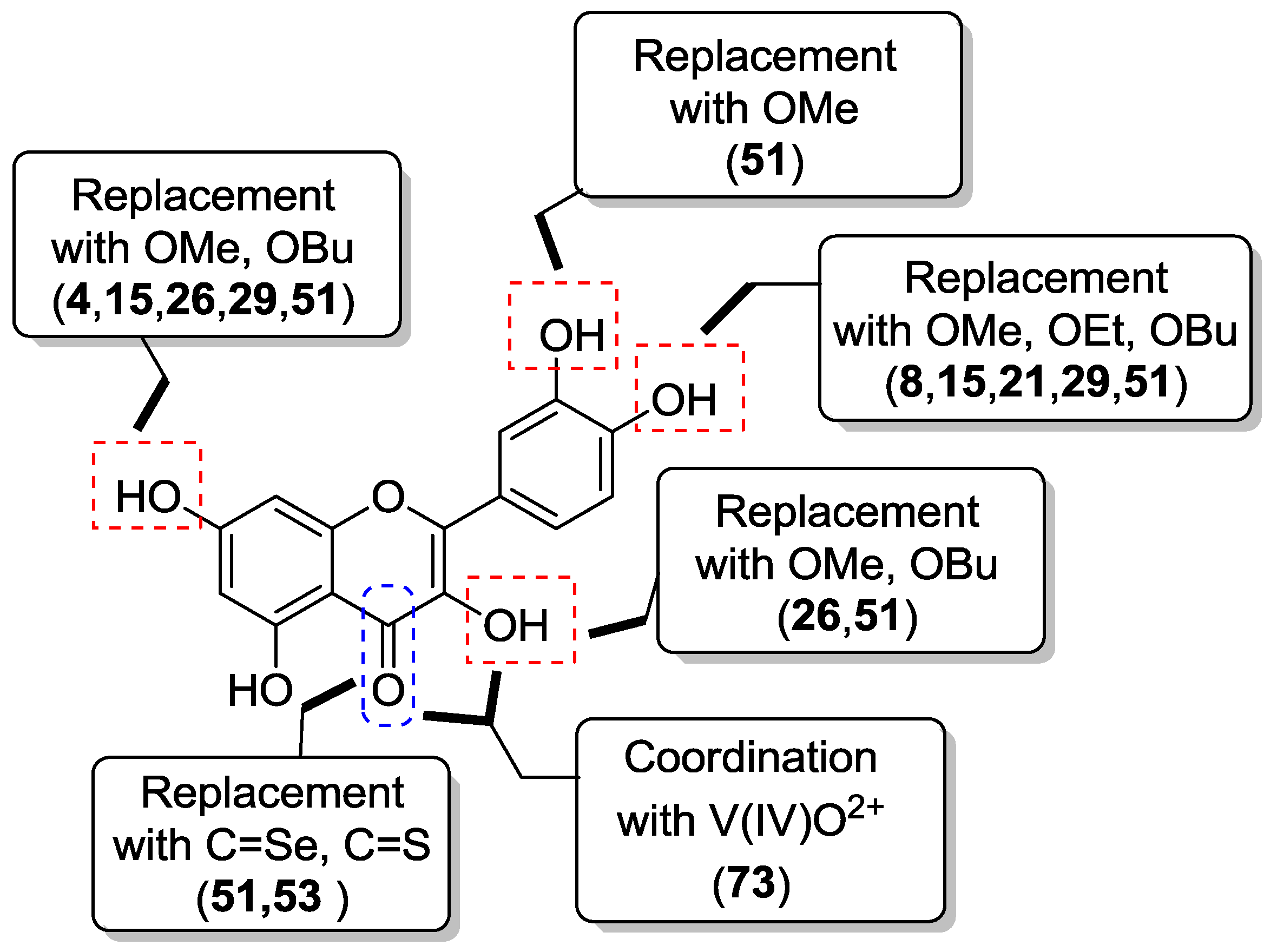
Phenolic hydroxyl groups of quercetin have been mostly manipulated by etherification (O-alkylation) and esterification (O-acylation), with the O-alkylation strategy being possibly accompanied by conversion of the C-4 carbonyl group into the corresponding thiocarbonyl or selenocarbonyl functions. Also, interchange of catecholic hydroxyl groups with bioisosteric moieties has been developed.
2.1.1. O-Alkylation
It has been reported that insertion of methoxy groups into a flavone molecule results in metabolically more stable derivatives with increased solubility, bioavailability and cancer cell antiproliferative activity, as well as reduced toxic side-effects [
37]. This information inspired several studies on the etherification of quercetin with either methyl or other alkyl groups in order to investigate their effect on either physico-chemical or anticancer-related properties.
