Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects

Abstract
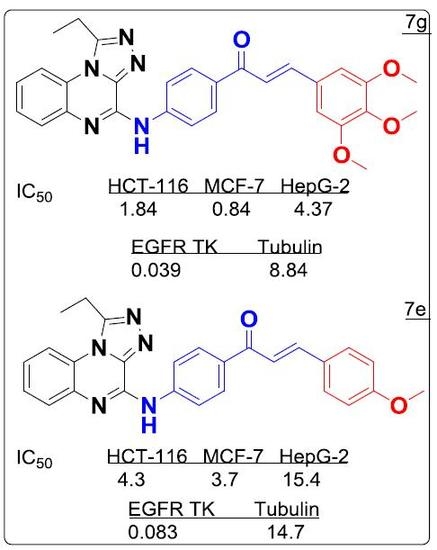
:
1. Introduction
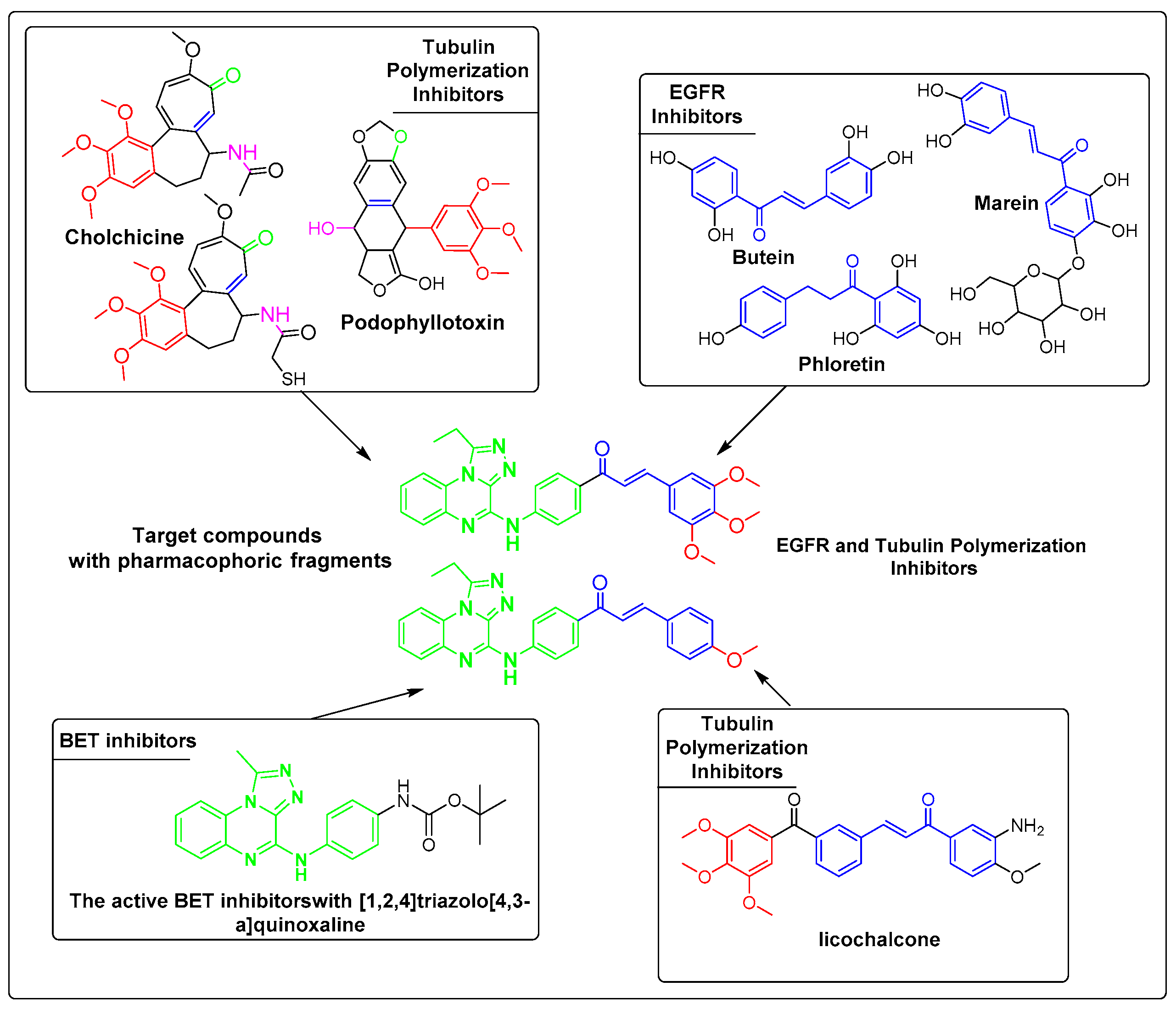
2. Rationale Study
