
Degradation of Methyl 2-Aminobenzoate (Methyl Anthranilate) by H2O2/UV: Effect of Inorganic Anions and Derived Radicals



Abstract
:1. Introduction
2. Results and Discussion
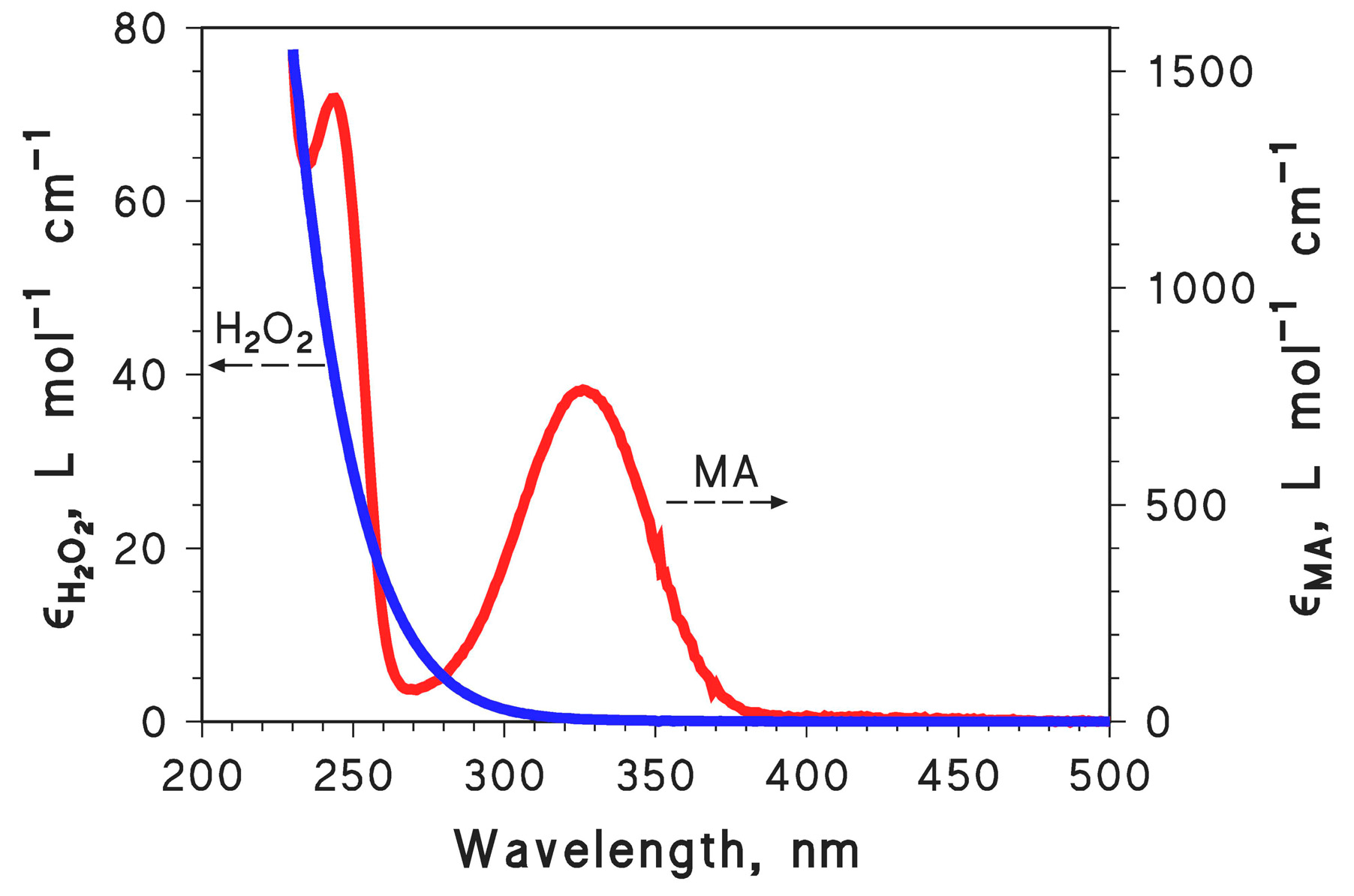
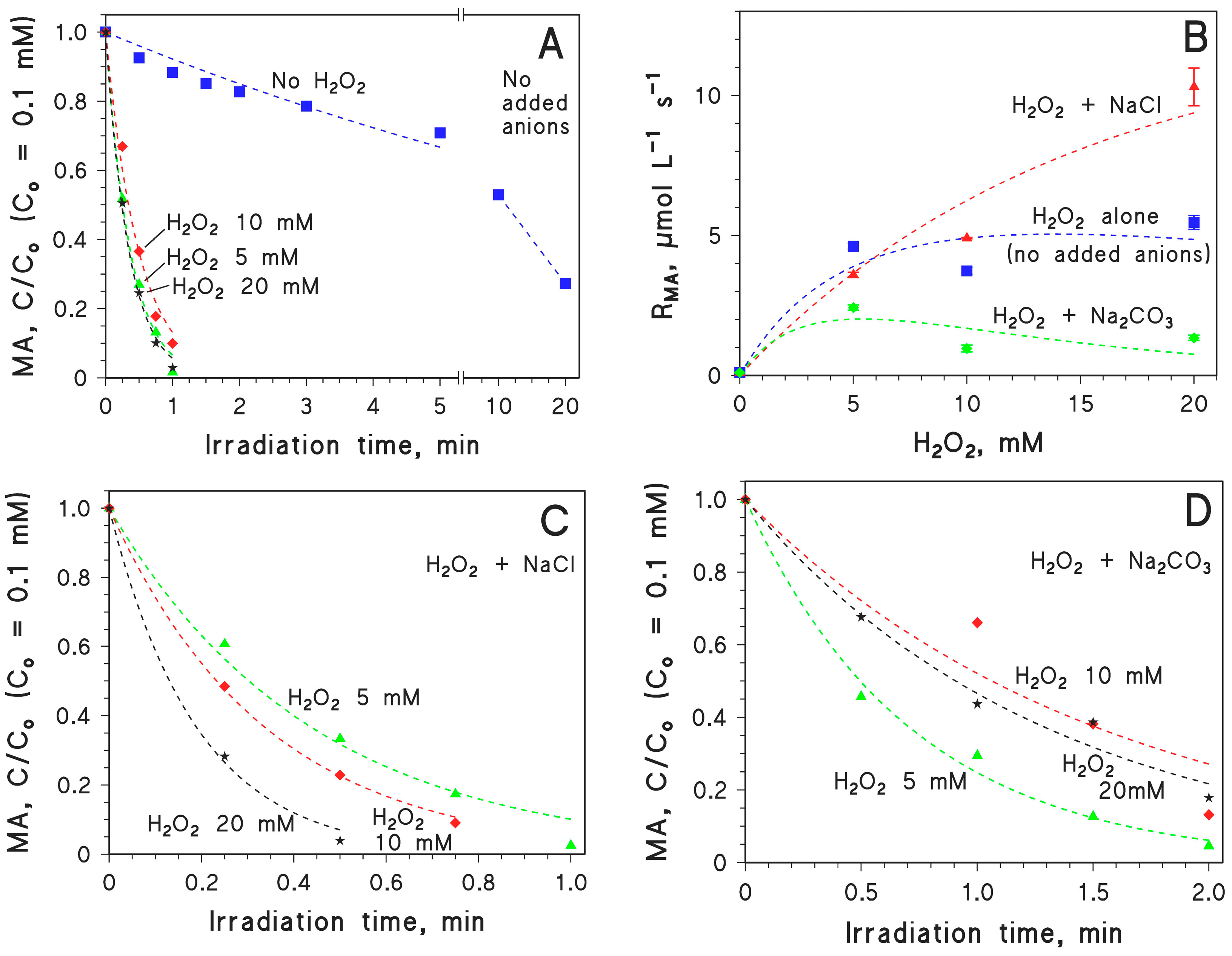
2.1. MA Photodegradation by UV and H2O2/UV
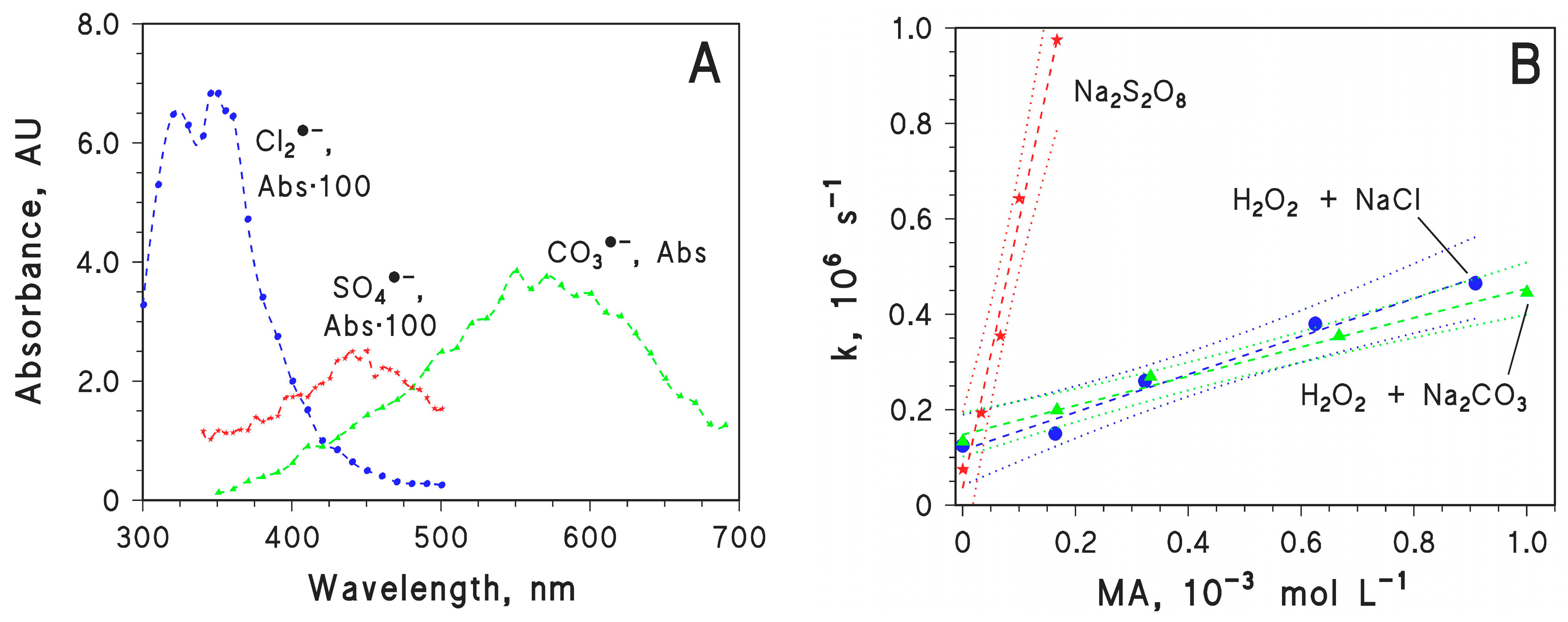
2.2. Effect of Inorganic Anions on MA Photodegradation