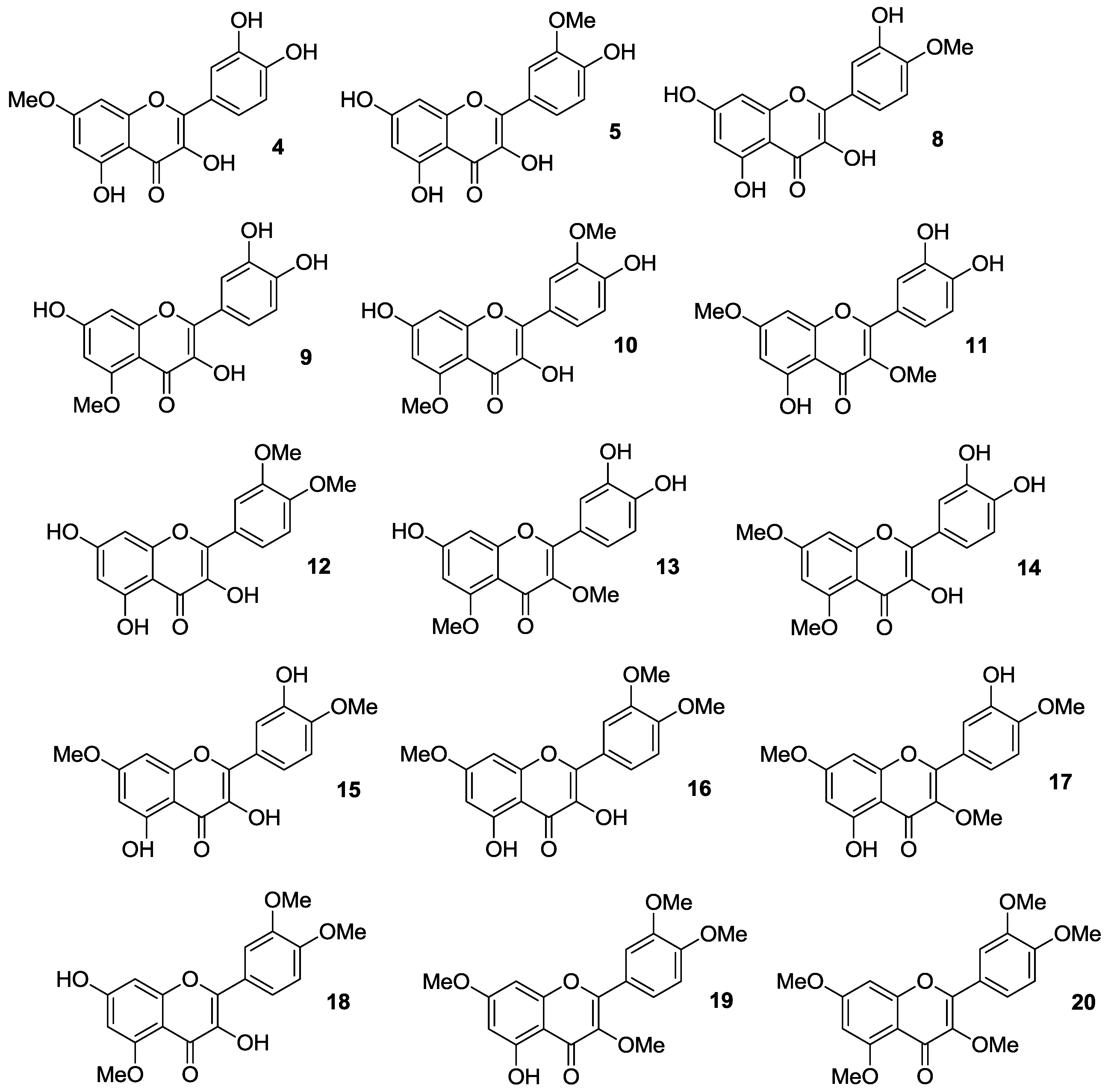
Thus, quercetin was converted into a series of monomethylated (
4,
5,
8,
9), dimethylated (
10–
15), trimethylated (
16–
18), tetramethylated (
19), and pentamethylated (
20) derivatives [
38,
39,
40,
41,
42], which are shown in
Figure 4.
Synthetically, the mono-protected compounds were prepared by suitable protection/deprotection steps of the phenolic hydroxyl groups in quercetin, with methyl iodide/K2CO3 system in N,N-dimethylformamide (DMF) being conveniently used at the time of installing the methyl ether moiety. Conversely, direct treatment of the flavonol starting material with methyl iodide and potassium carbonate in either DMF or acetone was carried out to yield the di-, tri- tetra- and penta-functionalized analogs.
In early studies [
38,
39], it was found that 3,3′,4′,7-tetra-
O-methylated quercetin
19 and 3,3′,4′,5,7-penta-
O-methylated quercetin
20 could represent potential anti-multidrug resistance (MDR) agents due to their ability to influence the effects of breast cancer resistance protein (BCRP), which is known to determine resistance in cancer cells. Importantly, both 3′,4′-OMe substitution and the presence of 5-OH group were essential for optimum BCRP inhibition, whereas this activity decreased upon methylation of C-5 phenolic hydroxyl group.
In particular, investigations in Madin-Darby canine kidney (MDCK) BCRP cells evidenced that
19 and
20 were able to inhibit BCRP as a result of Hoechst 33342 and pheophorbide A accumulation, contrary to quercetin, which gave no inhibitory effect (
Table 1) [
39].
Later, Shi et al. prepared the
O-methylated compounds
4,
5 and
8–
20, and evaluated their ability to inhibit cancer cell growth using a high-throughput screening (HTS) approach in an in vitro human disease-oriented cancer cell line, including melanoma (LOX-IMVI and M14), neck and head (M4E), cervical (HeLa), human breast cancer (SKBR) as well as human lung cancers (A549, H157, H460, 1792, 1944, H266, H522, Hop62, 1299, 292G, and Calu1) [
40,
41]. These investigations demonstrated that selective masking of the phenolic hydroxyl groups in quercetin is pivotal in determining antiproliferative activity. As a rule of thumb, it was possible to maintain inhibitory effects against all the cancer cell lines by methylation at the 4′-OH and/or 7-OH positions, while the coexistence of 3′- and 4′-OMe groups improved activity. Also, additional introduction of a methoxy moiety may enhance the inhibition of cancer cell growth, with 3′,4′,7-trimethoxyquercetin (
16) being more potent than 3′,4′-dimethoxyquercetin (
12).
The antiproliferative action of 3,7-
O-dimethylquercetin (
11), 3,4′,7-
O-trimethylquercetin (
17), and 3,3′,4′,7-
O-tetramethylquercetin (
19) against human androgen-refractory (DU-145 and PC-3) and androgen-sensitive (LNCaP) prostate cancer cell lines were examined as well [
42], showing that methylation barely determined a weak enhancement of activity compared to parent quercetin.