3. Results and Discussion
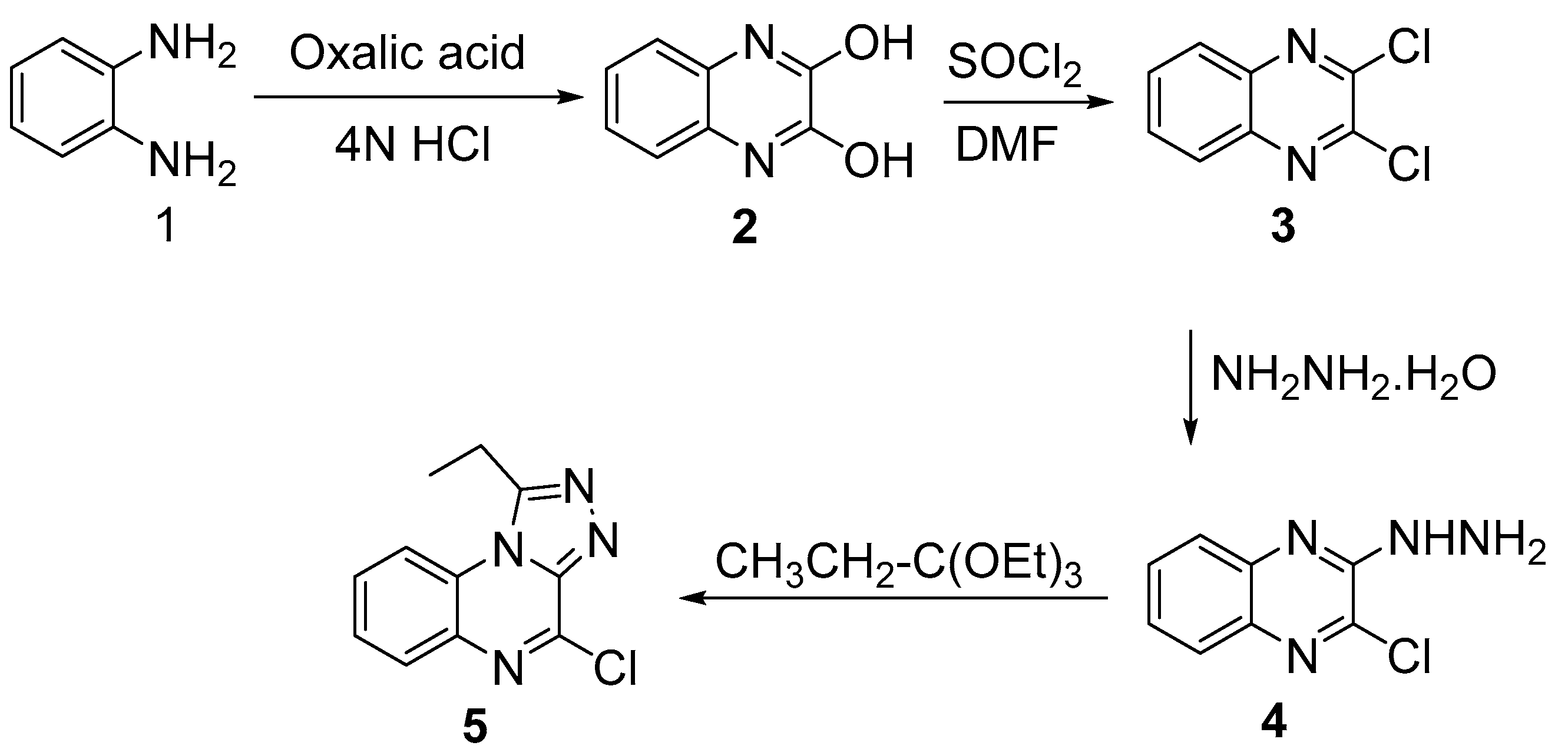
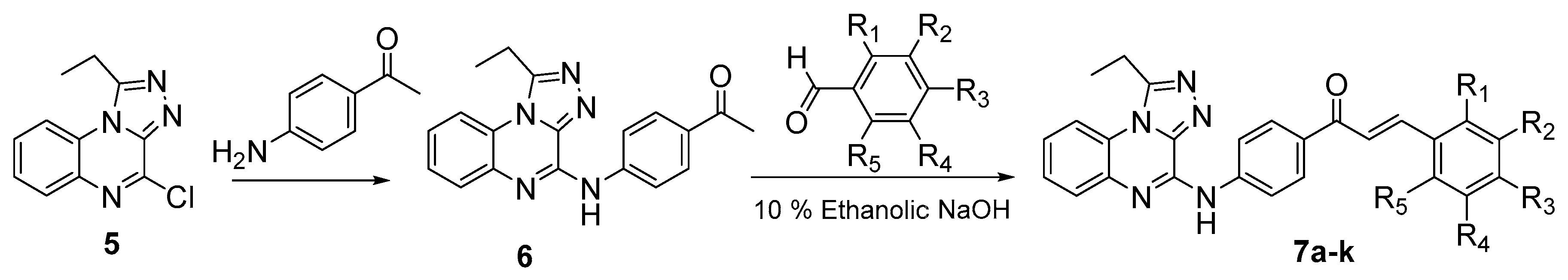
