve the oral bioavailability and effects of novel anticancer drugs berberine and betulinic acid. PLoS ONE 2014, 9, e89919. [Google Scholar] [CrossRef] [PubMed]
- Soica, C.; Danciu, C.; Savoiu-Balint, G.; Borcan, F.; Ambrus, R.; Zupko, I.; Bojin, F.; Coricovac, D.; Ciurlea, S.; Avram, S.; et al. Betulinic acid in complex with a gamma-cyclodextrin derivative decreases proliferation and in vivo tumor development of non-metastatic and metastatic B164A5 cells. Int. J. Mol. Sci. 2014, 15, 8235–8255. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.F.; Song, C.K.; Viernstein, H.; Unger, F.; Liang, Z.S. Apoptosis of human breast cancer cells induced by microencapsulated betulinic acid from sour jujube fruits through the mitochondria transduction pathway. Food Chem. 2013, 138, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Dehelean, C.A.; Feflea, S.; Ganta, S.; Amiji, M. Anti-angiogenic effects of betulinic acid administered in nanoemulsion formulation using chorioallantoic membrane assay. J. Biomed. Nanotechnol. 2011, 7, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, G.J.; Wang, S.J.; Li, X.T.; Xu, Y.P.; Wang, S.P.; Xiang, J.D.; Pan, S.R.; Cao, G.X.; Ye, W.C. Quantitative analysis of 23-hydroxybetulinic acid in mouse plasma using electrospray liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.E.; Blum, R.; Doto, J.; Galbraith, H.; Ballow, C. Multiple-dose pharmacokinetics and safety of bevirimat, a novel inhibitor of HIV maturation, in healthy volunteers. Clin. Pharmacokinet. 2007, 46, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Goto, M.; Yang, X.; Morris-Natschke, S.L.; Huang, L.; Chen, C.H.; Lee, K.H. Fluorinated betulinic acid derivatives and evaluation of their anti-HIV activity. Bioorg. Med. Chem. Lett. 2016, 26, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shin, Y.G.; Levine, B.S.; Smith, A.C.; Tomaszewski, J.E.; vanBreemen, R.B. Quantitative analysis of betulinic acid inmouse, rat and dog plasma using electrospray liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2089–2092. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Jia, J.; Yin, Z.; Gao, Y.; Sha, L.; Lai, Y.; Ping, Q.; Zhang, Y. Ethylene glycol-linked amino acid diester prodrugs of oleanolic acid for PepT1-mediated transport: Synthesis, intestinal permeability and pharmacokinetics. Mol. Pharm. 