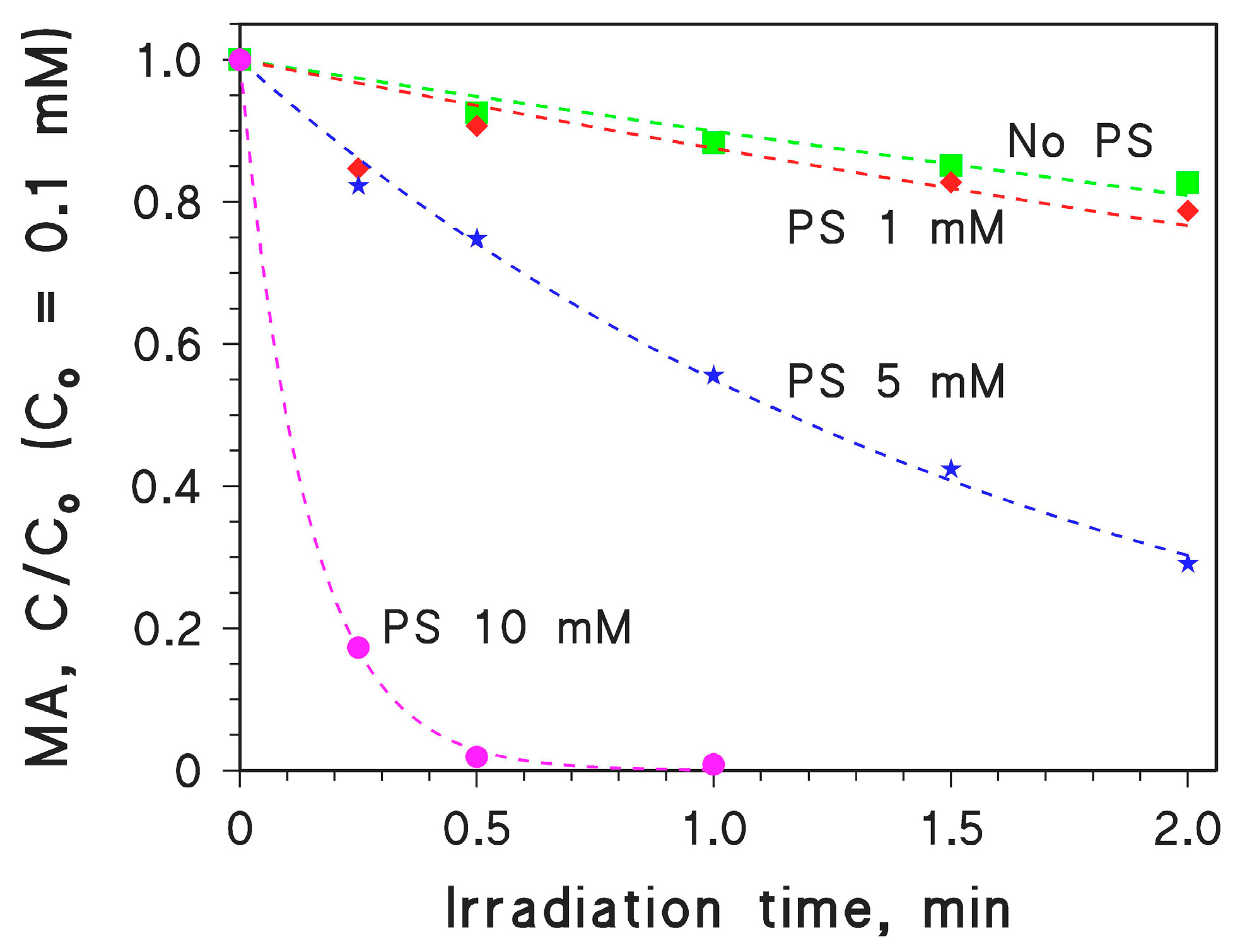
2.3. MA Photodegradation by Persulphate/UV
2.4. Second-Order Reaction Rate Constants of MA with Cl2·−, CO3·− and SO4·−

2.5. MA Photodegradation Intermediates
3. Methods
3.1. Reagents and Materials
3.2. Irradiation Experiments
3.3. Identification of Photodegradation Intermediates
3.4. Laser Flash Photolysis Experiments
3.5. Model Assessment of Toxicity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| kMA | pseudo-first order rate constant of MA photodegradation. |
| kX+Y | second-order reaction rate constant between the species X and the compound Y. |
| RMA | initial photodegradation rate of MA. |
| εY,λ | molar absorption coefficient of the compound Y at the wavelength λ. |
| p°(λ) | incident spectral photon flux density of the used lamp at the wavelength λ. |
| formation rate of the species X in the presence of the compound Y under irradiation. | |
| fraction of radiation absorbed by the compound Y at the wavelength λ, inside the solution S. |
References
- Sigma-Aldrich. Available online: http://www.sigmaaldrich.com/catalog/product/aldrich/w268208 (accessed on 3 March 2017).