3.1. Chemistry
3.2. Cytotoxicity Screening
3.3. EGFR Inhibitory Assay
3.4. Tubulin Assay
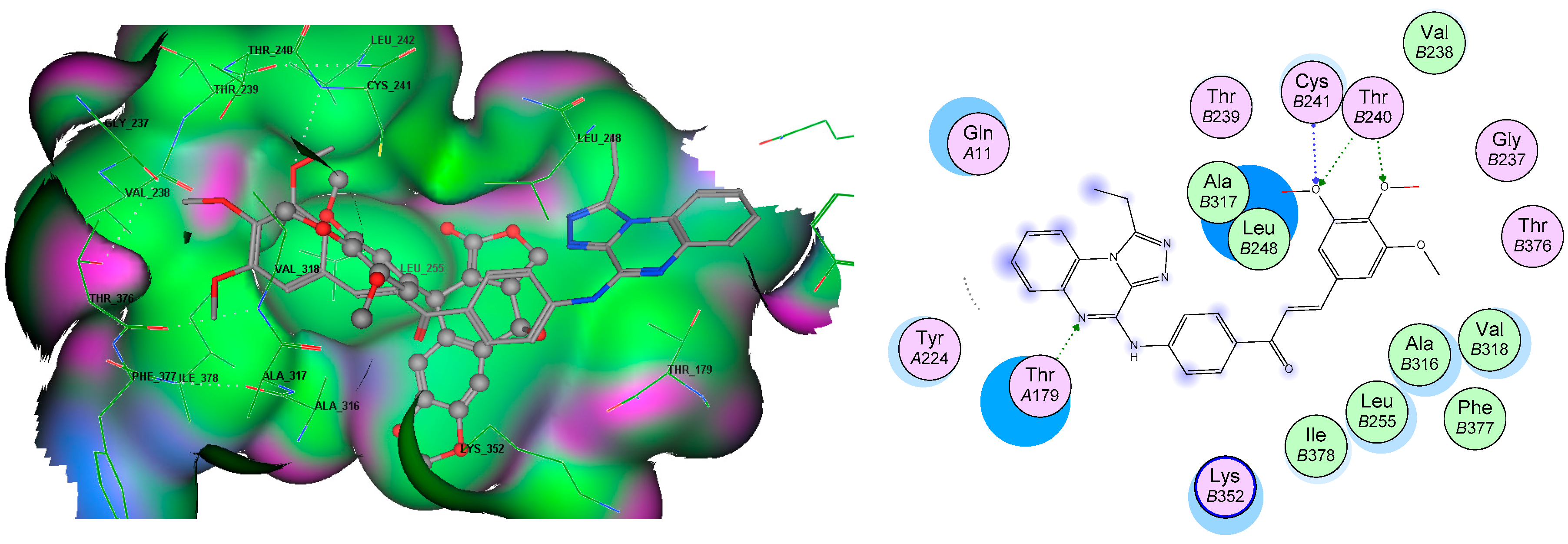
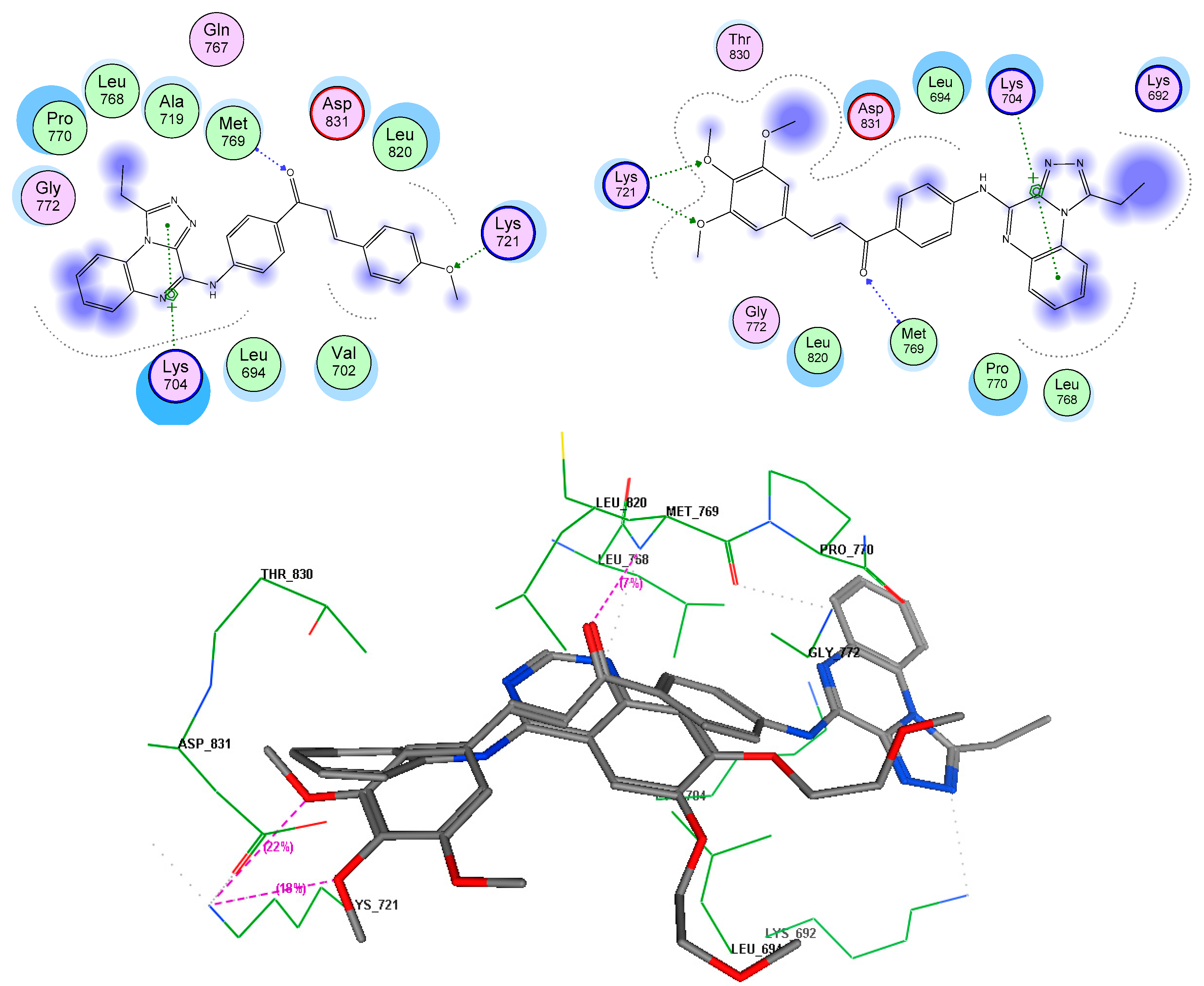