2012, 9, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chang, Q.; Chan, C.K.; Meng, Z.Y.; Wang, G.N.; Sun, J.B.; Wang, Y.T.; Tong, H.H.; Zheng, Y. Formulation development and bioavailability evaluation of a self-nanoemulsified drug delivery system of oleanolic acid. AAPS PharmSciTech 2009, 10, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yang, X.; Du, P.; Zhang, H.; Zhang, T. Dual strategies to improve oral bioavailability of oleanolic acid: Enhancing water-solubility, permeability and inhibiting cytochrome P450 isozymes. Eur. J. Pharm. Biopharm. 2016, 99, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Huang, X.; Dou, J.; Zhai, G.; Su, L. Self-microemulsifying drug delivery system for improved oral bioavailability of oleanolic acid: Design and evaluation. Int. J. Nanomed. 2013, 8, 2917–2926. [Google Scholar]
- Li, W.; Das, S.; Ng, K.Y.; Heng, P.W.S. Formulation, biological and pharmacokinetic studies of sucrose ester-stabilized nanosuspensions of oleanolic acid. Pharm. Res. 2011, 28, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yang, Y.; Sun, Y.; Cheng, L.; Zhao, S.; Xu, H.; Fawcett, J.P.; Sun, X.; Gu, J. Pharmacokinetic study of calenduloside E and its active metabolite oleanolic acid in beagle dog using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 951–952, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Guo, B.; Li, Y.; Geng, Y.; Zhao, F.; Zhang, T. Enhancement of dissolution rate and oral bioavailability in beagle dogs of oleanolic acid by adsorbing onto porous silica using supercritical carbon dioxide. J. Drug Deliv. Sci. Technol. 2014, 24, 380–385. [Google Scholar] [CrossRef]
- Kanellos, P.T.; Kaliora, A.C.; Gioxari, A.; Christopoulou, G.O.; Kalogeropoulos, N.; Karathanos, V.T. Absorption and bioavailability of antioxidant phytochemicals and increase of serum oxidation resistance in healthy subjects following supplementation with raisins. Plant Foods Hum. Nutr. 2013, 68, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-González, M.; Colom, H.; Lozano-Mena, G.; Juan, M.E.; Planas, J.M. Population pharmacokinetics of maslinic acid, a triterpene from olives, after intravenous and oral administration in rats. Mol. Nutr. Food Res. 2014, 58, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Hu, X.M.; Yi, Y.M.; Wan, J. Preparation and body distribution of freeze-dried powder of ursolic acid phospholipid nanoparticles. Drug Dev. Ind. Pharm. 2009, 35, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.-Q.; Du, X.-Y.; Huang, X.-N.; Qiao, H. Enhanced oral bioavailability of ursolic acid nanoparticles via antisolvent precipitation with TPGS1000 as a stabilizer. J. Drug Deliv. Sci. Technol. 2015, 29, 210–217. [Google Scholar] [CrossRef]
