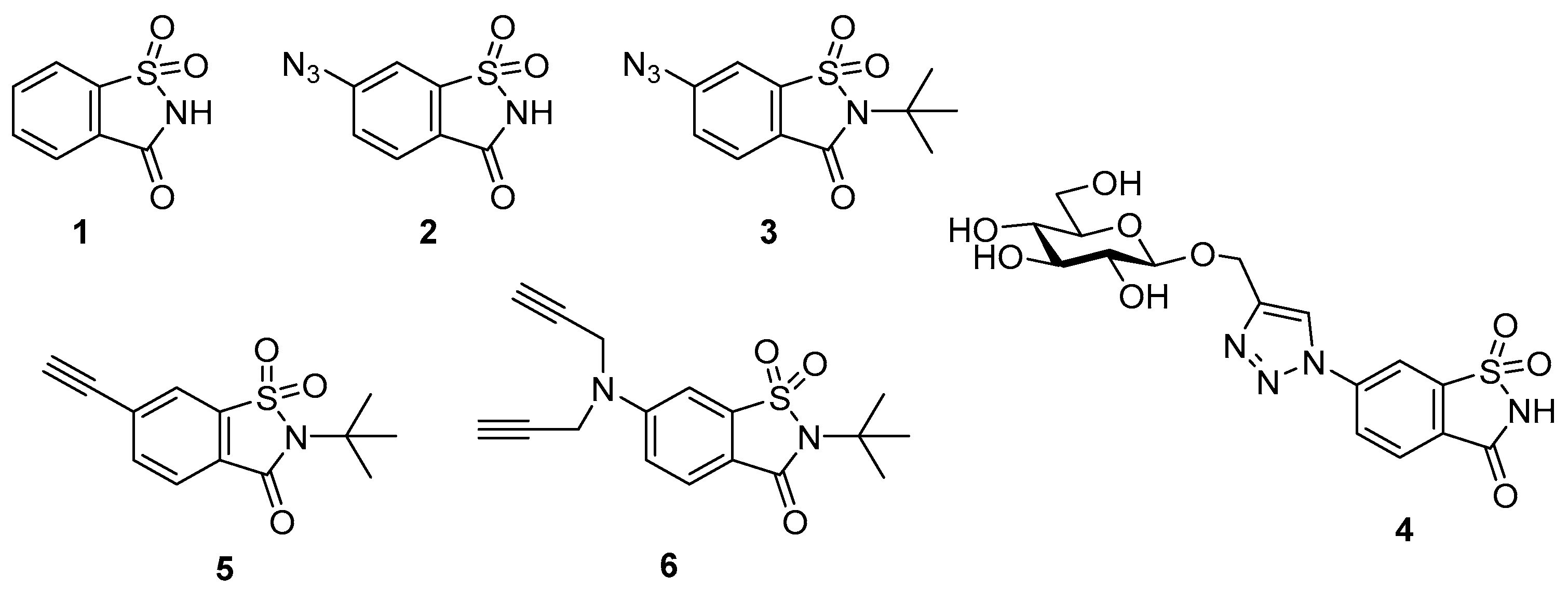
2. Results and Discussion
In our previous study using 6-azidosaccharins, we established that it was preferable to undertake CuAAC reactions with
N-
t-butyl protected 6-azidosaccharin
3, followed by removal of the
t-butyl protecting group, over the more direct synthetic approach of using 6-azidosaccharin
2 [
10]. Specifically, the ease of synthesis and isolated yield substantially improved when using
3 owing to the simplified reaction workup and product purification. We attributed these advantages to the blockade of metal complex formation between
3 and Cu
2+ (from the CuSO
4 used for CuAAC) by the
N-
t-butyl protecting group. Building on this experience we selected
N-t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide (
N-
t-butyl-protected 6-ethynylsaccharin
5) as the target building block for CuAAC in the present study (
Figure 1). Additionally,
N-
t-butyl-6-
N,N-bis(prop-2-yn-1-yl)amino-1,2-benzisothiazole-3-one-1,1-dioxide
6 was selected as the bis-alkyne rather than the unprotected form without the
t-butyl protecting group (
Figure 1).
The synthetic route to alkynes
5 and
6 and the earlier reported azide
3 have a common precursor,
N-
t-butyl-6-aminosaccharin
7 [
10] (
Scheme 1). Iodination of
7 with sodium nitrite and potassium iodide was achieved following a literature procedure used for iodination of similar aromatic and heterocyclic compounds to give the
N-protected 6-iodosaccharin
8 in an 83% yield [
21]. The Sonogashira cross-coupling reaction between
8 and ethynyltrimethylsilane
l generated the trimethylsilyl protected alkyne
9 in high yield. Removal of the trimethylsilyl group of
9 under standard conditions of K
2CO
3 in methanol proceeded, however these conditions additionally caused ring opening at the C-N bond of the heterocycle. The successful removal of this silyl group was instead achieved using mild acidic reaction conditions (tetrabutylammonium fluoride (TBAF)/1% AcOH in tetrahydrofuran (THF)) to afford the target alkyne
5 in an almost quantitative yield. Next, to install the two terminal alkyne groups of
6, the amino saccharin compound
7 [
10] was treated with propargyl bromide (2.2 equivalents (equiv)) in the presence of Cs
2CO
3.
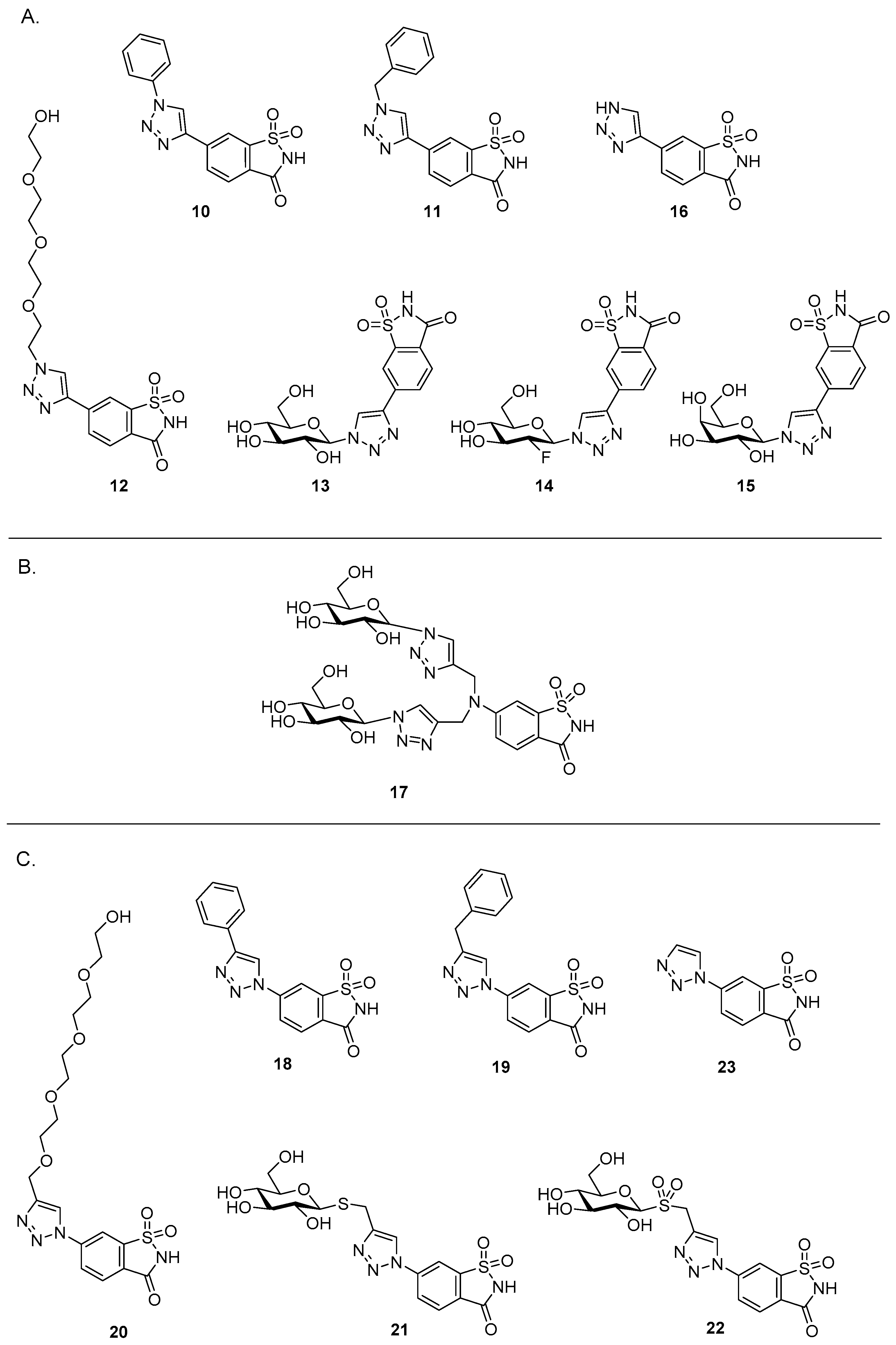
The target triazole derivatives of compound
1 for this study are shown in
Figure 3. Compounds derived from alkyne
5 and azides
a–
g include the phenyl derivative
10, benzyl derivative
11, tetraethylene glycol (PEG) derivative
12, sugar derivatives
13–
15, and unsubstituted triazole derivative
16 (
Figure 3A). The ‘sugar coated’ bis-triazole saccharin derivative
17 (generated from
6 and azide
d/d′) comprises two glucose moieties (
Figure 3B). The novel target triazoles derived from azide
8 and alkynes
h–
l include the benzyl derivative
19, PEG derivative
20, sugar derivatives
21 and
22, and unsubstituted triazole
16 (
Figure 3C). We have previously reported the phenyl derivative
18 [
10].