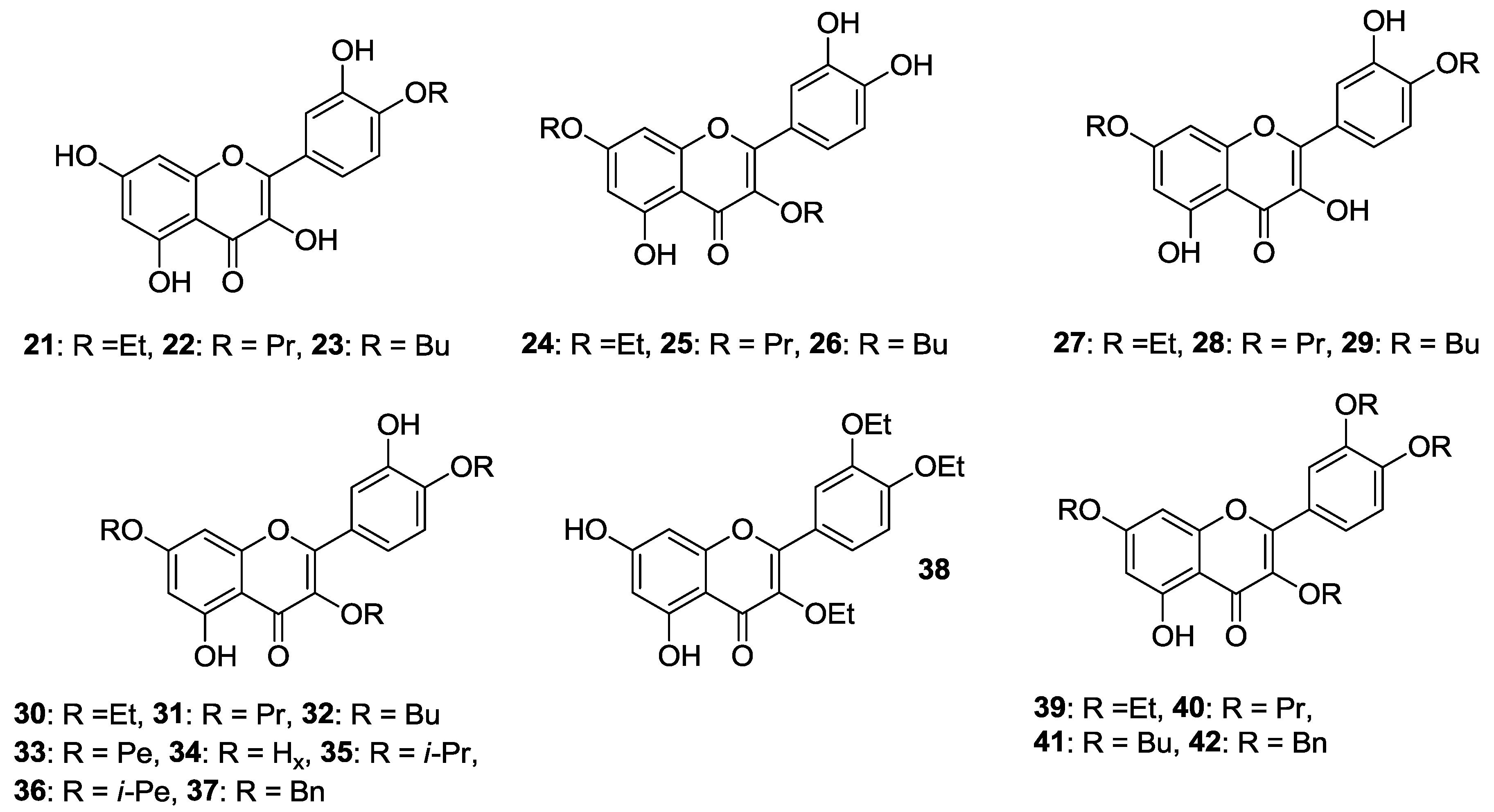
Besides, the preparation of 4′-
O-monoalkylated (
21–
23), 3,7-
O-dialkylated (
24–
26), 4′,7-
O-dialkylated (
27–
29), 3,4′,7-
O-trialkylated (
30–
37), 3,3′,4′-
O-trialkylated (
38), and 3,3′,4′,7-
O-tetraalkylated (
39–
42) derivatives of quercetin (
Figure 5) was achieved in the same way as the quercetin methyl ether compounds [
41,
42]. Ensuing in vitro biological evaluation by the abovementioned HTS method lead Shi et al. to demonstrate that cancer cell growth inhibitory activities were retained when etherification of 3-OH and 4′-OH was carried out using the long propyl chain or the short ethyl one, respectively [
41]. On the contrary, introduction of two
n-butyloxy moieties into the 3,7 or 4′,7 sites enhanced the antiproliferative action.
Speaking of these studies, cytotoxicity data of the most representative
O-alkylated quercetin derivatives are listed in
Table 2.
With particular regard to human prostate cancer cells, Al-Jabban et al. concluded that antiproliferative activity strongly depended on either length or steric hindrance of the introduced alkyl chain [
42]. Indeed, cancer cell growth greatly dropped when linear long or bulky alkyl groups were simultaneously introduced into C-3, C-4′ and C-7 hydroxyl groups, as observed for compounds (
31–
34) and (
35,
36), respectively. On the other hand, the derivative (
30) appended with the short, linear ethyl group showed a slightly increased activity, similarly to the corresponding methyl analog (
17,
Figure 4). However, no significant change in activity was detected for 3,3′,4′-
O-triethylquercetin (
38). Importantly, the potency of 3,7-
O-dialkylated derivatives (
24–
26) was 2–11 times higher than quercetin (
Table 3), with this behaviour being also observed for the corresponding dimethylated compound (
11,
Figure 4).
It should be mentioned that the structures of the 4′,7-
O-dialkylquercetins reported by Shi et al. [
41] have been found to be wrong by heteronuclear multiple bond correlation (HMBC) nuclear magnetic resonance (NMR) experiments, and were corrected as the corresponding 3,7-
O-dialkylated isomers [
42].
A recent work by Khan and coworkers evidenced that 3,4′,7-
O-triethylquercetin (
30,
Figure 5) was able to inhibit cell proliferation in colon (HCT-116) cancer cells (IC
50 = 50 μM, 24 h incubation). Moreover, it behaved as apoptosis-inducer in the same cancer cell line without affecting normal cells growth [
43]. It is worthwhile pointing out that
30 is supposed to take action through endoplasmic reticulum (ER) stress and a mitochondria-mediated pathway.
A three-step procedure involving peracetylation of quercetin followed by alkylation with a suitable alkyl chloride and base-mediated deacetylation gave access to 7-
O-butylquercetin
43 and 7-
O-geranylquercetin
44, which are shown in
Figure 5 [
44,
45]. These compounds showed a moderate solubility (180 μM) in Dulbecco’s modified eagle medium (DMEM) [
45], and exhibited much stronger antiproliferative effects than quercetin in estrogen receptor-positive human breast cancer cell line (MCF-7), likely due to their better accumulation capability [
44]. More precisely, the proliferation inhibitory activity of
43 and
44 depended on their apoptosis-inducing effects which were anyhow higher than those of quercetin. In this regard, it was demonstrated that the apoptotic process of MCF-7 cells occurred through a caspase-independent Endonuclease G (Endo G)-induced mitochondrial route, unlike quercetin.