- Yang, L.; Sun, Z.; Zu, Y.; Zhao, C.; Sun, X.; Zhang, Z.; Zhang, L. Physicochemical properties and oral bioavailability of ursolic acid nanoparticles using supercritical anti-solvent (SAS) process. Food Chem. 2012, 132, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yang, T.; Zhang, W.; Lu, M.; Ma, X.; Xiang, G. In vitro and in vivo antitumor effects of folate-targeted ursolic acid stealth liposome. J. Agric. Food Chem. 2014, 62, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Rush, W.R.; Murray, G.R.; Graham, D.J. The comparative steady-state bioavailability of the active ingredients of Madecassol. Eur. J. Drug Metab. Pharmacokinet. 1993, 18, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhao, Y.; Lu, W. Preparation, optimization, characterization and in vivo pharmacokinetic study of asiatic acid tromethamine salt-loaded solid lipid nanoparticles. Drug Dev. Ind. Pharm. 2016, 42, 1325–1333. [Google Scholar]
- Li, J.J.; Li, Y.; Bai, M.; Tan, J.F.; Wang, Q.; Yang, J. Simultaneous determination of corosolic acid and euscaphic acid in the plasma of normal and diabetic rat after oral administration of extract of Potentilla discolor Bunge by high-performance liquid chromatography/electrospray ionization mass spectrometry. Biomed. Chromatogr. 2014, 28, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, D.; Chen, X.; Li, Z.; Li, N.; Han, D.; Talbi, A.; Biwoele, T. Determination of corosolic acid, a natural potential anti-diabetes compound, in rat plasma by high-performance liquid chromatography-mass spectrometry and its application to pharmacokinetic and bioavailability studies. Planta Med. 2011, 77, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Liu, Z.; Ning, Z.; Liu, Y.; Song, Z.; Wang, C.; Lu, A. Prospects of boswellic acids as potential pharmaceutics. Planta Med. 2015, 81, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Xu, X.; Liu, Z.; Liu, J.; Hu, L. Synthesis and structure-activity relationships of boswellic acid derivatives as potent VEGFR-2 inhibitors. Bioorg. Med. Chem. 2015, 23, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Oavum, A.; Sharma, P.R.; Singh, S.K.; Shah, B.A. Synthesis of β-boswellic acid derivatives as cytotoxic and apoptotic agents. Bioorg. Med. Chem. Lett. 2016, 26, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, K.; Kolla, J.N.; Krishnaraju, A.V.; Yalamanchili, N.; Rao, C.V.; Golakoti, T.; Raychaudhuri, S.; Raychaudhuri, S.P. Cellular and molecular mechanisms of anti-inflammatory effect of Aflapin: A novel Boswellia serrata extract. Mol. Cell. Biochem. 