The reaction of
5 with azidobenzene
a [
22], benzylazide
b [
23], and PEG azide
c [
24] was carried out under typical CuAAC conditions (0.2 equiv of CuSO
4∙5H
2O and 0.4 equiv of sodium ascorbate,
t-BuOH:water 1:1, 45 °C) to generate triazoles
24–
26, respectively, in 74%–97% yield (
Scheme 2). Subsequent removal of the
N-
t-butyl group of
24–
26 was achieved following reflux in trifluoracetic acid (TFA) for 18 h to furnish the target derivatives of
1, triazoles
10–
12, respectively, in high yield (85%–99%). The reaction of alkyne
5 and 2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranosyl azide
d′ [
25] at 50 °C produced triazole
27 in high yield (90%). As we had concerns that the harsh basic conditions promote opening of the saccharin heterocyclic ring, the acetyl groups of
27 were hydrolysed using HCl in MeOH instead of the more usual Zemplén conditions of methoxide in MeOH [
26], to afford
28 in a 94% yield, however a lengthy reaction time of 90 h was required (
Scheme 2). The
N-
t-butyl group of
28 was removed with refluxing in TFA for 18 h to yield the target glycoconjugate
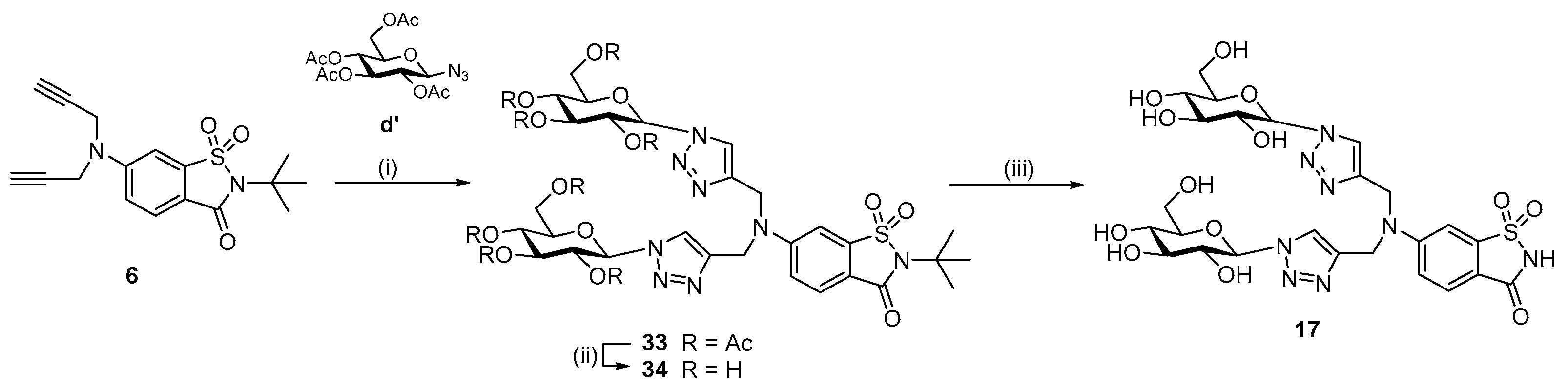
13 in high yield. The bis-triazole saccharin glycoconjugate
33 was prepared from bis-alkyne
6 and per-
O-acetylated glucosyl azide
d′ [
25] in high yield (87%) (
Scheme 3). Deacetylation of
33 under acidic conditions (HCl in MeOH, 90 h) gave the free sugar
34, and cleavage of the
N-
t-butyl group of
34 with TFA furnished the target bis-triazole saccharin glycoconjugate compound
17 (
Scheme 3). As the acidic conditions to remove the acetyl groups of
27 and
33 required a prolonged reaction time (90 h), this prompted us to investigate an alternate route to synthesise the glycoconjugates
14 and
15. This route employed free glycosyl azides
e and
f, instead of the corresponding per-
O-acetylated glycosyl azides, to eliminate the need for deprotection of the sugar hydroxyl groups following CuAAC, thus removing the dependence on this synthetic bottleneck [
25,
27,
28,
29]. The reaction of alkyne
5 and free glycosyl azides
e and
f [
25,
27,
28,
29] via CuAAC proceeded smoothly to form intermediates
29 and
30 (
Scheme 2). Subsequent removal of the
N-
t-butyl protecting groups of these intermediates by overnight refluxing in TFA afforded target glycoconjugates
14 and
15, respectively. Next, CuAAC of azidotrimethylsilane (TMSN
3)
g with alkyne
5 gave the monosubstituted triazole
31 as an inseparable mixture of thermodynamically stable tautomers [
30]. Cleavage of the
N-
t-butyl group of
31 using TFA furnished a mixture of
32a and
32b, where
1H nuclear magnetic resonance (NMR) and high resolution mass spectrometry (HRMS) analysis confirmed triazole N-alkylation, with the
t-butyl group on either the N-1 (
32a) or N-2 (
32b) of the triazole ring. Although the reaction of
5 with azidotrimethylsilane
g did not yield the intended target triazole
16, both
32a and
32b are novel compounds that retain the cyclic sulfonamide functional group of
1.
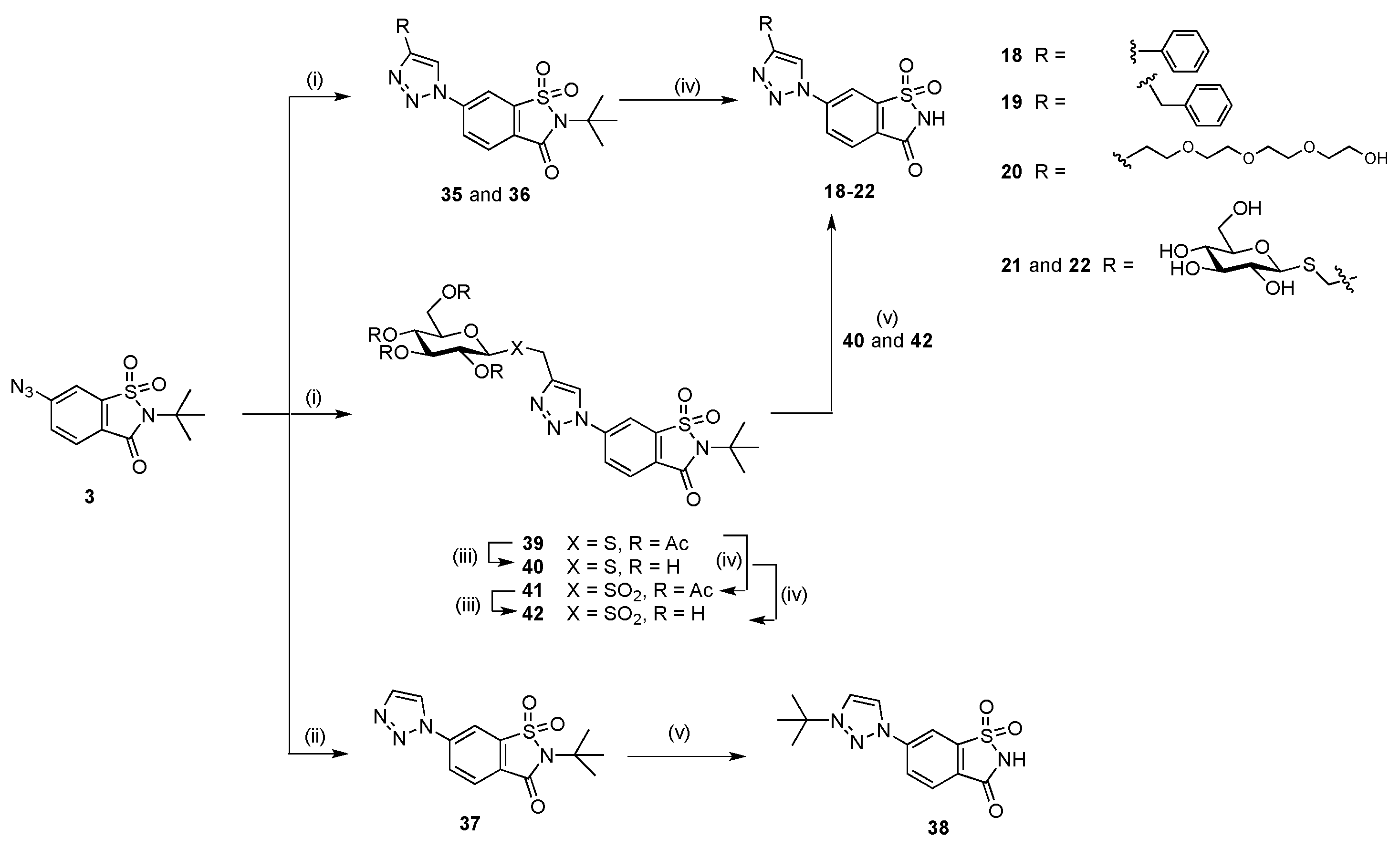
The target compounds prepared from the reaction of azidosaccharin
3 [
10] with alkynes
h–
l have similar physicochemical diversity to azides
a–
g reacted with the ethynylsaccharin
5 (
Figure 3). We have previously reported the synthesis of the phenyl derivative
18 while all other compounds are novel [
10]. Reaction of azide
3 with 3-phenyl-1-propyne (benzyl alkyne)
i and PEG alkyne
j [
31] under standard CuAAC conditions gave triazoles
35 and
36, respectively (
Scheme 4). Acid mediated cleavage of the
t-butyl protecting group in refluxing TFA furnished the target saccharin compounds
19 and
20, respectively. CuAAC of ethynyltrimethylsilane (TMSC≡CH)
l and azide
3 [
10] gave
37, a 1-substituted 1,2,3-triazole. Consistent with the outcome of deprotection of the related 1-substituted triazole
31, treatment of
37 with TFA removed the
N-
t-butyl group from the sulfonamide nitrogen, but furnished the alternate N-alkylation product
38, where the
t-butyl group is on N-3 of the 1,2,3-triazole instead of the desired target compound
16 (
Scheme 4).
1H-NMR and HRMS confirmed the formation of
38. Finally, the reaction of saccharin azide
3 and propargyl 2,3,4,6-tetra-
O-acetyl-thio-β-
d-glucopyranoside
k′ [
14] using CuAAC gave glycoconjugate
39 in high yield. Acidic cleavage of the acetyl groups of
39 (HCl in MeOH, 90 h) gave the free sugar derivative
40. Given the long 90 h reaction time, triazole
40 was also prepared directly from
3 utilising propargyl thio-β-
d-glucopyranoside
k [
32], as described for
14 and
15. Oxidation of
39 and
40 with
m-chloroperbenzoic acid (
mCPBA) gave sulfones
41 and
42 in high yields, respectively. Compound
42 was also prepared by the deacetylation of
41 under acidic conditions, and this demonstrated the versatility of protecting group manipulation in the presence of the saccharin core scaffold. The
N-
t-butyl protecting group of
40 and
42 was removed by refluxing in TFA for 18 h to afford
21 and
22, respectively (
Scheme 4).