- Fraternale, D.; Ricci, D.; Flamini, G.; Giomaro, G. Volatiles profile of red apple from Marche region (Italy). Rec. Nat. Prod. 2011, 5, 202–207. [Google Scholar]
- Dolbeer, R.A.; Clark, L.; Woronecki, P.; Seamans, T.W. Pen tests of methyl anthranilate as a bird repellent in water. In Proceedings of the Eastern Wildlife Damage Control Conference, Ithaca, NY, USA, 6–9 October 1991; Volume 5, pp. 112–116. [Google Scholar]
- Brown, J.E.; Luo, W.T.; Lorne, I.M.; Pankow, J.F. Candy flavorings in tobacco. N. Engl. J. Med. 2014, 370, 2250–2252. [Google Scholar] [CrossRef] [PubMed]
- M&U International. Methyl Anthranilate Material Safety Data Sheet. Available online: http://www.mu-intel.com/upload/msds/20141119004116.pdf (accessed on 19 December 2016).
- Clark, L.; Cummings, J.; Bird, S.; Aronov, E. Acute toxicity of the bird repellent, methyl anthranilate, to fry of Salmo salar, Oncorhynus mykiss, Ictalurus punctatus and Lepomis macrochirus. Pestic. Sci. 1993, 39, 313–317. [Google Scholar] [CrossRef]
- Mayo-Bean, K.; Moran, K.; Meylan, B.; Ranslow, P. Methodology Document for the ECOlogical Structure-Activity Relationship Model (ECOSAR) Class Program; US-EPA: Washington DC, 2012; 46 pp.
- US-EPA, ECOSAR Software (Ecological Structure Activity Relationships Predictive Model). Available online: https://www.epa.gov/tsca-screening-tools/ecological-structure-activity-relationships-ecosar-predictive-model (accessed on 20 December 2016).
- Avery, M.L. Evaluation of methyl anthranilate as a bird repellent in fruit crops. In Proceedings of the Fifteenth Vertebrate Pest Conference, Newport Beach, CA, USA, 3–5 March 1992; Volume 15, pp. 115–129. [Google Scholar]
- Dorr, B.; Clark, L.; Glahn, J.F.; Mezine, I. Evaluation of a methyl anthranilate-based bird repellent: Toxicity to channel catfish Ictalurus punctatus and effect on great blue heron Ardea herodias feeding behavior. J. World Aquacult. Soc. 1998, 29, 451–462. [Google Scholar] [CrossRef]
- US-EPA EPI Suite™-Estimation Program Interface. Available online: https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface (accessed on 15 December 2016).
- Zuccato, E.; Castiglioni, S.; Bagnati, R.; Melis, M.; Fanelli, R. Source, occurrence and fate of antibiotics in the Italian aquatic environment. J. Hazard. Mater. 2010, 179, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.D.; Ternes, T.A. Water analysis: Emerging contaminants and current issues. Anal. Chem. 2014, 86, 2813–2848. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.J.; Cramer, C.J.; Martin, N.H.; Mezyk, S.P.; O′Shea, K.E.; von Sonntag, C. Free radical mechanisms for the treatment of methyl tert-butyl ether (MTBE) via advanced oxidation/reductive processes in aqueous solutions. Chem. Rev. 2009, 109, 1302–1345. [Google Scholar] [CrossRef] [PubMed]
- Brillas, E.; Sires, I.; Oturan, M.A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 2009, 109, 6570–6631. [Google Scholar] [CrossRef] [PubMed]
- Pichat, P. (Ed.) Photocatalysis and Water Purification; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Gligorovski, S.; Strekowski, R.; Barbati, S.; Vione, D. Environmental implications of hydroxyl radicals (•OH). Chem. Rev. 2015, 115, 13051–13092. [Google Scholar] [CrossRef] [PubMed]