2011, 354, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Hüsch, J.; Bohnet, J.; Fricker, G.; Skarke, C.; Artaria, C.; Appendino, G.; Schubert-Zsilavecz, M.; Abdel-Tawab, M. Enhanced absorption of boswellic acids by a lecithin delivery form (Phytosome®) of Boswellia extract. Fitoterapia 2013, 84, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, K.; Jachak, S.M. Development and optimisation of 3-Acetyl-11-keto-β-boswellic acid loaded poly-lactic-co-glycolic acid-nanoparticles with enhanced oral bioavailability and in vivo anti-inflammatory activity in rats. J. Pharm. Pharmacol. 2015, 67, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Satija, S.; Nanda, A.; Garg, M. Nanotechnologies for boswellic acids. Am. J. Drug Discov. Dev. 2014, 4, 1–11. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | log Pow | Reference |
|---|---|---|
| Lupeol | 7.45 | Predicted by ChemAxon software |
| Betulin | 6.17 | Predicted by ChemAxon software |
| Betulinic acid | 6.73 | [53] |
| Oleanolic acid | 6.47 | [53] |
| Maslinic acid | 5.52 | Predicted by ChemAxon software |
| Ursolic acid | 6.43 | [53] |
| Asiatic acid | 5.80 | [54] |
| Corosolic acid | 5.51 | Predicted by ChemAxon software |
| β-boswellic acid | 6.58 | Predicted by ChemAxon software |
| Compound | Papp Value × 10−6 cm/s |
|---|---|
| 11-keto-β-boswellic acid | 29.54 |
| 3-acetyl-11-keto-β-boswellic acid | 17.83 |
| β-boswellic acid | 4.47 |
| 3-acetyl-β-boswellic acid | 6.18 |
| α-boswellic acid | 5.52 |
| 3-acetyl-α-boswellic acid | 4.72 |
| Compound | Species (Sample) | Dose (mg/kg) | Route of Administration | AUC0→∞ (µg·h/mL) | Cmax (µg/mL) | Tmax (h) | Reference |
|---|---|---|---|---|---|---|---|
| Betulinic acid | Mice (serum) | 500 | intraperitoneal | 39.9 | 4.00 | 0.22 | [73] |
| Betulinic acid | Mice (serum) | 250 | intraperitoneal | 18.4 | 2.21 | 0.15 | [73] |
| Betulinic acid | Mice (skin) | 500 | intraperitoneal | 3504.0 | 300.9 | 3.90 | [73] |
| Betulinic acid | Rat (plasma) | 100 | oral | 7.26 ± 1.65 | 1.16 ± 0.22 | 2.36 ± 0.38 | [74] |
| BA-SD | Rat (plasma) | 100 | oral | 53.86 ± 7.79 | 4.54 ± 0.25 | 3.17 ± 0.85 | [74] |
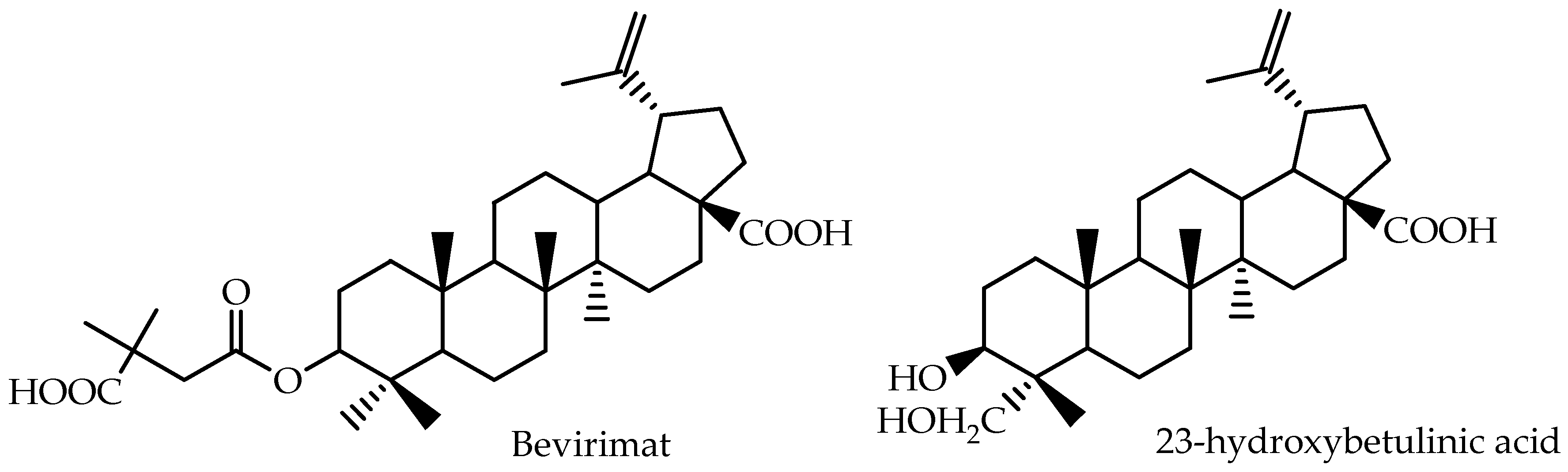
| 23-Hydroxybetulinic acid | Mouse (plasma) | 200 | intragastric | 24.9 | 3.1 | 2 | [78] |