3. Materials and Methods
3.1. General Chemistry
All starting materials and reagents were purchased from commercial suppliers. All solvents were available commercially dried or dried prior to use. Reaction progress was monitored by thin layer chromatography (TLC) using silica gel-60 F254 plates (Merck Millipore, Darmstadt, Germany) with detection by short wave ultraviolet (UV) fluorescence (λ = 254 nm) and staining with 5%
w/
v dodecamolybdophosphoric acid in ethanol or vanillin staining (5 g of vanillin in a mixture of EtOH:H
2O:H
2SO
4 = 85:10:2.75) with subsequent heating. Silica gel flash chromatography was performed using silica gel 60 Å (230–400 mesh) (Merck Millipore, Darmstadt, Germany). NMR (
1H,
13C,
19F, gradient correlation spectroscopy (gCOSY), and heteronuclear single quantum coherence (HSQC) spectra were recorded on either a 400 or 500 MHz spectrometer at 30 °C.
1H-NMR spectra were obtained at 500 MHz and were referenced to the residual solvent peak (CDCl
3 δ 7.26 ppm, dimethylsulfoxide (DMSO)-
d6 δ 2.50 ppm).
13C-NMR spectra were recorded at 125 MHz and were referenced to the internal solvent (CDCl
3 δ 77.0 ppm, DMSO-
d6 δ 39.5 ppm).
19F-NMR spectra were recorded at 376 MHz. Multiplicity is indicated as follows: s (singlet); d (doublet); t (triplet); m (multiplet); dd (doublet of doublet); ddd (doublet of doublet of doublet); b (broad). Coupling constants are reported in hertz (Hz). Melting points are uncorrected. Low and high resolution mass spectra (MS) were recorded using electrospray ionization (ESI) in positive ion and/or negative ion modes as stated. All MS analysis samples were prepared as solutions in methanol. The purity of all compounds was ≥95% as determined by HPLC with UV.
1H-,
13C-, and
19F-NMR spectra of all novel compounds are provided in the
supporting information.
N-
t-Butyl-6-amino-1,2-benzisothiazole-3-one-1,1-dioxide
7,
N-
t-butyl-6-azido-1,2-benzisothiazole-3-one-1,1-dioxide
3, and phenyl derivative
18 were synthesised using methods we have previously reported [
10]. Azide and alkyne building blocks that were not commercially available were prepared in accordance with the literature, including: azidobenzene
a [
22], benzylazide
b [
23], PEG azide
c [
24], 2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranosyl azide
d′ [
25], 2-deoxy-2-fluoro-β-
d-glucopyranosyl azide
e [
28], 2-deoxy-2-fluoro-β-
d-glycopyranosyl azide
f [
28], propargyl 2,3,4,6-tetra-
O-acetyl-thio-β-
d-glucopyranoside
k′ [
14], propargyl thio-β-
d-glucopyranoside
k [
32], and PEG alkyne
j [
31].
3.2. General Procedure 1—CuAAC
A mixture of azide (1.0 equiv) and alkyne (1.0 equiv) was prepared in t-butyl alcohol/H2O (1:1, 6–10 mL). To the mixture was added a solution of sodium ascorbate (0.4 equiv) in water (0.25 mL) followed by a solution of CuSO4.5H2O (0.2 equiv) in water (0.25 mL). The resulting suspension was stirred vigorously at the temperature and time indicated below. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel using the eluent conditions described below.
3.3. N-t-Butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide (5)
TBAF (1.0 M in THF, 0.328 mL, 0.328 mmol) was added to a solution of N-t-butyl-6-trimethylsilylethynyl-1,2-benzisothiazole-3-one-1,1-dioxide (9) (0.100 g, 0.298 mmol) and acetic acid (0.051 mL, 0.894 mmol) in THF (5 mL). The reaction mixture was stirred for 5 min, then quenched by the addition of water (20 mL) and extracted into EtOAc (3 × 30 mL). The combined organic fractions were washed with brine (30 mL), dried with MgSO4, filtered, and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:9) to give the title compound 5 (0.076 g, 97%) as a pale yellow solid. m.p. 162–164 °C (EtOAc/hexane); 1H-NMR (500 MHz, CDCl3) δ 1.76 (s, 9H, tBu), 3.40 (s, 1H, CHalkyne), 7.83 (dd, J = 1.3, 7.9 Hz, 1H, Ar-H), 7.90 (dd, J = 0.7, 1.4 Hz, 1H, Ar-H), 7.93 (dd, J = 0.7, 7.9 Hz, 1H, Ar-H); 13C-NMR (125 MHz, CDCl3) δ 27.9 (C(CH3)3), 61.6 (C(CH3)3), 80.9 (PhCCH), 83.1 (PhCCH), 123.6 (Ar-CH), 124.6 (Ar-CH), 126.8 (Ar-C), 129.1 (Ar-C), 137.5 (Ar-CH), 138.2 (Ar-C), 159.4 (C=O); HRMS-ESI [M + Na]+ Calcd. for C13H13NNaO3S: 286.0508. Found: 286.0529.
3.4. N-t-Butyl-6-N,N-bis(prop-2-yn-1-yl)amino-1,2-benzisothiazole-3-one-1,1-dioxide (6)
To a solution of
N-
t-butyl-6-amino-1,2-benzisothiazole-3-one-1,1-dioxide
7 [
10] (0.100 g, 0.393 mmol) in DMF (5 mL) was added cesium carbonate (0.256 g, 0.786 mmol) and the solution was cooled to 0 °C. Propargyl bromide 80% solution in toluene (0.096 mL, 0.865 mmol) was added dropwise and the solution was left to stir for 48 h. The solvent was removed in vacuo. The residue was dissolved into EtOAc (50 mL) and washed with water (3 × 40 mL). The combined organic fractions were washed with brine (50 mL), dried (MgSO
4), and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:9 to 1:2) to give the title compound
6 (0.098 g, 75%) as a white solid. m.p. 178–180 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.37 (s, 9H,
tBu), 3.27 (t,
J = 2.3 Hz, 2H, CH
2CC
H), 4.43 (d,
J = 2.5 Hz, 4H, NCH
2), 7.29 (dd,
J = 2.4, 8.8 Hz, 1H, Ar-H), 7.42 (d,
J = 2.3 Hz, 1H, Ar-H), 7.85 (d,
J = 8.7 Hz, 1H, Ar-H);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.5 (C(
CH
3)
3), 40.3 (NCH
2), 59.8 (
C(CH
3)
3), 75.6 (CH
2C
CH), 78.7 (CH
2CCH), 103.7 (Ar-CH), 114.2 (Ar-C), 118.6, 125.5 (Ar-CH), 139.4, 151.9 (Ar-C), 159.8 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
17H
18N
2NaO
3S: 353.0930. Found: 353.0944.
3.5. N-t-Butyl-6-iodo-1,2-benzisothiazole-3-one-1,1-dioxide (8)
To a solution of
p-toluenesulfonic acid,
pTsOH·H
2O (4.49 g, 23.6 mmol) in CH
3CN (20 mL) was added
N-
t-butyl-6-amino-1,2-benzisothiazole-3-one-1,1-dioxide (
7) [
10] (2.00 g, 7.86 mmol). The resulting suspension of amine salt was cooled to 10–15 °C and to this was added, dropwise, a solution of NaNO
2 (1.09 g, 15.7 mmol) and KI (3.26 g 19.7 mmol) in H
2O (5 mL). The reaction mixture was stirred for 10 min, then warmed to r.t. and stirred for 1 h. To the reaction mixture was added H
2O (10 mL), NaHCO
3 (1.0 M; until pH = 9–10) and Na
2S
2O
3 (2.0 M, 5 mL). The solution was extracted with EtOAc (3 × 50 mL) and the combined organic extracts were washed with brine (100 mL), dried (MgSO
4), and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:2) to give the title compound
8 (2.39 g, 83%) as a light brown solid. m.p. 166–168 °C (EtOAc/hexane);
1H-NMR (500 MHz, CDCl
3) δ 1.75 (s, 9H,
tBu), 7.69 (dd,
J = 0.6, 8.0 Hz, 1H, Ar-H), 8.11 (dd,
J = 1.4, 8.0 Hz, 1H, Ar-H), 8.16 (dd,
J = 0.5, 1.4 Hz, 1H, Ar-H);
13C-NMR (125 MHz, CDCl
3) δ 27.8 (C(
CH
3)
3), 61.6 (
C(CH
3)
3), 101.1 (Ar-C), 125.8 (Ar-CH), 126.8 (Ar-C), 129.0 (Ar-CH), 139.0 (Ar-C), 143.3 (Ar-CH), 159.6 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
11H
12INNaO
3S: 387.9475. Found: 387.951.
3.6. N-t-Butyl-6-trimethylsilylethynyl-1,2-benzisothiazole-3-one-1,1-dioxide (9)
N-t-Butyl-6-iodo-1,2-benzisothiazole-3-one-1,1-dioxide (8) (1.00 g, 2.74 mmol), Pd(PPh3)Cl2 (0.077 g, 0.110 mmol) and CuI (0.026 g, 0.137 mmol) were dried together under high vacuum and then flushed with argon. Triethylamine (20 mL) was added and the reaction mixture was stirred and heated to 40 °C. To this was added ethynyltrimethylsilane l (0.468 mL, 3.29 mmol) and the solution was stirred for 2 h. The reaction mixture was filtered through a pad of Celite and washed with EtOAc (100 mL). The residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:19 to 1:9) to give the title compound 9 (0.871 g, 95%) as a light brown solid. m.p. 114–115 °C (EtOAc/hexane); 1H-NMR (500 MHz, CDCl3) δ 0.27 (s, 9H, Si(CH3)3), 1.76 (s, 9H, tBu), 7.78 (dd, J = 1.3, 7.9 Hz, 1H, Ar-H), 7.87 (dd, J = 0.7, 1.3 Hz, 1H, Ar-H), 7.90 (dd, J = 0.7, 7.9 Hz, 1H, Ar-H); 13C-NMR (125 MHz, CDCl3) δ −0.3 (Si(CH3)3), 27.9 (C(CH3)3), 61.5 (C(CH3)3), 101.7 (Si(CH3)3C), 101.8 (Si(CH3)3CC), 123.4 (Ar-CH), 124.5 (Ar-CH), 126.3 (Ar-C), 130.2 (Ar-C), 137.2 (Ar-CH), 138.1 (Ar-C), 159.6 (C=O); HRMS-ESI [M + Na]+ Calcd. for C16H21NNaO3SSi: 358.0904. Found: 358.0899.