It is worthy of note that compounds 43 and 44 did not affect normal breast epithelial (MCF-10A) cells and were also effective in estrogen receptor-negative MDA-MB-231 breast cancer cells. Furthermore, 43 and 44 were proposed to possess reversal activities on MDR cancer cells, but no evidence in support of this hypothesis was furnished.
Further studies revealed that
44 had strong cytotoxicity on human colon (CaCo-2), human lung (NCI-H446 and A549) as well as human gastric (MGC-803 and SGC-7901) cancer cells thereby revealing potential antiproliferative properties [
45]. In all cases, the observed activity proved to be higher as compared to quercetin. For the sake of clarity, the most relevant biological data regarding compounds
43 and
44 are detailed in
Table 4.
In order to extend previous results on quercetin conjugates bearing a pivaloxymethyl (POM) promoiety at the 3 or the 7 position [
46], the 3,7-bis-
O-pivaloxymethyl (POM) quercetin (
45,
Figure 5) was prepared by sequential K
2CO
3-promoted alkylation of quercetin diphenylmethylketal with pivaloxymethyl iodide (POM-I) and deprotection [
47]. In-depth studies evidenced that
45 had great stability in Dulbecco’s modified eagle medium complete (cDMEM) (
Table 5) and efficient uptake inside cells wherein it was selectively hydrolyzed to the corresponding 3-
O-POM-quercetin, with no trace of other metabolites (i.e., 7-
O-POM-quercetin or quercetin) being detected.
Significant cytostatic activity of 45 was observed in MCF-7, HCT-116, and DU-145 cancer cell lines, as compared to quercetin which gave no inhibition of cell proliferation. Importantly, 45 displayed a cancer cell specific cytostatic effect, and no action was demonstrated on normal human diploid fibroblast (HS 27) cell line. Mechanistically, it has been proposed that the quercetin-POM conjugate 45 operates via a different pathway against quercetin, with cell cycle arrest taking place in the G0/G1 phase.
In a proof-of-concept study, Chong et al. demonstrated that 7-
O-POM-quercetin (
46,
Figure 5) was able to reverse MDR in drug-resistant MES-SA/Dx5 cells derived from the drug-sensitive human uterine sarcoma (MES-SA) cell line (
Table 6) [
48,
49].
Mechanistically, it was evidenced competition of 46 with verapamil binding to the P-glycoprotein (P-gp), which is a major MDR target. Moreover, 46 proved to be considerably more potent than quercetin and as active as verapamil in inhibiting the drug efflux mediated by P-gp.
Importantly, 46 evidenced accumulation inside MES-SA/Dx5 cells wherein it persisted along with its hydrolyzed product quercetin and quercetin metabolites (glucuronide and sulfate) for more than 48 h. As a result, the intracellular levels of 46 were adequately high to elicit the increased MDR-reversal effect as compared to quercetin.
Suitably protected quercetin derivatives were reacted with iodomethyl isopropyl carbonate (POC-I)/K
2CO
3 system in either DMF or DMF/acetone mixture and the compounds obtained were then deprotected to afford the quercetin conjugates
47–
49 (
Figure 5) bearing an isopropyloxycarbonylmethoxy (POC) group at 3-OH and/or 7-OH [
50]. These species were deeply studied with regard to solubility, stability, permeability and intracellular metabolism. Compounds (
47) and (
49) were poorly soluble in phosphate-buffered saline (PBS) differently to
48 which proved to dissolve well in the same medium even at high concentrations. Anyhow, complete dissolution of all derivatives was observed in cDMEM (up to 100 μM concentration). With regard to stability, it has been demonstrated that quercetin-POC conjugates were almost as stable as the quercetin-POM derivatives [
46,
47]. All compounds featured high stability in PBS (
t1/2 >96 h) (
Table 7), while either decomposition or hydrolysis occurred in cell-free culture medium. Thus, the 3,7-bis-
O-POC derivative
49 was hydrolyzed into 3-
O-POC compound
48, whereas the 7-functionalized analog
47 gave rise to decomposition and/or hydrolysis to the mother quercetin. Among the series, 3-
O-POC
48 showed the best stability profile in term of resistance to both decomposition and hydrolysis.
Besides, membrane permeability assays assessed that the 7-conjugated derivative
47 behaved as quercetin, while 3-
O-POC-quercetin
48 was the less permeable. In any case, the permeability of
48 is worthwhile noting as the corresponding 3-
O-POM conjugate was totally impermeable [
46]. Remarkably, no data could be obtained for
49 due to its low solubility in PBS at the concentration (25 μM) used for the membrane permeability test.
Cell-line-dependent hydrolytic and metabolic profiles were observed for quercetin derivatives
47–
49. On the one hand, they were smoothly converted to quercetin and its metabolites in MCF-7 cell line, with quercetin glucuronide being predominantly formed in all cases, according to literature data [
51]. It should be highlighted that these results were completely different from those observed for the quercetin-POM analogs, which have been shown to be less prone to both intracellular hydrolysis and metabolism [
46,
47]. In addition, 3-
O-POC-quercetin
48 was easily hydrolyzed and metabolized contrary to 3-
O-POM-quercetin and 3,7-bis-
O-POM-quercetin
45 [
46,
47].
On the other hand,
47–
49 underwent slow hydrolysis and low metabolism in HCT-116 cells. In particular, 7-
O-POC-quercetin (
47) hydrolyzed to quercetin but neither of its metabolites was detected, while 3-
O-POC-quercetin (
48) proved to be very stable (up to 12 h) giving no trace of quercetin. This metabolic profile was also typical of 3,7-bis-
O-POC-quercetin (
49), but its hydrolysis hastened (
t1/2 ≅ 3 h) in relation to cell-free medium (
t1/2 = 24 h,
Table 7). In this case, 3-
O-POC-quercetin (
48) was formed as the exclusive hydrolysis product.
Cytotoxic activities of 47–49 were strictly related to their stability properties. As a matter of fact, low antiproliferative effects against MCF-7 cells were observed for the POC-protected quercetins likely due to enhanced passive transport, intracellular hydrolysis, and metabolism. More precisely, compounds 47–49 were as active as quercetin. On the contrary, 47 and 49 displayed higher cytotoxicity than quercetin in HCT-116 cells, with 49 being more effective than 47. Given the slow hydrolysis and metabolism of 47 and 49 in HCT-116 cells, both these compounds and their hydrolyzed products were present at concentrations high enough to enhance cytotoxicity. Importantly, 48 was not cytotoxic at all.