| Bevirimat | Human (plasma) | 200 | oral | 1113.7 ± 216.7 | 58.0 ± 10.83 | 1.50 (0.8–3.0) | [79] |
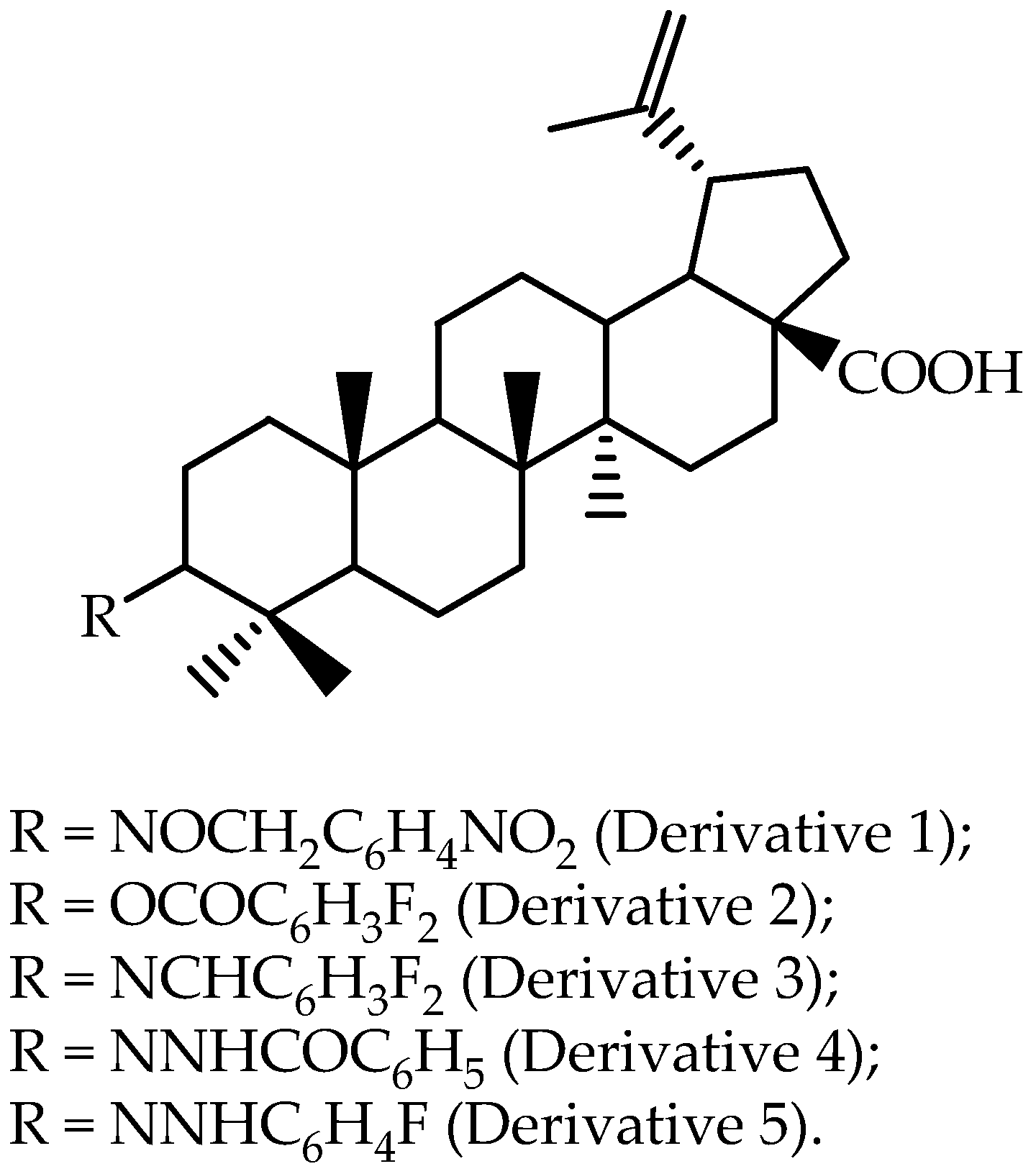
| Derivative 1 | Rat (plasma) | 10 | intravenous | 43.6 ± 6.3 | 101.5 ± 21.7 | 0.05 ± 0.0 | [63] |
| Compound | Specie (Sample) | Dose (mg/kg) | Route of Administration | AUC0→∞ (µg·min/mL) | Cmax (µg/mL) | Tmax (h) | Reference |
|---|---|---|---|---|---|---|---|
| Oleanolic acid | rat (plasma) | 0.5 | intravenous | 16.0 ± 1.9 | N.A. | N.A. | [64] |
| Oleanolic acid | rat (plasma) | 1 | intravenous | 32.6 ± 10.4 | N.A. | N.A. | [64] |
| Oleanolic acid | rat (plasma) | 2 | intravenous | 71.6 ± 12.7 | N.A. | N.A. | [64] |
| Oleanolic acid | rat (plasma) | 10 | oral | N.A. | N.A. | N.A. | [64] |
| Oleanolic acid | rat (plasma) | 25 | oral | 5.9 ± 5.5 | 0.074 ± 0.06 | 0.42 ± 0.30 | [64] |
| Oleanolic acid | rat (plasma) | 50 | oral | 10.7 ± 10.0 | 0.132 ± 0.12 | 0.35 ± 0.28 | [64] |
| Oleanolic acid | rat (plasma) | 300 | intragastric | N.A.; AUC0→24 (µg·h/mL) = 4.98 ± 0.42 | 0.47 ± 0.034 | 0.50 | [82] |
| Oleanolic acid prodrug 5a | rat (plasma) | 300 | intragastric | N.A.; AUC0→24 (µg·h/mL) = 10.99 ± 0.65 | 0.73 ± 0.067 | 0.83 ± 0.22 | [82] |
| Oleanolic acid prodrug 6f | rat (plasma) | 300 | intragastric | N.A.; AUC0→24 (µg·h/mL) = 10.14 ± 1.14 | 0.72 ± 0.070 | 0.58 ± 0.17 | [82] |
| Oleanolic acid prodrug 7a | rat (plasma) | 300 | intragastric | N.A.; AUC0→24 (µg·h/mL) = 17.68 ± 3.07 | 1.43 ± 0.17 | 1.25 ± 1.37 | [67] |
| Oleanolic acid prodrug 9b | rat (plasma) | 300 | intragastric | N.A.; AUC0→24 (µg·h/mL) = 16.88 ± 2.84 | 1.23 ± 0.24 | 1.67 ± 1.81 | [67] |
| Formula B | oral | N.A.; AUC0→t (ng·min/mL) = 40,216.98 ± 31,860.38 | 0.16 ± 0.11 | 0.80 ± 0.45 | [65] | ||
| Formula F | oral | N.A.; AUC0→t (ng·min/mL) = 31,067.44 ± 17,840.92 | 0.39 ± 0.18 | 0.21 ± 0.16 | [65] | ||
| Formula G | oral | N.A.; AUC0→t (ng·min/mL) = 32,657.41 ± 11,832.92 | 0.34 ± 0.16 | 0.26 ± 0.15 | [65] | ||
| Commercial oleanolic acid tablet | rat (plasma) | 50 | oral | N.A.; AUC0→t (ng·min/mL) = 14,974.89 ± 10,906.19 | 0.10 ± 0.06 | 0.80 ± 0.45 | [83] |
| OA SEDDS | rat (plasma) | 50 | oral | N.A.; AUC0→t (ng·min/mL) = 36,041.38 ± 28,965.03 | 0.09 ± 0.04 | 1.5 ± 1.21 | [83] |
| Oleanolic acid | rat (plasma) | 50 | oral | N.A.; AUC0→t (ng·min/mL) = 15,576 ± 1378.8 | 0.059 ± 0.01 | 0.313 ± 0.12 | [84] |
| OPCH | rat (plasma) | 50 | oral | N.A.; AUC0→t (ng·min/mL) = 21,636 ± 1147.8 | 0.078 ± 0.01 | 0.46 ± 0.001 | [84] |