3.7. 6-(1-Phenyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (10)
N-t-Butyl protected 24 (0.090 g, 0.235 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo. EtOAc was added to the residue and the solid was collected by filtration to give the title compound 10 (0.065 g, 85%) as a white solid. m.p. greater than 300 °C (EtOAc); 1H-NMR (500 MHz, (CD3)2SO) δ 7.55 (t, J = 7.4 Hz, 1H, Ar-H), 7.66 (t, J = 7.9 Hz, 2H, Ar-H), 7.94 (d, J = 7.6 Hz, 2H, Ar-H), 8.13 (d, J = 8.0 Hz, 1H, Ar-H), 8.50 (dd, J = 1.4, 8.0 Hz, 1H, Ar-H), 8.60 (d, J = 1.4 Hz, 1H, Ar-H), 9.64 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 117.2, 120.1 (Ar-CH), 122.2 (CHtriazole), 125.8 (Ar-CH), 126.8 (Ar-C), 129.1, 130.1, 130.5 (Ar-CH), 136.3, 137.0, 140.7 (Ar-C), 145.0 (Ctriazole), 160.7 (C=O); HRMS-ESI [M − H]− Calcd. for C15H9N4O3S: 325.0389. Found: 325.0378.
3.8. 6-(1-Benzyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (11)
N-t-Butyl protected 25 (0.080 g, 0.202 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 1:4) and gave the title compound 11 (0.068 g, 99%) as a white solid. m.p. 247–249 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ 5.67 (s, 2H, CH2), 7.33–7.43 (m, 5H, Ar-H), 7.66 (d, J = 8.2 Hz, 1H, Ar-H), 8.09–8.12 (m, 2H, Ar-H), 8.87 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 53.2 (CH2), 115.7, (Ar-CH), 122.9 (CHtriazole), 123.3, 128.0, 128.2, 128.8 (Ar-CH), 133.5, 133.6, 135.7, 145.5 (Ar-C), 146.0 (Ctriazole), 167.1 (C=O); HRMS-ESI [M − H]− Calcd. for C16H11N4O3S: 339.0546. Found: 339.0534.
3.9. 6-(1-[2-[2-[2-(2-Hydroxyethoxy)ethoxy]ethoxy]ethyl]-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (12)
N-t-Butyl protected 26 (0.087 g, 0.180 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo and the residue purified by reverse phase (RP-18) column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 1:9) to give the title compound 12 (0.074 g, 96%) as a colourless oil which solidified to a white gum upon standing. 1H-NMR (500 MHz, (CD3)2SO) δ 3.34–3.37 (m, 2H, CH2), 3.42–3.52 (m, 10H, CH2), 3.54–3.57 (m, 2H, CH2), 3.88 (dd, J = 4.6, 5.6 Hz, 2H, CH2), 4.61 (t, J = 5.1 Hz, 1H, OH), 7.94 (d, J = 7.9 Hz, 1H, Ar-H), 8.32 (dd, J = 1.4, 7.9 Hz, 1H, Ar-H), 8.40 (d, J = 1.4 Hz, 1H, Ar-H), 8.86 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 49.9, 60.2, 68.5, 69.60, 69.64, 69.7, 69.8, 72.3 (CH2), 116.7, (Ar-CH), 124.0 (CHtriazole), 125.2 (Ar-CH), 128.0 (Ar-C), 129.9 (Ar-CH), 136.7, 141.9 (Ar-C), 144.2 (Ctriazole), 162.2 (C=O); HRMS-ESI [M + Na]+ Calcd. for C17H22N4NaO7S: 425.1136. Found: 425.1132.
3.10. 6-(1-β-d-Glucopyranosyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (13)
N-t-Butyl protected 28 (0.060 g, 0.128 mmol) was refluxed in TFA (1.5 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 1:19) to give the title compound 13 (0.050 g, 94%) as a white solid. m.p. 227–229 °C (MeOH/H2O); 1H-NMR (500 MHz, (CD3)2SO) δ 3.26 (t, J = 9.2 Hz, 1H, H-4), 3.42–3.49 (m, 2H, H-3, H-6), 3.50–3.55 (m, 1H, H-5), 3.71–3.78 (m, 2H, H-2, H-6), 5.64 (d, J = 9.2 Hz, 1H, H-1), 8.08 (d, J = 8.0 Hz, 1H, Ar-H), 8.45 (dd, J = 1.4, 8.0 Hz, 1H, Ar-H), 8.57 (d, J = 1.3 Hz, 1H, Ar-H), 9.23 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 60.7 (C-6), 69.6 (C-4), 72.5 (C-2), 76.6 (C-3), 80.0 (C-5), 87.8 (C-1), 117.0, (Ar-CH), 123.0 (CHtriazole), 125.7 (Ar-CH), 126.9 (Ar-C), 130.3 (Ar-CH), 137.2, 140.9 (Ar-C), 144.2 (Ctriazole), 161.0 (C=O); HRMS-ESI [M − H]− Calcd. for C15H15N4O8S: 411.0616. Found: 411.0601.
3.11. 6-(1-[2-Deoxy-2-fluoro-β-d-glucopyranosyl]-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (14)
N-t-Butyl protected 29 (0.080 g, 0.170 mmol) was refluxed in TFA (1.5 mL) for 18 h. The solvent was removed in vacuo and the residue purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 1:9) to give the title compound 14 (0.057 g, 81%) as a white solid. m.p. 274–276 °C (MeOH/H2O); 1H-NMR (500 MHz, (CD3)2SO) δ 3.35 (dd, J = 8.9, 9.8 Hz, 1H, H-4), 3.49 (dd, J = 5.7, 12.2 Hz, 1H, H-6), 3.67 (ddd, J = 2.0, 5.8, 9.9 Hz, 1H, H-5), 3.73 (dd, J = 2.0, 12.3 Hz, 1H, H-6), 3.82 (dt, J = 8.9, 15.6 Hz, 1H, H-3), 4.80 (dt, J = 9.0, 51.0 Hz, 1H, H-2), 6.20 (dd, J = 2.4, 9.1 Hz, 1H, H-1), 8.09 (d, J = 8.1 Hz, 1H, Ar-H), 8.43 (dd, J = 1.4, 8.1 Hz, 1H, Ar-H), 8.55 (d, J = 1.4 Hz, 1H, Ar-H), 9.35 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 60.3 (C-6), 69.3 (d, J = 8.0 Hz, C-4), 74.1 (d, J = 15.9 Hz, C-3), 79.9 (C-5), 84.2 (d, J = 24.2 Hz, C-1), 91.2 (d, J = 186.8 Hz, C-2), 117.2 (Ar-H), 123.1 (CHtriazole), 125.7 (Ar-CH), 127.1 (Ar-C), 130.1 (Ar-CH), 136.7, 140.1 (Ar-C), 144.7 (Ctriazole), 160 (C=O); 19F-NMR (376 MHz, (CD3)2SO) δ -193.6 (ddd, J = 2.4, 15.6, 51.1 Hz); HRMS-ESI [M − H]− Calcd. for C15H14FN4O7S: 413.0573. Found: 413.0563.
3.12. 6-(1-β-d-Galactopyranosyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (15)
N-t-Butyl protected 30 (0.080 g, 0.171 mmol) was refluxed in TFA (1.5 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:19, product eluting at 1:32) to give the title compound 15 (0.024 g, 34%) as a white solid. m.p. 232–235 °C (MeOH/H2O); 1H-NMR (500 MHz, (CD3)2SO) δ 3.50–3.58 (m, 2H, 6-CH2), 3.60 (dd, J = 3.1, 9.4 Hz, 1H, H-3), 3.76–3.82 (m, 2H, H-4, H-5), 4.09 (t, J = 9.2 Hz, 1H, H-2), 5.57 (d, J = 9.1 Hz, 1H, H-1), 7.95 (d, J = 8.0 Hz, 1H, Ar-H), 8.30 (dd, J = 1.5, 7.9 Hz, 1H, Ar-H), 8.49 (d, J = 1.4 Hz, 1H, Ar-H), 9.16 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 60.5 (C-6), 68.4 (C-4), 69.4 (C-2), 73.5 (C-3), 78.5 (C-5), 88.4 (C-1), 116.7 (Ar-CH), 122.5 (CHtriazole), 124.9 (Ar-CH), 128.9 (Ar-C), 129.7 (Ar-CH), 136.1, 142.5 (Ar-C), 144.6 (Ctriazole), 162.9 (C=O); HRMS-ESI [M − H]− Calcd. for C15H15N4O8S: 411.0616. Found: 411.0608.
3.13. 6-N,N-Bis([1-β-d-glucopyranosyl-1H-1,2,3-triazol-4-yl]methyl)amino-1,2-benzisothiazole-3-one-1,1-dioxide (17)
N-t-Butyl protected 34 (0.070 g, 0.095 mmol) was refluxed in TFA (2.0 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 1:9) to give the title compound 17 (0.057 g, 84%) as a white solid. m.p. 184–186 °C (MeOH/H2O); 1H-NMR (500 MHz, (CD3)2SO) δ 3.22 (t, J = 9.0 Hz, 2H, H-4), 3.38 (t, J = 8.9 Hz, 2H, H-3), 3.41–3.47 (m, 4H, H-5, H-6), 3.66–3.71 (m, 2H, H-6), 3.75 (t, J = 9.1 Hz, 2H, H-2), 4.48–4.72 (m, 2H, OH), 4.80–4.89 (m, 4H, NCH2), 5.03–5.46 (m, 6H, OH), 5.53 (d, J = 9.3 Hz, 2H, H-1), 7.30 (dd, J = 2.4, 8.9 Hz, 1H, Ar-H), 7.54 (d, J = 2.3 Hz, 1H, Ar-H), 7.68 (d, J = 8.8 Hz, 1H, Ar-H), 8.38 (s, 2H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 45.2 (NCH2), 60.7 (C-6), 69.6 (C-4), 72.1 (C-2), 76.9 (C-3), 80.0 (C-5), 87.5 (C-1), 102.8, (Ar-CH), 114.6 (Ar-C), 116.6 (Ar-C), 122.6 (CHtriazole), 125.7 (Ar-CH), 142.4 (Ar-C), 142.8 (Ctriazole), 152.8 (Ar-C), 161.4 (C=O); HRMS-ESI [M + Na]+ Calcd. for C25H32N8NaO13S: 707.1702. Found: 707.1756.