- Doré, M. Chemistry of Oxidants and Water Treatments; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Ganiyu, S.O.; van Hullebusch, E.D.; Cretin, M.; Esposito, G.; Oturan, M.A. Coupling of membrane filtration and advanced oxidation processes for removal of pharmaceutical residues: A critical review. Separ. Purif. Technol. 2015, 156, 891–914. [Google Scholar] [CrossRef]
- Wang, J.L.; Wang, S.Z. Removal of pharmaceuticals and personal care products (PPCPs) from wastewater: A review. J. Environ. Manag. 2016, 182, 620–640. [Google Scholar] [CrossRef] [PubMed]
- Rozas, O.; Vidal, C.; Baeza, C.; Jardim, W.F.; Rossner, A.; Mansilla, H.D. Organic micropollutants (OMPs) in natural waters: Oxidation by UV/H2O2 treatment and toxicity assessment. Water Res. 2016, 98, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Minero, C.; Pellizzari, P.; Maurino, V.; Pelizzetti, E.; Vione, D. Enhancement of dye sonochemical degradation by some inorganic anions present in natural waters. Appl. Catal. B Environ. 2008, 77, 308–316. [Google Scholar] [CrossRef]
- Avetta, P.; Pensato, A.; Minella, M.; Malandrino, M.; Maurino, V.; Minero, C.; Hanna, K.; Vione, D. Activation of persulfate by irradiated magnetite: Implications for the degradation of phenol under heterogeneous photo-Fenton-like conditions. Environ. Sci. Technol. 2015, 49, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Matzek, L.W.; Carter, K.E. Activated persulfate for organic chemical degradation: A review. Chemosphere 2016, 151, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Braslavsky, S.E. Glossary of terms used in photochemistry, 3rd edition (IUPAC Recommendations 2006). Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Jayson, G.G.; Parsons, B.J.; Swallow, A.J. Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. J. Chem. Soc., Faraday I 1973, 1597–1607. [Google Scholar]
- Vione, D.; Maurino, V.; Minero, C.; Calza, P.; Pelizzetti, E. Phenol chlorination and photochlorination in the presence of chloride ions in homogeneous aqueous solution. Environ. Sci. Technol. 2005, 39, 5066–5075. [Google Scholar] [CrossRef] [PubMed]
- Canonica, S.; Kohn, T.; Mac, M.; Real, F.J.; Wirz, J.; Von Gunten, U. Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds. Environ. Sci. Technol. 2005, 39, 9182–9188. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Hori, H.; Yamamoto, A.; Hayakawa, E.; Taniyasu, S.; Yamashita, N.; Kutsuna, S. Efficient decomposition of environmentally persistent perfluorocarboxylic acids by use of persulfate as a photochemical oxidant. Environ. Sci. Technol. 2005, 39, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Criquet, J.; Leitner, N.K.V. Degradation of acetic acid with sulfate radical generated by persulfate ions photolysis. Chemosphere 2009, 77, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, M.G.; de la Cruz, A.A.; Dionysiou, D.D. Degradation of microcystin-LR using sul fate radicals generated through photolysis, thermolysis and e− transfer mechanisms. Appl. Catal. B: Environ. 2010, 96, 290–298. [Google Scholar] [CrossRef]