| OPCH with KCZ | rat (plasma) | 50 | oral | N.A.; AUC0→t (ng·min/mL) = 42,462 ± 1812.6 | 0.131 ± 0.01 | 0.25 ± 0.00 | [84] |
| SMEDDS | rat (plasma) | 50 | oral | 106.51 ± 9.47 | 0.209 ± 0.04 | 2.00 ± 1.00 | [85] |
| Oleanolic acid tablet | rat (plasma) | 50 | oral | 21.00 ± 4.42 | 0.077 ± 0.01 | 2.75 ± 0.50 | [85] |
| OANS | rat (plasma) | 2 | intravenous | 121.49 ± 27.37 | 21.98 ± 5.79 | N.A. | [86] |
| OANS | rat (plasma) | 10 | oral | 21.35 ± 3.89 | 0.39 ± 0.17 | 0.21 ± 0.07 | [86] |
| OANS | rat (plasma) | 20 | oral | 44.06 ± 7.25 | 0.81 ± 0.25 | 0.35 ± 0.13 | [86] |
| OA coarse suspension | rat (plasma) | 20 | oral | 6.74 ± 3.42 | 0.06 ± 0.04 | 0.21 ± 0.07 | [86] |
| Calenduloside E | Beagle dogs (plasma) | 4.2 | oral | N.A.; AUC0→t (ng·h/mL) = 83.51 ± 26.91 | 0.013 ± 0.004 | 1.33 ± 0.52 | [87] |
| Calenduloside E | Beagle dogs (plasma) | 2.1 | intravenous | N.A.; AUC0→t (ng·h/mL) = 395.19 ± 167.79 | 1.057 ± 0.591 | 0.083 ± 0.00 | [87] |
| Commercial oleanolic acid tablet | Beagle dogs (plasma) | 6.6 | oral | N.A.; AUC0→24 (ng·h/mL) = 128.87 ± 37.55 | 0.03 ± 0.005 | 1.50 ± 0.45 | [88] |
| OA-silica capsules | Beagle dogs (plasma) | 6.6 | oral | N.A.; AUC0→24 (ng·h/mL) = 228.51 ± 20.35 | 0.07 ± 0.01 | 1.17 ± 0.26 | [88] |
| Compound | Specie (Sample) | D (mg/kg) | Route of Administration | AUC0→12 (µg·h/mL) | Cmax (µg/mL) | Tmax (h) | Reference |
|---|---|---|---|---|---|---|---|
| Ursolic acid | rat (plasma) | 10 | oral | 1.37 ± 0.43 | 1.17 ± 0.27 | 0.75 ± 0.07 | [92] |
| Ursolic acid nanoparticles freshly prepared | rat (plasma) | 10 | oral | 36.57 ± 1.90 | 9.32 ± 0.46 | 0.5 ± 0.04 | [92] |
| Ursolic acid | rat (plasma) | 100 | oral | 0.98 ± 0.05 | 0.29 ± 0.27 | 1.2 ± 0.3 | [93] |
| Ursolic acid nanoparticles | rat (plasma) | 100 | oral | 2.84 ± 0.11 | 1.27 ± 0.12 | 1.1 ± 0.2 | [93] |
| Ursolic acid | mice (plasma) | 20 | intravenous | N.A.; AUC (mg·h/L) = 36.88 ± 2.16 | 43,820 ± 4490 | N.A. | [93] |
| Ursolic acid PEGylated liposome | mice (plasma) | 20 | intravenous | N.A.; AUC (mg·h/L) = 316.11 ± 3.48 | 87,150 ± 10480 | N.A. | [94] |
| Ursolic acid FR-targeted liposome | mice (plasma) | 20 | intravenous | N.A.; AUC (mg·h/L) = 218.32 ± 12.73 | 109.30 ± 8300 | N.A. | [94] |
| Compound | Dose (mg) | Route of Administration | Condition | AUC0→∞ (ng·h /mL) (Mean Value) | Cmax (ng/mL) (Mean Value) | Tmax (h) (Mean Value) | Reference |
|---|---|---|---|---|---|---|---|