3.14. 6-(4-Benzyl-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (19)
N-t-Butyl protected 35 (0.080 g, 0.202 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 1:9 to 1:4) to give the title compound 19 (0.044 g, 64%) as a light brown solid. m.p. 177–179 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ 4.12 (s, 2H, CH2), 7.21–7.26 (m, 1H, Ar-H), 7.31–7.34 (m, 4H, Ar-H), 8.10 (d, J = 8.3 Hz, 1H, Ar-H), 8.43 (dd, J = 1.9, 8.4 Hz, 1H, Ar-H), 8.65 (d, J = 1.9 Hz, 1H, Ar-H), 8.86 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 31.2 (CH2) 111.9, (Ar-CH), 121.5 (CHtriazole), 124.8, 126.3, 126.4 (Ar-CH), 128.0 (Ar-C), 128.5, 128.6 (Ar-CH), 138.8, 140.8, 142.3 (Ar-C), 147.8 (Ctriazole), 161.4 (C=O); HRMS-ESI [M − H]− Calcd. for C16H11N4O3S: 339.0546. Found: 339.0532.
3.15. 6-[4-(13-Hydroxy-2,5,8,11-tetraoxatridec-1-yl)-1H-1,2,3-triazol-1-yl]-1,2-benzisothiazole-3-one-1,1-dioxide (20)
N-t-Butyl protected 36 (0.080 g, 0.156 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 1:9) to give the title compound 20 (0.052 g, 73%) as a colourless oil. 1H-NMR (500 MHz, (CD3)2SO) δ 3.38–3.41 (m, 2H, CH2), 3.45–3.53 (m, 10H, CH2), 3.55–3.58 (m, 2H, CH2), 3.61–3.64 (m, 2H, CH2), 4.58 (t, J = 5.5 Hz, 1H, OH), 4.63 (s, 2H, CH2), 7.78 (d, J = 8.1 Hz, 1H, Ar-H), 8.18 (dd, J = 1.9, 8.1 Hz, 1H, Ar-H), 8.23 (d, J = 1.9 Hz, 1H, Ar-H), 8.98 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 60.2, 63.3, 69.1, 69.7, 69.74, 69.79, 69.8, 72.3 (CH2), 110.8, (Ar-CH), 122.7 (CHtriazole), 123.3, 124.2 (Ar-CH), 134.3, 138.5, 145.4 (Ar-C), 147.1 (Ctriazole), 166.7 (C=O); HRMS-ESI [M + Na]+ Calcd. for C18H24N4NaO8S: 479.1207. Found: 479.1202.
3.16. 6-(4-{[β-d-Glucopyranosyl]thiomethyl}-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (21)
N-t-Butyl protected 40 (0.100 g, 0.194 mmol) was refluxed in TFA (1.5 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 1:9) to give the title compound 21 (0.057 g, 64%) as a white solid. m.p. 176–178 °C (MeOH/H2O); 1H-NMR (500 MHz, (CD3)2SO) δ 3.03–3.10 (m, 2H, H-2, H-4), 3.16 (t, J = 8.6 Hz, 1H, H-3), 3.20 (ddd, J = 2.0, 6.4, 9.8 Hz, 1H, H-5), 3.46 (dd, J = 6.4, 11.9 Hz, 1H, H-6), 3.73 (dd, J = 2.0, 11.9 Hz, 1H, H-6), 3.95 (d, J = 14.4 Hz, 1H, SCH2), 4.09 (d, J = 14.4 Hz, 1H, SCH2), 4.34 (d, J = 9.6 Hz, 1H, H-1), 7.99 (d, J = 8.3 Hz, 1H, Ar-H), 8.31 (dd, J = 1.9, 8.3 Hz, 1H, Ar-H), 8.47 (d, J = 1.8 Hz, 1H, Ar-H), 8.92 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 22.9 (SCH2), 61.3 (C-6), 70.1 (C-4), 73.2 (C-2), 78.1 (C-3), 81.0 (C-5), 84.0 (C-1), 111.5, (Ar-CH), 122.0 (CHtriazole), 124.3, 125.6 (Ar-CH), 130.3, 140.0, 143.9 (Ar-C), 146.2 (Ctriazole), 163.2 (C=O); HRMS-ESI [M + H]+ Calcd. for C16H19N4O8S2: 459.0638. Found: 459.0665.
3.17. 6-(4-{[β-d-Glucopyranosyl]sulfonylmethyl}-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (22)
N-t-Butyl protected 42 (0.120 g, 0.220 mmol) was refluxed in TFA (1.5 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by RP-18 column chromatography (MeOH/H2O = 0:1 to 1:9, product eluting at 5:95) to give the title compound 22 (0.046 g, 43%) as a white solid. m.p. 186–189 °C (MeOH/H2O); 1H-NMR (500 MHz, (CD3)2SO) δ 3.08 (dd, J = 8.8, 9.8 Hz, 1H, H-4), 3.29 (t, J = 8.8 Hz, 1H, H-3), 3.43 (ddd, J = 1.9, 6.8, 9.9 Hz, 1H, H-5), 3.51 (dd, J = 6.8, 12.1 Hz, 1H, H-6), 3.59 (t, J = 9.1 Hz, 1H, H-2), 3.80 (dd, J = 1.8, 12.2 Hz, 1H, H-6), 4.51 (d, J = 9.5 Hz, 1H, H-1), 4.70 (d, J = 14.7 Hz, 1H, SCH2), 4.85 (d, J = 14.7 Hz, 1H, SCH2), 8.14 (d, J = 8.3 Hz, 1H, Ar-H), 8.42 (dd, J = 1.9, 8.4 Hz, 1H, Ar-H), 8.65 (d, J = 1.8 Hz, 1H, Ar-H), 9.15 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 48.3 (SCH2), 61.1 (C-6), 69.2 (C-2), 69.5 (C-4), 77.5 (C-3), 81.6 (C-5), 88.6 (C-1), 112.4, (Ar-CH), 124.7 (CHtriazole), 125.3, 126.4 (Ar-CH), 128.4, 136.9, 140.7 (Ar-C), 142.2 (Ctriazole), 161.3 (C=O); HRMS-ESI [M + Na]+ Calcd. for C16H18N4NaO10S2: 513.0356. Found: 513.0404.
3.18. N-t-Butyl-6-(1-phenyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (24)
The title compound
24 was prepared from
N-t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide
5 (0.150 g, 0.570 mmol) and azidobenzene
a [
22] (0.068 g, 0.570 mmol) at 45 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (EtOAc/hexane = 9:1 to 1:1) gave the title compound
24 (0.162 g, 74%) as a yellow solid. m.p. 207–208 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ1.71 (s, 9H,
tBu), 7.53–7.56 (m, 1H, Ar-H), 7.64–7.68 (m, 2H, Ar-H), 7.91–7.95 (m, 2H, Ar-H), 8.15 (d,
J = 8.0 Hz, 1H, Ar-H), 8.51 (dd,
J = 1.4, 8.0 Hz, 1H, Ar-H), 8.61 (d,
J = 1.4 Hz, 1H, Ar-H), 9.65 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 60.5 (
C(CH
3)
3), 116.7, 120.0 (Ar-CH), 122.3 (CH
triazole), 125.1 (Ar-C), 125.7, 129.1, 130.0, 130.8 (Ar-CH), 136.3, 137.2, 138.1 (Ar-C), 144.8 (C
triazole), 159.2 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
19H
18N
4NaO
3S: 405.0992. Found: 405.0993.
3.19. N-t-Butyl-6-(1-benzyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (25)
The title compound
25 was prepared from
N-t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide
5 (0.099 g, 0.376 mmol) and benzylazide
b [
23] (0.050 g, 0.376 mmol) at 45 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (EtOAc/hexane = 9:1 to 1:2) gave the title compound
25 (0.144 g, 97%) as a white solid. m.p. 156–157 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ1.69 (s, 9H,
tBu), 5.70 (s, 2H, CH
2), 7.33–7.44 (m, 5H, Ar-H), 8.09 (d,
J = 8.1 Hz, 1H, Ar-H), 8.46 (dd,
J = 1.4, 8.1 Hz, 1H, Ar-H), 8.58 (d,
J = 1.4 Hz, 1H, Ar-H), 8.98 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 53.3 (CH
2), 60.5 (
C(CH
3)
3), 116.6, (Ar-CH), 124.1 (CH
triazole), 124.9 (Ar-C), 125.5, 128.1, 128.3, 128.9, 130.7 (Ar-CH), 135.5, 137.6, 138.1 (Ar-C), 144.3 (C
triazole), 159.3 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
20H
20N
4NaO
3S: 419.1148. Found: 419.1141.
3.20. N-t-Butyl-6-(1-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethyl]-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (26)
The title compound
26 was prepared from
N-t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide
5 (0.100 g, 0.380 mmol) and PEG azide
c [
24] (0.083 g, 0.380 mmol) at 45 °C in 16 h according to general procedure 1. Purification of the crude product by flash chromatography (MeOH/EtOAc = 0:1 to 5:95) gave the title compound
26 (0.167 g, 91%) as a pale-yellow oil.
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.71 (s, 9H,
tBu), 3.34–3.37 (m, 2H, CH
2), 3.41–3.48 (m, 6H, CH
2), 3.48–3.52 (m, 2H, CH
2), 3.54–3.57 (m, 2H, CH
2), 3.88 (dd,
J = 4.5, 5.6 Hz, 2H, CH
2), 4.53 (t,
J = 5.1 Hz, 1H, OH), 4.63 (t,
J = 5.1 Hz, 2H, CH
2), 8.11 (d,
J = 8.1 Hz, 1H, Ar-H), 8.46 (dd,
J = 1.4, 8.1 Hz, 1H, Ar-H), 8.58 (d,
J = 1.4 Hz, 1H, Ar-H), 8.92 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 50.0, 60.1 (CH
2), 60.5 (
C(CH
3)
3), 68.5, 69.60, 69.62, 69.69, 69.73, 72.3 (CH
2), 116.5, (Ar-CH), 124.4 (CH
triazole), 124.8 (Ar-C), 125.5, 130.7 (Ar-CH), 137.8, 138.1 (Ar-C), 143.8 (C
triazole), 159.3 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
21H
30N
4NaO
7S: 505.1727. Found: 505.1739.