In addition to alkyl halides, 2,3-dichloro-1,4-naphtoquinone has been employed to alkylate quercetin using
N,
N-diisopropylethylamine as the base giving chloronaphtoquinone quercetin (
50, CHNQ), which is depicted in
Figure 5. This compound featured a 3-fold higher cytotoxicity than quercetin in colorectal (HCT-116 and HT-29) cancer cells (
Table 8), and strong apoptosis-inducing effects, too, have been observed [
52].
Furthermore, likely due to the presence of the 1,4-napthoquinone framework, treatment of cells with 50 resulted in a potent generation of oxidative stress leading to reactive oxygen species (ROS)-induced autophagy in vitro. In particular, the authors highlighted that complete autophagy occurred in HCT-116 cells, while incomplete autophagy took place in the HT-29 ones. Herein, CHNQ promoted LC3 lipidation, with the formation of acidic vacuoles being not observed.
It should be pointed out that conversion of quercetin into an isomeric chloronaphtoquinone derivative has been previously reported by Danihelová et al. [
53]. This structural modification led to lower the antioxidant properties of quercetin, but enhanced the cancer cell inhibitory activities. However, the chloronaphtoquinone derivative also featured cytolytic effects towards non-cancer murine fibroblast (NIH-3T3) at a concentration of 100 μM, but the total degeneration of cancer cells (HeLa) took place at lower concentrations (50 μM).
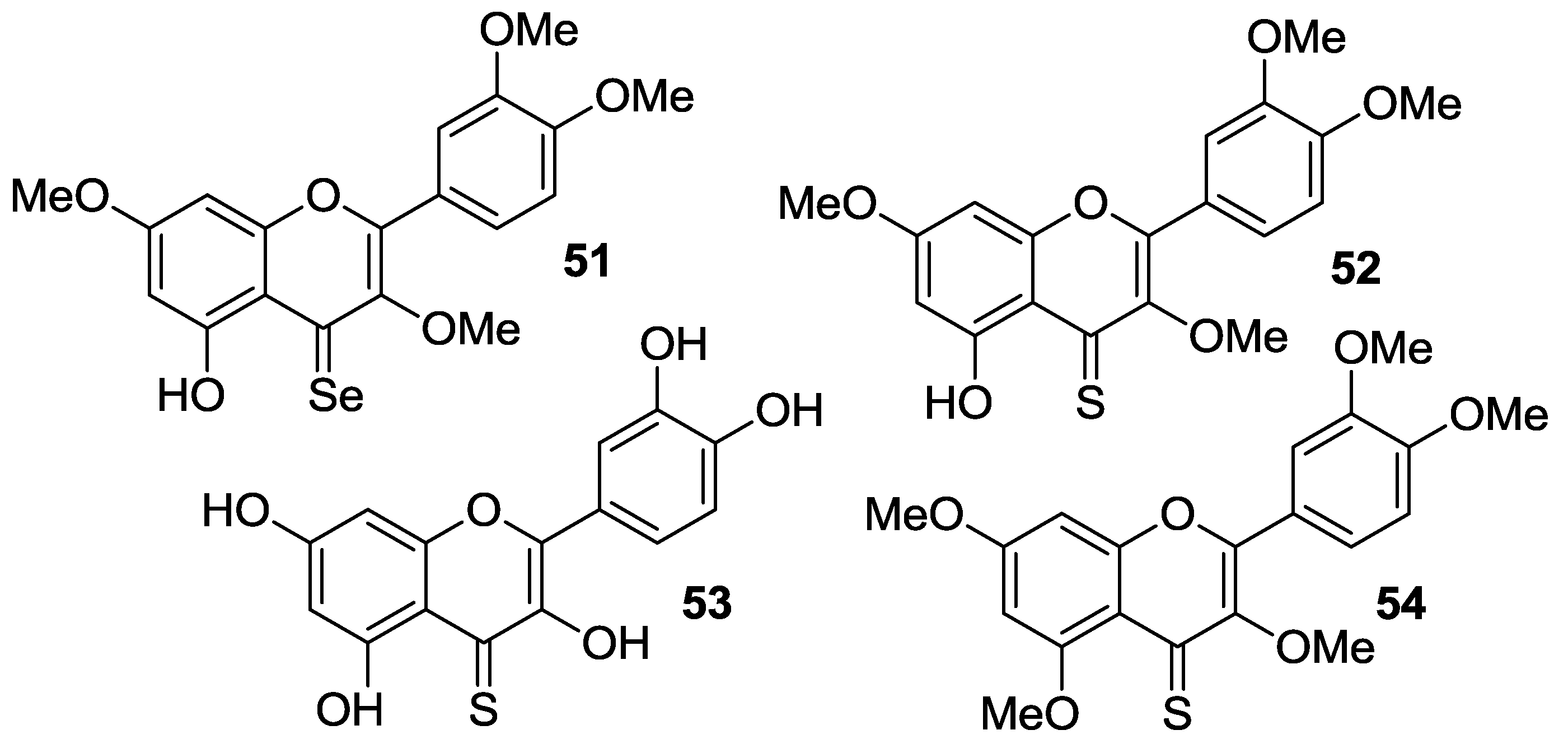
2.1.2. O-Alkylation and C-4 Carbonyl Group Modification
The
O-alkylation of quercetin has been conveniently coupled with the bioisosteric conversion of the C-4 carbonyl group into the corresponding thiocarbonyl or selenocarbonyl moieties. So, synthetic routes involving suitable methylation of quercetin followed by either oxygen/sulfur or oxygen/selenium exchange and deprotection have been carried out to provide the sulfur- and seleno-compounds
51–
54, which are shown in
Figure 6.
Martins et al. reported the synthesis of analogs
51–
53 via chalcogenation of the quercetin-derived 3,3′,4′,7-
O-tetramethyl compound (
19,
Figure 4) [
54]. All the compounds have been tested on nine human cancer cell lines, namely melanoma cells (A375), colorectal adenocarcinoma cells (HCT-15), pancreatic adenocarcinoma cells (BxPC3), MCF-7 cells and the multidrug-resistant variant MCF-7/ADR, cervical adenocarcinoma cells (A431) and the corresponding cisplatin-resistant one (A431/Pt), cisplatin-sensitive and cisplatin-resistant ovarian adenocarcinoma cells (2008 and C13*). For the purpose of comparison, the same cancer cell lines were used in parallel experiments with quercetin,
19, and cisplatin.