- Herrmann, H.; Hoffmann, D.; Schaefer, T.; Braeuer, P.; Tilgner, A. Tropospheric aqueous-phase free-radical chemistry: Radical sources, spectra, reaction kinetics and prediction tools. ChemPhysChem 2010, 11, 3796–3822. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.H.; Cai, Z.L.; Katsumura, Y.; Chitose, N.; Muroya, Y. Reinvestigation of the acid-base equilibrium of the (bi)carbonate radical and pH dependence of its reactivity with inorganic reactants. Radiat. Phys. Chem. 1999, 55, 15–23. [Google Scholar] [CrossRef]
- Wu, Y.L.; Bianco, A.; Brigante, M.; Dong, W.B.; de Sainte-Claire, P.; Hanna, K.; Mailhot, G. Sulfate radical photogeneration using Fe-EDDS: Influence of critical parameters and naturally occurring scavengers. Environ. Sci. Technol. 2015, 49, 14343–14349. [Google Scholar] [CrossRef] [PubMed]
- Martell, A.E.; Smith, R.M.; Motekaitis, R.J. Critically Selected Stability Constants of Metal Complexes Database, Version 4.0; NIST: Gaithersburg, MD, USA, 1997. [Google Scholar]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2•/O2•− radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Merritt, C.R.B. Mass Spectrometry: Part A; Merritt, C., Jr., McEwen, C.N., Eds.; Marcel Dekker: New York, NY, USA, 1979. [Google Scholar]
- Barker, J. Mass Spectrometry: Analytical Chemistry by Open Learning, 2nd ed.; John Wiley and Sons: West Sussex, UK, 1999. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [H2O2], mM | [S2O82−], mM | [Cl−], mM | [CO32−], mM | kMA, min−1 (±σ) |
|---|---|---|---|---|
| 0 | / | / | / | 0.081 ± 0.007 |
| 5 | / | / | / | 2.72 ± 0.12 |
| 10 | / | / | / | 2.03 ± 0.13 |
| 20 | / | / | / | 2.87 ± 0.09 |
| 5 | / | 100 | / | 2.29 ± 0.16 |
| 10 | / | 100 | / | 2.97 ± 0.06 |
| 20 | / | 100 | / | 5.30 ± 0.34 |
| 5 | / | / | 100 | 1.40 ± 0.08 |
| 10 | / | / | 100 | 0.652 ± 0.121 |
| 20 | / | / | 100 | 0.764 ± 0.049 |
| / | 0 | / | / | 0.106 ± 0.007 |
| / | 1 | / | / | 0.133 ± 0.031 |
| / | 5 | / | / | 0.598 ± 0.018 |
| / | 10 | / | / | 7.09 ± 0.16 |
| kX+Y, M−1 s−1 | MA | H2O2 | HO2− |
|---|---|---|---|
| Cl2·− | 4.0 × 108 | 1.4 × 105 [30] | n/a |
| CO3·− | 3.1 × 108 | 8 × 105 [30] | 3 × 107 [30] |
| ·OH | ~109 | 2.7 × 107 [26] | 7.5 × 109 [26] |
| Compound Acronym | LC Retention Time, (min) | MS2 Fragments, m/z (% Relative Abundance) |
|---|---|---|
| P1 | 10.6 | 141 (8), 137 (7), 109 (100), 81 (38) |
| P2 | 11.0 | 136 (100), 137 (30), 107 (3) |
| P3 | 11.4 | 136 (100), 137 (25), 107 (25) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzafame, G.M.; Sarakha, M.; Fabbri, D.; Vione, D. Degradation of Methyl 2-Aminobenzoate (Methyl Anthranilate) by H2O2/UV: Effect of Inorganic Anions and Derived Radicals. Molecules 2017, 22, 619. https://doi.org/10.3390/molecules22040619
Lanzafame GM, Sarakha M, Fabbri D, Vione D. Degradation of Methyl 2-Aminobenzoate (Methyl Anthranilate) by H2O2/UV: Effect of Inorganic Anions and Derived Radicals. Molecules. 2017; 22(4):619. https://doi.org/10.3390/molecules22040619
Chicago/Turabian StyleLanzafame, Grazia Maria, Mohamed Sarakha, Debora Fabbri, and Davide Vione. 2017. "Degradation of Methyl 2-Aminobenzoate (Methyl Anthranilate) by H2O2/UV: Effect of Inorganic Anions and Derived Radicals" Molecules 22, no. 4: 619. https://doi.org/10.3390/molecules22040619
APA StyleLanzafame, G. M., Sarakha, M., Fabbri, D., & Vione, D. (2017). Degradation of Methyl 2-Aminobenzoate (Methyl Anthranilate) by H2O2/UV: Effect of Inorganic Anions and Derived Radicals. Molecules, 22(4), 619. https://doi.org/10.3390/molecules22040619