3.21. N-t-Butyl-6-(1-[2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl]-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (27)
The title compound
27 was prepared from
N-t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide
5 (0.100 g, 0.380 mmol) and 2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranosyl azide
d′ [
25] (0.142 g, 0.380 mmol) at 50 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (EtOAc/hexane = 2:1 to 1:1) gave the title compound
27 (0.217 g, 90%) as a white solid. m.p. 112–115 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.71 (s, 9H,
tBu), 1.83 (s, 3H, OCOCH
3), 1.99 (s, 3H, OCOCH
3), 2.02 (s, 3H, OCOCH
3), 2.05 (s, 3H, OCOCH
3), 4.11 (dd,
J = 2.3, 12.6 Hz, 1H, H-6), 4.19 (dd,
J = 5.4, 12.7 Hz, 1H, H-6), 4.46 (ddd,
J = 2.3, 5.4 10.1 Hz, 1H, H-5), 5.16 (dd,
J = 9.3, 10.1 Hz, 1H, H-4), 5.57 (t,
J = 9.3 Hz, 1H, H-2), 5.64 (t,
J = 9.5 Hz, 1H, H-3), 6.49 (d,
J = 9.0 Hz, 1H, H-1), 8.14 (dd,
J = 0.6, 8.0 Hz, 1H, Ar-H), 8.44 (dd,
J = 1.5, 8.1 Hz, 1H, Ar-H), 8.58 (d,
J = 1.3 Hz, 1H, Ar-H), 9.35 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 19.9, 20.2, 20.4, 20.5 (OCO
CH
3), 27.3 (C(
CH
3)
3), 60.6 (
C(CH
3)
3), 61.7 (C-6), 67.5 (C-4), 70.4 (C-2), 71.9 (C-3), 73.4 (C-5), 84.0 (C-1), 116.7, (Ar-CH), 123.2 (CH
triazole), 125.3 (Ar-C), 125.7, 131.0 (Ar-CH), 136.9, 138.1 (Ar-C), 144.6 (C
triazole), 159.2 (C=O), 168.6, 169.3, 169.5, 170.0 (O
COCH
3); HRMS-ESI [M + H]
+ Calcd. for C
27H
33N
4O
12S: 637.1810. Found: 637.1811.
3.22. N-t-Butyl-6-(1-β-d-glucopyranosyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (28)
To a solution of 27 (0.149 g, 0.234 mmol) in methanol (9.2 mL) was added HCl (0.8 mL). The reaction was stirred at rt for 90 h and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 0:1 to 1:9) to give the title compound 28 (0.103 g, 94%) as a white solid. m.p. 169–170 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ 1.70 (s, 9H, tBu), 3.26 (t, J = 9.3 Hz, 1H, H-4), 3.45 (t, J = 8.9 Hz, 1H, H-3), 3.47–3.55 (m, 2H, H-5, H-6), 3.70–3.78 (m, 2H, H-2, H-6), 5.65 (d, J = 9.2 Hz, 1H, H-1), 8.13 (d, J = 8.1 Hz, 1H, Ar-H), 8.49 (dd, J = 1.4, 8.0 Hz, 1H, Ar-H), 8.61 (d, J = 1.3 Hz, 1H, Ar-H), 9.26 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 27.3 (C(CH3)3), 60.5 (C(CH3)3), 60.7 (C-6), 69.6 (C-4), 72.5 (C-2), 76.6 (C-3), 80.0 (C-5), 87.8 (C-1), 116.6 (Ar-CH), 123.1 (CHtriazole), 125.0 (Ar-C), 125.6, 130.7 (Ar-CH), 137.5, 138.1 (Ar-C), 144.1 (Ctriazole), 159.3 (C=O); HRMS-ESI [M + H]+ Calcd. for C19H25N4O8S: 469.1388. Found: 469.1397.
3.23. N-t-Butyl-6-(1-[2-deoxy-2-fluoro-β-d-glucopyranosyl]-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (29)
The title compound
29 was prepared from
N-t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide
5 (0.057 g, 0.217 mmol) and 2-deoxy-2-fluoro-β-
d-glucopyranosyl azide
e [
28] (0.045 g, 0.217 mmol) at 50 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (MeOH/CH
2Cl
2 = 1:9) gave the title compound
29 (0.103 g, quant.) as a white solid. m.p. 142–144 °C (MeOH/CH
2Cl
2);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.71 (s, 9H,
tBu), 3.32–3.38 (m, 1H, H-4), 3.46–3.51 (m, 1H, H-6), 3.67 (ddd,
J = 2.0, 5.7, 9.9 Hz, 1H, H-5), 3.73 (ddd,
J = 2.0, 5.6, 12.1 Hz, 1H, H-6), 3.82 (dtd,
J = 5.4, 8.9, 14.4 Hz, 1H, H-3), 4.72 (t,
J = 5.8 Hz, 1H, OH-6), 4.79 (dt,
J = 9.0, 51.0 Hz, 1H, H-2), 5.52 (d,
J = 5.6 Hz, 1H, OH-4), 5.88 (d,
J = 5.4 Hz, 1H, OH-3), 6.20 (dd,
J = 2.4, 9.1 Hz, 1H, H-1), 8.14 (d,
J = 8.1 Hz, 1H, Ar-H), 8.47 (dd,
J = 1.5, 8.1 Hz, 1H, Ar-H), 8.59 (d,
J = 1.4 Hz, 1H, Ar-H), 9.37 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 60.3 (C-6), 60.5 (
C(CH
3)
3), 69.3 (d,
J = 8.0 Hz, C-4), 74.1 (d,
J = 16.0 Hz, C-3), 79.9 (C-5), 84.2 (d,
J = 24.3 Hz, C-1), 91.2 (d,
J = 186.8 Hz, C-2), 116.7 (Ar-H), 123.2 (CH
triazole), 125.3 (Ar-C), 125.7, 131.0 (Ar-CH), 137.1, 138.1, (Ar-C), 144.5 (C
triazole), 159.2 (C=O);
19F-NMR (376 MHz, (CD
3)
2SO) δ -193.6 (ddd,
J = 1.7, 15.6, 51.3 Hz); HRMS-ESI [M + Na]
+ Calcd. for C
19H
23FN
4NaO
7S: 493.1164. Found: 493.1166.
3.24. N-t-Butyl-6-(1-β-d-galactopyranosyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (30)
The title compound
30 was prepared from
N-
t-butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide
5 (0.103 g, 0.390 mmol) and β-
d-galactopyranosyl azide
f [
25] (0.080 g, 0.390 mmol) at 50 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (MeOH/CH
2Cl
2 = 0:1 to 1:9) gave the title compound
30 (0.161 g, 88%) as a white solid. m.p. 195–196 °C (MeOH/CH
2Cl
2);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.70 (s, 9H,
tBu), 3.49–3.58 (m, 2H, 6-CH
2), 3.58–3.63 (m, 1H, H-3), 3.77–3.83 (m, 2H, H-4, H-5), 4.08 (td,
J = 5.8, 9.2, 9.3 Hz, 1H, H-2), 4.71 (t,
J = 5.6 Hz, 1H, OH-6), 4.76 (d,
J = 4.3 Hz, 1H, OH-4), 5.11 (d,
J = 5.5 Hz, 1H, OH-3), 5.32 (d,
J = 5.8 Hz, 1H, OH-2), 5.59 (d,
J = 9.1 Hz, 1H, H-1), 8.11 (d,
J = 8.1 Hz, 1H, Ar-H), 8.52 (dd,
J = 1.5, 8.1 Hz, 1H, Ar-H), 8.67 (d,
J = 1.3 Hz, 1H, Ar-H), 9.24 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 60.5 (
C(CH
3)
3 and C-6), 68.4 (C-4), 69.5 (C-2), 73.4 (C-3), 78.5 (C-5), 88.4 (C-1), 116.7 (Ar-CH), 123.0 (CH
triazole), 125.0 (Ar-C), 125.5, 130.1 (Ar-CH), 137.6, 138.1 (Ar-C), 144.1 (C
triazole), 159.3 (C=O); HRMS-ESI [M + H]
+ Calcd. for C
19H
25N
4O
8S: 469.1388. Found: 469.1340.
3.25. N-t-Butyl-6-1H-1,2,3-triazol-4-yl-1,2-benzisothiazole-3-one-1,1-dioxide (31)
N-t-Butyl-6-ethynyl-1,2-benzisothiazole-3-one-1,1-dioxide 5 (0.150 g, 0.570 mmol) and azidotrimethylsilane g (0.151 mL, 1.14 mmol) were dissolved in tert-butyl alcohol/H2O (1:1, 8 mL). To the reaction mixture was added a solution of sodium ascorbate (0.045 g, 0.228 mmol) in water (0.25 mL) followed by a solution of CuSO4·5H2O (0.028 g, 0.114 mmol) in water (0.25 mL). The suspension was stirred vigorously at 45 °C overnight. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (EtOAc/hexane = 2:3) to give the title compound 31 (0.086 g, 50%) as a white solid. m.p. greater than 300 °C (EtOAc/hexane); 1H-NMR (500 MHz, (CD3)2SO) δ 1.70 (s, 9H, tBu), 8.10 (d, J = 8.1 Hz, 1H, Ar-H), 8.46 (dd, J = 1.4, 8.1 Hz, 1H, Ar-H), 8.62 (d, J = 1.4 Hz, 1H, Ar-H), 8.75 (s, 1H, CHtriazole), 15.5 (brs, 1H, NH); 13C-NMR (125 MHz, (CD3)2SO) δ 27.3 (C(CH3)3), 60.5 (C(CH3)3), 116.9, (Ar-CH), 125.0 (Ar-C), 125.5 (Ar-CH), 127.3 (CHtriazole), 131.1 (Ar-CH), 137.7, 138.1 (Ar-C), 143.5 (Ctriazole), 159.3 (C=O); HRMS-ESI [M − H]− Calcd. for C13H13N4O3S: 305.0703. Found: 305.0690.
3.26. 6-(1-t-Butyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (32a) and 6-(2-t-Butyl-1H-1,2,3-triazol-4-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (32b)
N-t-Butyl protected 31 (0.085 g, 0.277 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 1:9) to give the title compounds 32a and 32b (0.068 g, 80%) as white solids. m.p. 203–204 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ1.67 (s, 9H, tBu), 7.97 (d, J = 8.0 Hz, 0.7H, Ar-H), 8.00 (d, J = 8.3 Hz, 0.3H, Ar-H), 8.30 (dd, J = 1.4, 7.8 Hz, 0.7H, Ar-H), 8.39 (dd, J = 1.4, 7.8 Hz, 0.3H, Ar-H), 8.47–8.50 (m, 1H, Ar-H), 8.53 (s, 0.7H, CHtriazole), 9.08 (s, 0.3H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 29.1, 29.4 (C(CH3)3), 59.6, 63.2 (C(CH3)3), 116.7, 117.2 (Ar-CH), 121.5 (CHtriazole), 125.1, 125.2 (Ar-CH), 127.6, 128.6 (Ar-C), 129.7, 130.4 (Ar-CH), 132.3 (CHtriazole), 136.1, 137.1, 141.7, 142.0 (Ar-C), 143.9, 144.2 (Ctriazole), 162.0, 162.3 (C=O); HRMS-ESI [M − H]− Calcd. for C13H13N4O3S: 305.0714. Found: 305.0715.