| Capsule Wok Vel™ (11-keto-β-boswellic acid) | 333 | oral | N.A. | N.A.; AUC0→∞ (µmol/mL·h) = 27.33 × 10−3 | N.A.; AUC0→∞ (µmol/mL) = 2.72 × 10−3 | 4.5 | [19] |
| B. serrata dry extract (β-boswellic acid) | 786 | oral | fasting | 6697.1 | 188.2 | 4.0 | [18] |
| B. serrata dry extract (β-boswellic acid) | 786 | oral | food | 23,316.7 | 1120.1 | 8.0 | [18] |
| B. serrata dry extract (11-keto-β-boswellic acid) | 786 | oral | fasting | 1660.72 | 83.8 | 3.5 | [18] |
| B. serrata dry extract (11-keto-β-boswellic acid) | 786 | oral | food | 3037.15 | 227.1 | 4.0 | [18] |
| B. serrata dry extract (acetyl-11-keto-β-boswellic acid) | 786 | oral | fasting | 153.6 | 6.0 | 2.0 | [18] |
| B. serrata dry extract (acetyl-11-keto-β-boswellic acid) | 786 | oral | food | 748.9 | 28.8 | 3.0 | [18] |
| B. serrata dry extract (α-boswellic acid) | 786 | oral | food | 9695 | 316.7 | 8.0 | [18] |
| B. serrata dry extract (acetyl-α-boswellic acid) | 786 | oral | food | N.A.; AUC0→t (ng·h /mL) = 1636 | 118.5 | 8.0 | [18] |
| Boswelan capsule (11-keto-β-boswellic acid) | 800 | oral | fasting | 859.4 | 156.7 | 2.4 | [17] |
| Boswelan capsule (11-keto-β-boswellic acid) | 800 | oral | food | 1179.2 | 205.7 | 2.5 | [17] |
| Boswelan capsule (3-acetyl-11-keto-β-boswellic acid) | 800 | oral | fasting | 72.2 | 30.3 | 1.9 | [17] |
| Boswelan capsule (3-acetyl-11-keto-β-boswellic acid) | 800 | oral | food | 112.1 | 32.8 | 2.1 | [17] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
J. C. Furtado, N.A.; Pirson, L.; Edelberg, H.; M. Miranda, L.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; André, C.M. Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies. Molecules 2017, 22, 400. https://doi.org/10.3390/molecules22030400
J. C. Furtado NA, Pirson L, Edelberg H, M. Miranda L, Loira-Pastoriza C, Preat V, Larondelle Y, André CM. Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies. Molecules. 2017; 22(3):400. https://doi.org/10.3390/molecules22030400
Chicago/Turabian StyleJ. C. Furtado, Niege A., Laetitia Pirson, Hélène Edelberg, Lisa M. Miranda, Cristina Loira-Pastoriza, Véronique Preat, Yvan Larondelle, and Christelle M. André. 2017. "Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies" Molecules 22, no. 3: 400. https://doi.org/10.3390/molecules22030400
APA StyleJ. C. Furtado, N. A., Pirson, L., Edelberg, H., M. Miranda, L., Loira-Pastoriza, C., Preat, V., Larondelle, Y., & André, C. M. (2017). Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies. Molecules, 22(3), 400. https://doi.org/10.3390/molecules22030400