Thus, the selenium compound
51 showed a 9-fold and 3-fold higher cytotoxicity than quercetin and cisplatin, respectively, while
19 proved to be 3-fold less cytotoxic than quercetin (
Table 9).
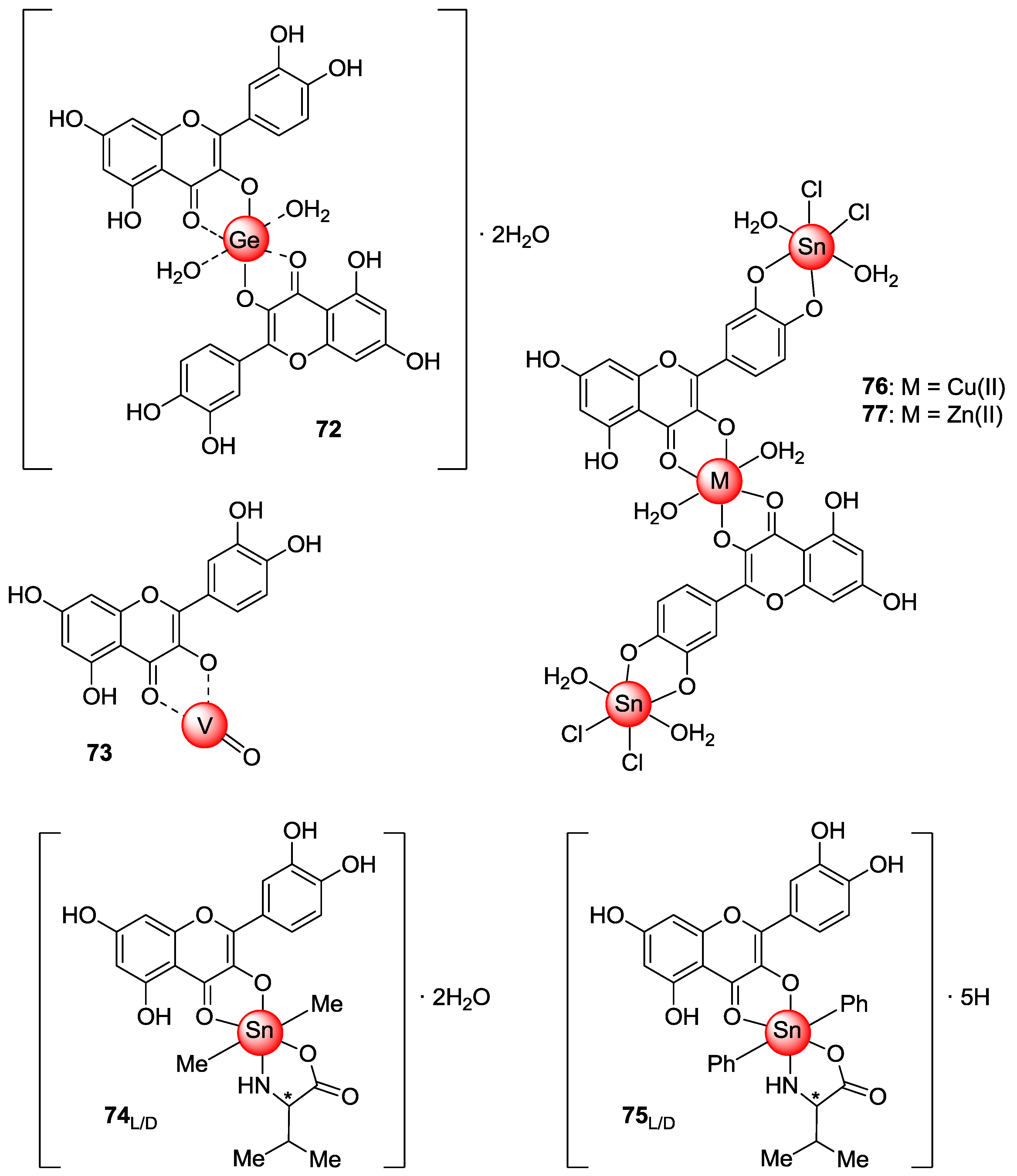
This result suggested that the selenocarbonyl moiety rather than the flavonoid core was responsible for the observed biological effects. On the contrary, unreproducible data were observed for the sulfur-containing derivatives 52 and 53 as a result of their low stability in solution. Furthermore, protected selenoquercetin 51 was able to overcome cisplatin-resistance due to comparable cytotoxic action against both the cisplatin-sensitive and cisplatin resistant cell lines. Remarkably, preliminary studies aimed at understanding the mechanism of cytotoxicity in MCF-7 cells evidenced that 51 hampered thioredoxin reductase (TrxR) activity, whilst it lacks efficacy to affect the glutathione peroxidase (GPx)/glutathione reductase (GR) redox system.
Exhaustive
O-methylation of quercetin to the 3,3′,4′,5,7-
O-pentamethyl derivative (
20,
Figure 4) followed by thionation gave access to compound
54, which was eventually deprotected yielding
53 [
55,
56]. It has been shown that both compounds possess in vitro antiangiogenic and antiproliferative properties [
55]. As a matter of fact, they were able to inhibit the migration of human umbilical endothelial vascular cells (HUVECs) promoted by the vascular endothelial growth factor (VEGF). However, the antiangiogenic activity of thiocarbonyl compounds
53 and
54 was inferior to that of both quercetin and
20, with
54 being more active than
53. Besides,
53 and
54 had antiproliferative activity towards MCF-7 cancer cell line and the doxorubicin-resistant variant MCF-7/DX, but much higher concentrations (approximately a 10 to 100-fold increase) were required compared to those determining the antiangiogenic action. In particular, the observed antiproliferative effects were in the order quercetin >
53 >
20 >
54. Overall, derivative
20 was found to be the top of the line as it optimally balanced the high antiangiogenic activity with minimal toxicity. Further antiproliferative studies using breast cancer cell line MDA-MB-231 evidenced that the 4-thio compounds, and in particular
53, had greater effects than
20 and the parent quercetin [
56]. However,
53 did not reach the IC
50 at 10 μM. Moreover,
20,
53, and
54 proved to be less active than quercetin towards both MCF-7 and MCF-7/DX cell lines, with the lowest activity being observed in the latter. This result was supposed to depend on the fact that these compounds are likely to act as substrates of the P-gp efflux pump, which is known to be over expressed in the MCF-7/DX cell line. It is worthwhile noting that in all cases no stability problems of
53 were pointed out, in contrast with others [
54].