3.27. N-t-Butyl-6-N,N-bis([1-{2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl}-1H-1,2,3-triazol-4-yl]methyl) amino-1,2-benzisothiazole-3-one-1,1-dioxide (33)
The title compound
33 was prepared from saccharin bis-alkyne
6 (0.108 g, 0.327 mmol) and 2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranosyl azide
d′ [
25] (0.244 g, 0.654 mmol) at 45 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (EtOAc/hexane = 1:1 to 7:3) gave the title compound
33 (0.308 g, 87%) as a white solid. m.p. 197–199 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.63 (s, 9H,
tBu), 1.73 (s, 6H, OCOCH
3), 1.95 (s, 6H, OCOCH
3), 1.99 (s, 6H, OCOCH
3), 2.02 (s, 6H, OCOCH
3), 4.07 (dd,
J = 2.4, 12.5 Hz, 2H, H-6), 4.13 (dd,
J = 5.5, 12.6 Hz, 2H, H-6), 4.36 (ddd,
J = 2.5, 5.4 10.2 Hz, 2H, H-5), 4.84 (s, 4H, NCH
2), 5.15 (dd,
J = 9.1, 10.1 Hz, 2H, H-4), 5.54 (t,
J = 9.3 Hz, 2H, H-3), 5.59 (t,
J = 9.2 Hz, 2H, H-2), 6.34 (d,
J = 8.8 Hz, 2H, H-1), 7.18 (dd,
J = 2.4, 9.0 Hz, 1H, Ar-H), 7.44 (d,
J = 2.4 Hz, 1H, Ar-H), 7.68 (d,
J = 8.8 Hz, 1H, Ar-H), 8.42 (s, 2H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 19.7, 20.2, 20.4, 20.5 (OCO
CH
3), 27.4 (C(
CH
3)
3), 45.5 (NCH
2), 59.6 (
C(CH
3)
3), 61.7 (C-6), 67.5 (C-4), 70.1 (C-2), 72.0 (C-3), 73.2 (C-5), 83.8 (C-1), 102.5 (Ar-CH), 113.0 (Ar-C), 117.4 (Ar-CH), 122.5 (CH
triazole), 125.4 (Ar-CH), 139.6 (Ar-C), 143.6 (C
triazole), 152.8 (Ar-C), 159.9 (C=O), 168.4, 169.3, 169.5, 170.0 (O
COCH
3); HRMS-ESI [M + Na]
+ Calcd. for C
45H
56N
8NaO
21S: 1099.3173. Found: 1099.3144.
3.28. N-t-Butyl-6-N,N-bis([1-β-d-glucopyranosyl-1H-1,2,3-triazol-4-yl]methyl)amino-1,2-benzisothiazole- 3-one-1,1-dioxide (34)
To a solution of 33 (0.110 g, 0.102 mmol) in methanol (9.2 mL) was added HCl (0.8 mL). The reaction was stirred at rt for 90 h and the solvent was removed in vacuo. The residue was purified by RP-18 column chromatography (MeOH/H2O = 5:95 to 1:1, product eluting at 1:1) to give the title compound 34 (0.069 g, 92%) as a white solid. m.p. 203–205 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ 1.65 (s, 9H, tBu), 3.19–3.25 (m, 2H, H-4), 3.35–3.47 (m, 6H, H-3, H-5, H-6), 3.66–3.77 (m, 4H, H-2, H-6), 4.61 (t, J = 5.6 Hz, 2H, OH-6), 4.80–4.89 (m, 4H, NCH2), 5.15 (d, J = 5.5 Hz, 2H, OH-4), 5.27 (d, J = 4.9 Hz, 2H, OH-3), 5.36 (d, J = 6.0 Hz, 2H, OH-2), 5.53 (d, J = 9.2 Hz, 2H, H-1), 7.34 (dd, J = 2.4, 9.0 Hz, 1H, Ar-H), 7.56 (d, J = 2.3 Hz, 1H, Ar-H), 7.73 (d, J = 8.8 Hz, 1H, Ar-H), 8.36 (s, 2H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 27.5 (C(CH3)3), 45.2 (NCH2), 59.6 (C(CH3)3), 60.7 (C-6), 69.6 (C-4), 72.1 (C-2), 76.9 (C-3), 79.9 (C-5), 87.5 (C-1), 102.2 (Ar-CH), 112.7 (Ar-C), 117.2 (Ar-CH), 122.6 (CHtriazole), 125.7 (Ar-CH), 139.7 (Ar-C), 142.7 (Ctriazole), 153.0 (Ar-C), 159.9 (C=O); HRMS-ESI [M + Na]+ Calcd. for C29H40N8NaO13S: 763.2328. Found: 763.2366.
3.29. N-t-Butyl-6-(4-benzyl-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (35)
The title compound
35 was prepared from
N-t-butyl-6-azido-1,2-benzisothiazole-3-one-1,1-dioxide
3 [
10] (0.150 g, 0.535 mmol) and 3-phenyl-1-propyne
i (0.067 mL, 0.535 mmol) at 45 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (EtOAc/hexane = 1:4 to 1:2) gave the title compound
35 (0.185 g, 87%) as a light brown solid. m.p. 171–173 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.70 (s, 9H,
tBu), 4.12 (s, 2H, CH
2), 7.21–7.26 (m, 1H, Ar-H), 7.31–7.34 (m, 4H, Ar-H), 8.20 (d,
J = 8.4 Hz, 1H, Ar-H), 8.52 (dd,
J = 1.9, 8.4 Hz, 1H, Ar-H), 8.78 (d,
J = 1.9 Hz, 1H, Ar-H), 8.88 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 31.2 (CH
2), 60.8 (
C(CH
3)
3), 111.7, (Ar-CH), 121.5 (CH
triazole), 125.1 (Ar-C), 125.5, 126.4, 126.6, 128.5, 128.6 (Ar-CH), 138.6, 138.7, 141.5 (Ar-C), 148.0 (C
triazole), 158.7 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
20H
20N
4NaO
3S: 419.1148. Found: 419.1144.
3.30. N-t-Butyl-6-[4-(13-hydroxy-2,5,8,11-tetraoxatridec-1-yl)-1H-1,2,3-triazol-1-yl]-1,2-benzisothiazole-3-one-1,1-dioxide (36)
The title compound
36 was prepared from
N-t-butyl-6-azido-1,2-benzisothiazole-3-one-1,1-dioxide
3 [
10] (0.100 g, 0.357 mmol) and PEG alkyne
j [
31] (0.083 mL, 0.357 mmol) at 45 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (MeOH/CH
2Cl
2 = 0:1 to 5:95) gave the title compound
36 (0.135 g, 74%) as a pale yellow oil.
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.71 (s, 9H,
tBu), 3.38–3.41 (m, 2H, CH
2), 3.44–3.54 (m, 10H, CH
2), 3.56–3.59 (m, 2H, CH
2), 3.63–3.66 (m, 2H, CH
2), 4.55 (t,
J = 5.4 Hz, 1H, OH), 4.66 (s, 2H, CH
2), 8.23 (d,
J = 8.4 Hz, 1H, Ar-H), 8.55 (dd,
J = 1.9, 8.4 Hz, 1H, Ar-H), 8.82 (d,
J = 1.9 Hz, 1H, Ar-H), 9.09 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 60.2 (CH
2), 60.8 (
C(CH
3)
3), 63.3, 69.2, 69.68, 69.73, 69.8, 72.3 (CH
2), 111.9, (Ar-CH), 122.9 (CH
triazole), 125.3 (Ar-C), 125.8, 126.7 (Ar-CH), 138.6, 141.4 (Ar-C), 145.8 (C
triazole), 158.7 (C=O); HRMS-ESI [M + Na]
+ Calcd. for C
22H
32N
4NaO
8S: 535.1833. Found: 535.1845.
3.31. N-t-Butyl-6-1H-1,2,3-triazol-1-yl-1,2-benzisothiazole-3-one-1,1-dioxide (37)
N-
t-Butyl-6-azido-1,2-benzisothiazole-3-one-1,1-dioxide
3 [10] (0.150 g, 0.535 mmol) and ethynyltrimethylsilane
l (0.152 mL, 1.07 mmol) were dissolved in
tert-butyl alcohol/H
2O (1:1, 8 mL). To the reaction mixture was added a solution of sodium ascorbate (0.042 g, 0.214 mmol) in water (0.25 mL) followed by a solution of CuSO
4.5H
2O (0.027 g, 0.107 mmol) in water (0.25 mL). The suspension was stirred vigorously at 45 °C overnight. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:4 to 1:1) to give the title compound
37 (0.073 g, 45%) as a pale yellow solid. m.p. 187–188 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.71 (s, 9H,
tBu), 8.09 (d,
J = 1.2 Hz, 1H, CH
triazole), 8.24 (d,
J = 8.4 Hz, 1H, Ar-H), 8.56 (dd,
J = 1.9, 8.4 Hz, 1H, Ar-H), 8.82 (d,
J = 1.9 Hz, 1H, Ar-H), 9.11 (d,
J = 1.3 Hz, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 27.3 (C(
CH
3)
3), 60.8 (
C(CH
3)
3), 116.9, (Ar-CH), 124.0 (CH
triazole), 125.3 (Ar-C), 125.9, 126.7 (Ar-CH), 135.1 (CH
triazole), 138.6, 141.4 (Ar-C), 158.7 (C=O); HRMS-ESI [M − H]
− Calcd. for C
13H
13N
4O
3S: 305.0714. Found: 305.0712.
3.32. 6-(3-t-Butyl-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (38)
N-t-Butyl protected 37 (0.080 g, 0.261 mmol) was refluxed in TFA (3 mL) for 18 h. The solvent was removed in vacuo. EtOAc was added to the residue and the solid collected by filtration to give the title compound 38 (0.058 g, 89%) as an off white solid. m.p. 166–168 °C (EtOAc); 1H-NMR (500 MHz, (CD3)2SO) δ 1.71 (s, 9H, tBu), 7.93 (d, J = 8.1 Hz, 1H, Ar-H), 8.26 (dd, J = 2.0, 8.1 Hz, 1H, Ar-H), 8.45 (d, J = 1.9 Hz, 1H, Ar-H), 9.39 (d, J = 1.7 Hz, 1H, CHtriazole), 9.72 (d, J = 1.7 Hz, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 28.5 (C(CH3)3), 66.6 (C(CH3)3), 113.3, 124.4, 125.4 (Ar-CH), 129.2, 129.8 (CHtriazole), 136.7, 136.8, 147.0 (Ar-C), 165.9 (C=O); HRMS-ESI [M + H]+ Calcd. for C13H13N4O3S: 305.0714. Found: 305.0702.
3.33. N-t-Butyl-6-(4-{2,3,4,6-tetra-O-acetyl-[β-d-glucopyranosyl]thiomethyl}-1H-1,2,3-triazol-1-yl)-1,2- benzisothiazole-3-one-1,1-dioxide (39)
The title compound
39 was prepared from N-
t-butyl-6-amino-1,2-benzisothiazole-3-one-1,1-dioxide
3 [
10] (0.300 g, 1.07 mmol) and propargyl 2,3,4,6-tetra-
O-acetyl-thio-β-
d-glucopyranoside
k′ [14] (0.431 g, 1.07 mmol) at 40 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (EtOAc/hexane = 2:3 to 1:1) gave the title compound
39 (0.656 g, 90%) as a white solid. m.p. 96–98 °C (EtOAc/hexane);
1H-NMR (500 MHz, (CD
3)
2SO) δ 1.71 (s, 9H,
tBu), 1.94 (s, 3H, OCOCH
3), 1.98 (s, 6H, 2 × OCOCH
3), 1.99 (s, 3H, OCOCH
3), 3.99–4.16 (m, 5H, H-5, SCH
2, CH
2-6), 4.91–5.00 (m, 3H, H-1, H-2, H-4), 5.31 (t,
J = 9.0 Hz, 1H, H-3), 8.24 (d,
J = 8.4 Hz, 1H, Ar-H), 8.52 (dd,
J = 1.9, 8.4 Hz, 1H, Ar-H), 8.78 (d,
J = 1.9 Hz, 1H, Ar-H), 8.95 (s, 1H, CH
triazole);
13C-NMR (125 MHz, (CD
3)
2SO) δ 20.3, 20.4, 20.4, 20.5 (OCO
CH
3), 23.1 (SCH
2), 27.3 (C(
CH
3)
3), 60.8 (
C(CH
3)
3), 61.8 (C-6), 68.1 (C-4), 69.6 (C-2), 72.9 (C-3), 74.4 (C-5), 80.9 (C-1), 111.9, (Ar-CH), 122.4 (CH
triazole), 125.3 (Ar-C), 125.7, 126.7 (Ar-CH), 138.6, 141.4 (Ar-C), 145.3 (C
triazole), 158.7 (C=O), 169.1, 169.2, 169.5, 170.0 (O
COCH
3); HRMS-ESI [M + Na]
+ Calcd. for C
28H
34N
4NaO
12S
2: 705.1507. Found: 705.1551.
3.34. N-t-Butyl-6-(4-{[β-d-glucopyranosyl]thiomethyl}-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (40)
The title compound 40 was prepared using two different synthetic routes, A and B.
The title compound
40 was prepared from
N-
t-butyl-6-amino-1,2-benzisothiazole-3-one-1,1-dioxide
3 [
10] (0.200 g, 0.714 mmol) and propargyl thio-β-
d-glucopyranoside (
k) [
32] (0.167 g, 0.714 mmol) at 40 °C in 2 h according to general procedure 1. Purification of the crude product by flash chromatography (MeOH/CH
2Cl
2 = 0:1 to 3:17) gave the title compound
40 (0.324 g, 88%) as a white solid.
To a solution of 39 (0.385 g, 0.564 mmol) in methanol (9.2 mL) was added HCl (0.8 mL). The reaction was stirred at r.t. for 90 h and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 0:1 to 1:9) to give the title compound 40 (0.255 g, 88%) as a white solid. m.p. 155–157 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ 1.71 (s, 9H, tBu), 3.03–3.11 (m, 2H, H-2, H-4), 3.15 (dd, J = 4.8, 8.6 Hz, 1H, H-3), 3.20 (ddd, J = 2.0, 6.5, 8.6 Hz, 1H, H-5), 3.42–3.48 (m, 1H, H-6), 3.73 (ddd, J = 2.0, 5.9, 11.9 Hz, 1H, H-6), 3.97 (d, J = 14.4 Hz, 1H, SCH2), 4.10 (d, J = 14.4 Hz, 1H, SCH2), 4.36 (d, J = 9.6 Hz, 1H, H-1), 4.70 (t, J = 5.8 Hz, 1H, OH-6), 4.97 (d, J = 5.3 Hz, 1H, OH-4), 5.05 (d, J = 4.8 Hz, 1H, OH-3), 5.18 (d, J = 5.9 Hz, 1H, OH-2), 8.22 (d, J = 8.4 Hz, 1H, Ar-H), 8.50 (dd, J = 1.9, 8.4 Hz, 1H, Ar-H), 8.76 (d, J = 1.9 Hz, 1H, Ar-H), 8.97 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 22.9 (SCH2), 27.3 (C(CH3)3), 60.8 (C(CH3)3), 61.3 (C-6), 70.1 (C-4), 73.1 (C-2), 78.1 (C-3), 81.0 (C-5), 84.1 (C-1), 111.8, (Ar-CH), 122.2 (CHtriazole), 125.2 (Ar-C), 125.7, 126.7 (Ar-CH), 138.6, 141.4 (Ar-C), 146.6 (Ctriazole), 158.7 (C=O); HRMS-ESI [M + H]+ Calcd. for C20H27N4O8S2: 515.1265. Found: 515.1269.
3.35. N-t-Butyl-6-(4-{2,3,4,6-tetra-O-acetyl-[β-d-glucopyranosyl]sulfonylmethyl}-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (41)
To a stirred solution of 39 (0.250 g, 0.366 mmol) in anhydrous CH2Cl2 (5 mL) at 0 °C was added mCPBA (0.737 g, 2.56 mmol) in anhydrous CH2Cl2 (2 mL) dropwise. The solution was allowed to warm to r.t. over 2 h. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with H2O (30 mL), brine (30 mL), dried (MgSO4), and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:1 to 3:2) to give the title compound 41 (0.260 g, 99%) as a white solid. m.p. 172–174 °C (EtOAc/hexane); 1H-NMR (500 MHz, (CD3)2SO) δ 1.72 (s, 9H, tBu), 1.94 (s, 3H, OCOCH3), 1.95 (s, 3H, OCOCH3), 2.00 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 4.17–4.28 (m, 3H, H-5, CH2-6), 4.78–4.87 (m, 2H, SCH2), 5.01–5.06 (m, 1H, H-4), 5.14–5.20 (m, 1H, H-1), 5.37–5.45 (m, 2H, H-2, H-3), 8.26 (d, J = 8.4 Hz, 1H, Ar-H), 8.59 (dd, J = 2.0, 8.5 Hz, 1H, Ar-H), 8.89 (d, J = 2.0 Hz, 1H, Ar-H), 9.14 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 20.2, 20.29, 20.33, 20.5 (OCOCH3), 27.3 (C(CH3)3), 46.9 (SCH2), 60.9 (C(CH3)3), 61.4 (C-6), 65.7 (C-2), 67.2 (C-4), 72.5 (C-3), 74.9 (C-5), 84.9 (C-1), 112.3, (Ar-CH), 124.8 (CHtriazole), 125.6 (Ar-C), 126.1, 126.7 (Ar-CH), 135.8, 138.6 (Ar-C), 141.2 (Ctriazole), 158.7 (C=O), 168.6, 169.2, 169.5, 170.1 (OCOCH3); HRMS-ESI [M + Na]+ Calcd. for C28H34N4NaO14S2: 737.1405. Found: 737.1486.
3.36. N-t-Butyl-6-(4-{[β-d-glucopyranosyl]sulfonylmethyl}-1H-1,2,3-triazol-1-yl)-1,2-benzisothiazole-3-one-1,1-dioxide (42)
The title compound 42 was prepared using two different synthetic routes, A and B.
To a stirred solution of 40 (0.150 g, 0.292 mmol) in anhydrous CH2Cl2 (5 mL) at 0 °C was added mCPBA (0.587 g, 2.04 mmol) in anhydrous CH2Cl2 (2 mL) dropwise. The solution was allowed to warm to rt over 2 h and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 1:9 to 3:17) to give the title compound 42 (0.126 g, 79%) as a white solid.
To a solution of 41 (0.220 g, 0.308 mmol) in methanol (9.2 mL) was added HCl (0.8 mL). The reaction was stirred at rt for 90 h and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 = 0:1 to 1:9) to give the title compound 42 (0.117 g, 69%) as a white solid. m.p. 145–146 °C (MeOH/CH2Cl2); 1H-NMR (500 MHz, (CD3)2SO) δ 1.72 (s, 9H, tBu), 3.08 (ddd, J = 5.4, 8.8, 9.8 Hz, 1H, H-4), 3.28 (td, J = 5.5, 8.8 Hz, 1H, H-3), 3.42 (ddd, J = 1.9, 6.8, 9.8 Hz, 1H, H-5), 3.51 (ddd, J = 5.5, 6.9, 12.1 Hz, 1H, H-6), 3.60 (td, J = 6.1, 9.1 Hz, 1H, H-2), 3.80 (ddd, J = 1.9, 6.2, 12.2 Hz, 1H, H-6), 4.51 (d, J = 9.5 Hz, 1H, H-1), 4.73 (d, J = 14.7 Hz, 1H, SCH2), 4.85 (d, J = 14.7 Hz, 1H, SCH2), 4.99 (t, J = 5.8 Hz, 1H, OH-6), 5.17 (d, J = 5.5 Hz, 1H, OH-4), 5.24 (d, J = 5.5 Hz, 1H, OH-3), 5.55 (d, J = 6.1 Hz, 1H, OH-2), 8.25 (d, J = 8.4 Hz, 1H, Ar-H), 8.51 (dd, J = 2.0, 8.4 Hz, 1H, Ar-H), 8.81 (d, J = 1.9 Hz, 1H, Ar-H), 9.18 (s, 1H, CHtriazole); 13C-NMR (125 MHz, (CD3)2SO) δ 27.3 (C(CH3)3), 48.2 (SCH2), 60.9 (C(CH3)3), 61.0 (C-6), 69.2 (C-2), 69.5 (C-4), 77.5 (C-3), 81.6 (C-5), 88.7 (C-1), 112.2, (Ar-CH), 124.8 (CHtriazole), 125.5 (Ar-C), 126.0, 126.8 (Ar-CH), 137.0, 138.6 (Ar-C), 141.3 (Ctriazole), 158.7 (C=O); HRMS-ESI [M + Na]+ Calcd. for C20H26N4NaO10S2: 569.0983. Found: 569.0994.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