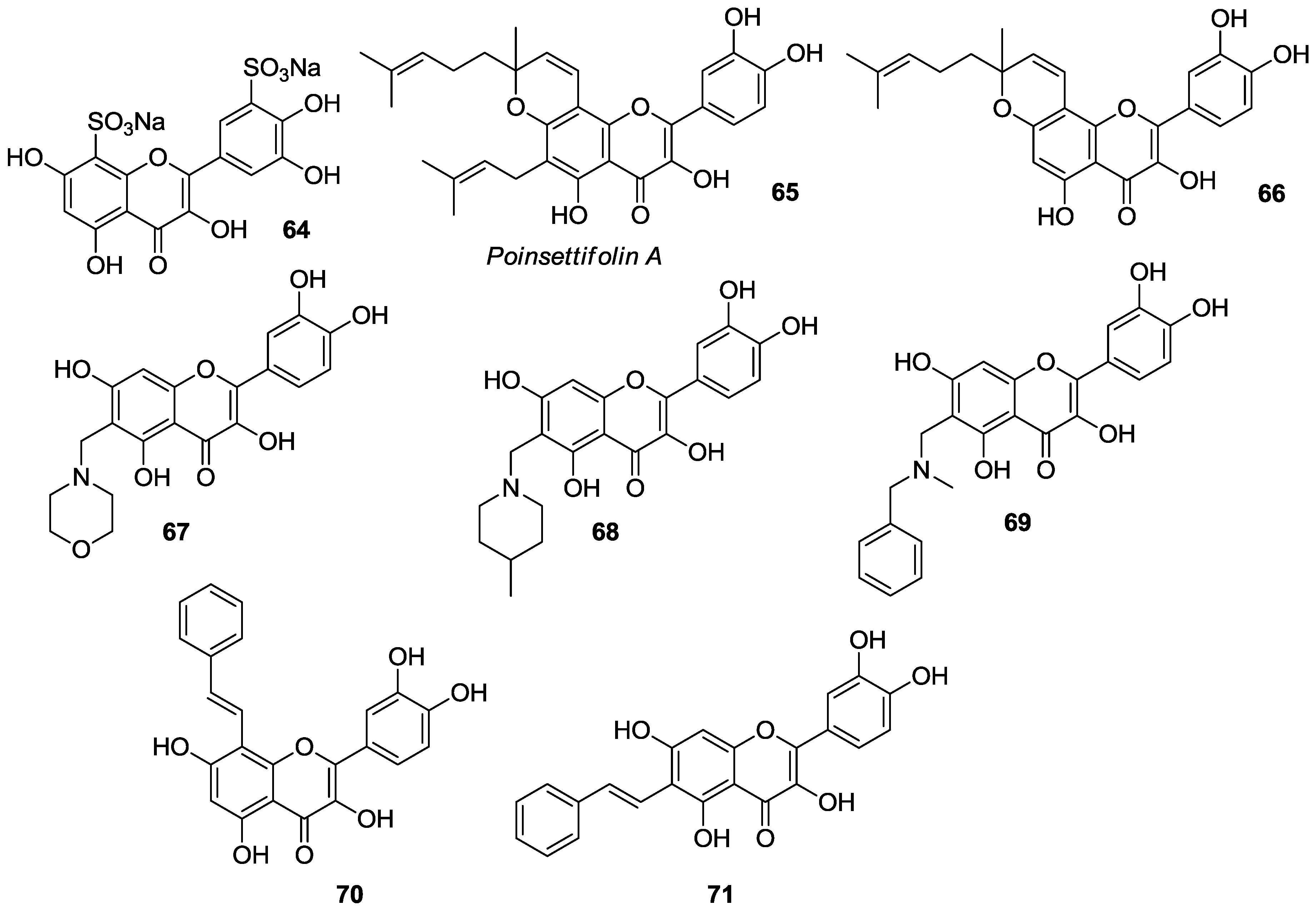
2.1.4. O-Acylation
Introduction of ester and urethane moieties into the quercetin scaffold produced compounds with improved cytotoxic action. This is likely due to a better bioavailability as a possible result of lipophilization [
53,
59,
60]. During the period 2012–2016, synthesis and anticancer activities of
O-acylated quercetin derivatives
56–
63 (
Figure 8) were reported.
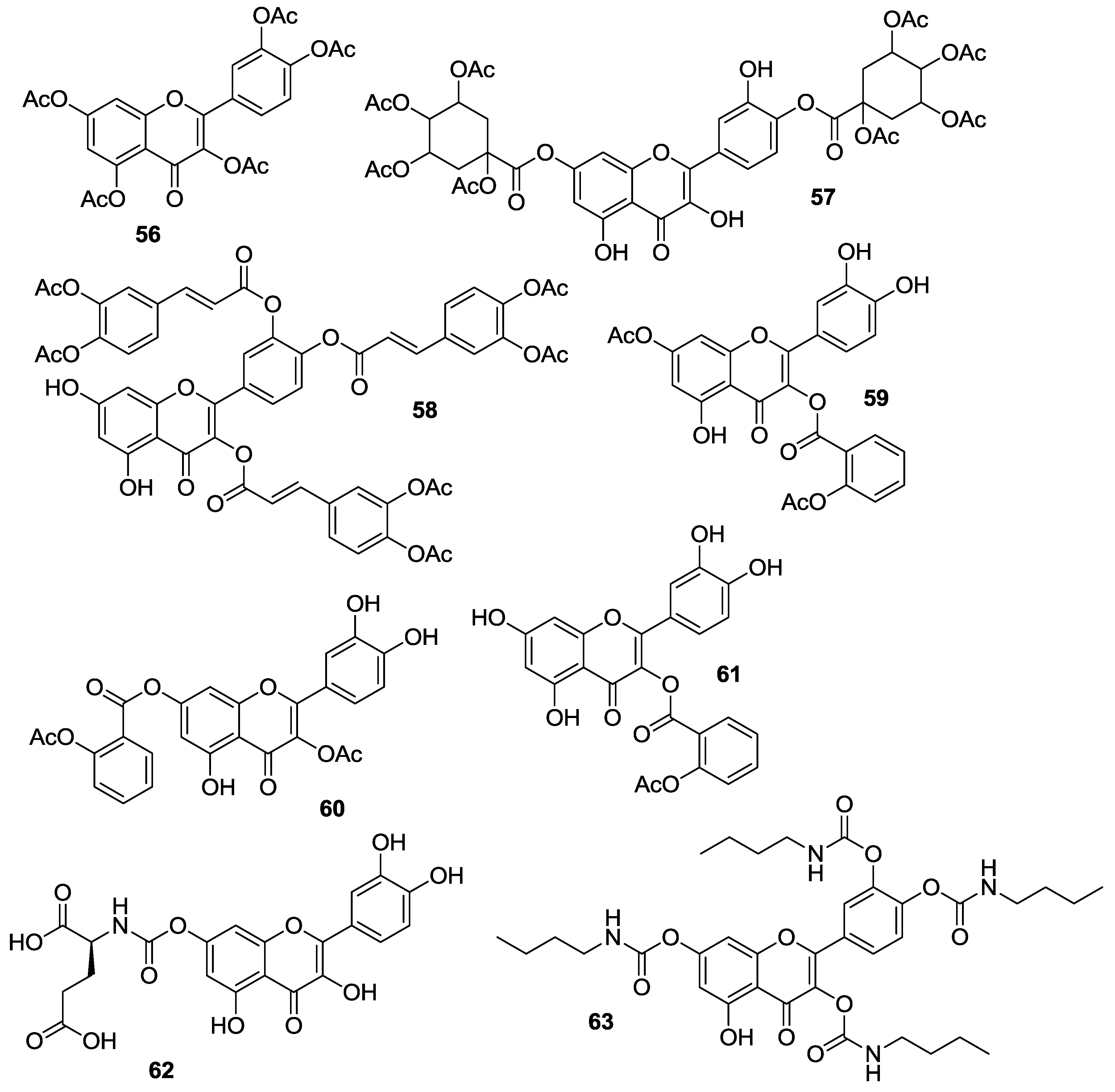
Danihelová et al. prepared fifteen quercetin-derived compounds through condensation or selective protection reactions followed by acylation with acyl chlorides [
53]. Among the series, pentaacetyl quercetin (
56), di(tetraacetylquinoyl)quercetin (
57), and tri(diacetylcaffeoyl)quercetin (
58) exhibited the highest cytotoxicity towards HeLa cells and the non-cancerous cell line NIH-3T3. Notably, all these compounds were more effective than quercetin regardless of cell type.
Conversion of quercetin into the corresponding diphenylmethylketal followed by esterification with aspirin at the 3- and 7-OH gave access to quercetin aspirinates
59–
61 showing higher cytotoxic effects against liver (HepG2) and promyelocytic leukemia (HL-60) cancer cells than quercetin [
61].
Results of the cytotoxic activities of compounds
56–
61 are reported in
Table 10.
It has been previously demonstrated that quercetin amino-acid conjugates possess properties (i.e., water solubility, hydrolytic stability, cell permeability) superior to those of quercetin [
62]. Based on this result, Kim et al. prepared six quercetin derivatives appended with alanine or glutamic acid residues at 3-
O and/or 7-
O sites [
63]. Key steps in the synthetic strategy concerned the selective protection/deprotection of quercetin hydroxyl groups and the coupling of the intermediates obtained with suitably protected alanine and glutamic acid compounds.
The quercetin-amino acid analogs were tested for their cytotoxicity and ability to modulate MDR using the MES-SA cell line and the corresponding drug-resistant MES-SA/Dx5, that is known to overexpress P-gp [
49]. At the concentration levels applied for MDR modulation (5 μM), the quercetin-amino acid conjugates showed no cytotoxic action in MES-SA cell line, likewise quercetin. Importantly, addition of the latter or the quercetin-amino acid derivatives did not affect the cytotoxic properties of a given anticancer agent, similarly to verapamil which has been used as the positive control. With regard to MES-SA/Dx5 cell line, MDR-reversal activity of the quercetin-amino acid compounds was strictly dependent on either the nature or position of the amino acid moieties. The 7-
O-functionalized compounds in the series displayed the most potent effects, and the best results were evidenced for the quercetin-7-
O-glutamic acid conjugate
62. As depicted in
Table 11, this compound proved to be 30.5-fold more active (fold-reversal, FR= 58.6) than quercetin (FR = 1.9) in reversing MDR towards doxorubicin, and was potent as much as the doxorubicin-resistance reversing drug verapamil (FR = 68.0). Additionally,
62 showed 13.8–14.8-times enhanced MDR-reversal effects against other anticancer drugs, including actinomycin D, vinblastine, and paclitaxel.
It is worthy of note that the MDR modulatory activity of 62 was not dictated by the stereochemistry of the amino acid promoiety. As a matter of fact, the synthetically prepared enantiomer of 62 was a potent MDR modulator (FR = 52.2), though less than 62 itself. Moreover, flow cytometric analysis and P-gp ATPase test evidenced that 62 inhibited the drug efflux by P-gp, and stimulated ATPase activity of P-gp by interaction with its drug-binding site, respectively.
Evaluation of the physico-chemical properties of compound
62 highlighted that the presence of the glutamic acid residue accounted for enhanced solubility, stability, and cellular uptake of quercetin. Indeed,
62 was markedly soluble in aqueous medium up to 400 μM, while quercetin solubility harshly dropped at concentration >100 μM. In comparison with quercetin, compound
62 possessed high stability both in PBS (
t1/2 > 72 h) and Roswell Park Memorial Institut (RPMI)-1640 complete culture medium (cRPMI), with decomposition occurring only after 9.3 h (
Table 12).
Importantly, the intracellular level of
62 remained adequately high for a prolonged period of time (6–24 h) due to slow metabolism to quercetin and quercetin metabolites (i.e., quercetin glucuronide and quercetin sulfate) that co-existed with
62. This result may profile the use of
62 as a safe MDR modulator given the riskless nature of quercetin. Quercetin was also reacted with
n-butyl-isocyanate to obtain the 3,3′,4′,7-
O-tetraacylated derivative
63. This compound proved to inhibit the proliferation of MCF-7 cells in vitro [
64], with the IC
50 value obtained being 36 μM compared to 128 μM for quercetin.

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}