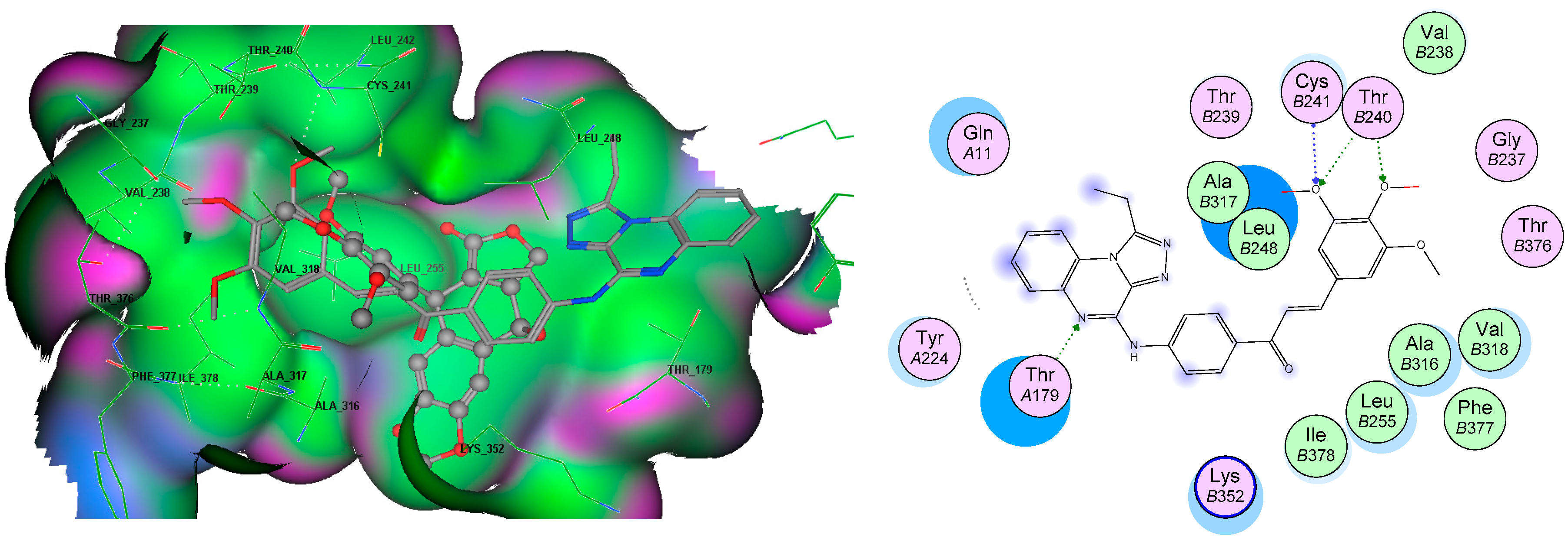
3.5. Molecular Docking Analyses
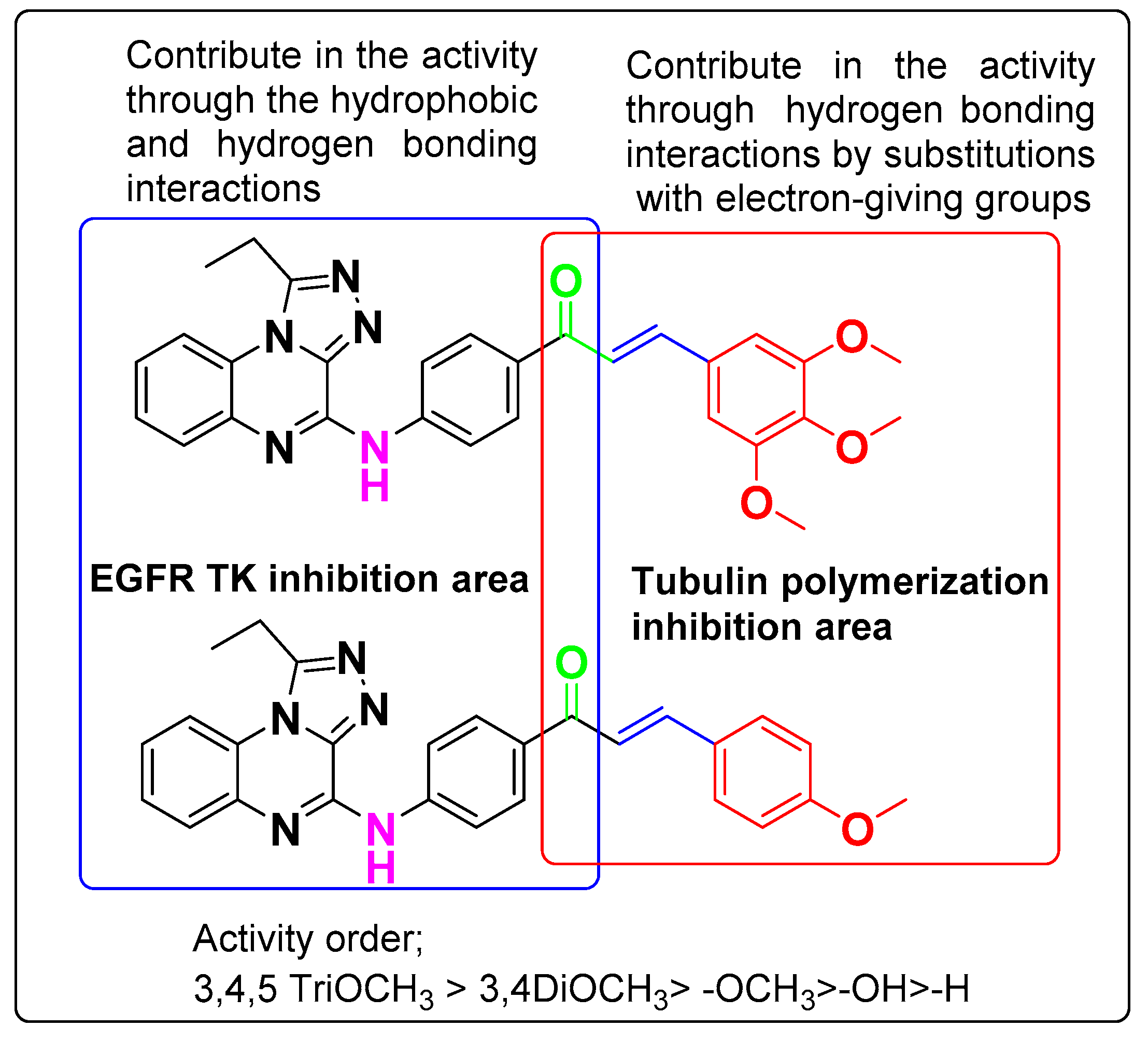
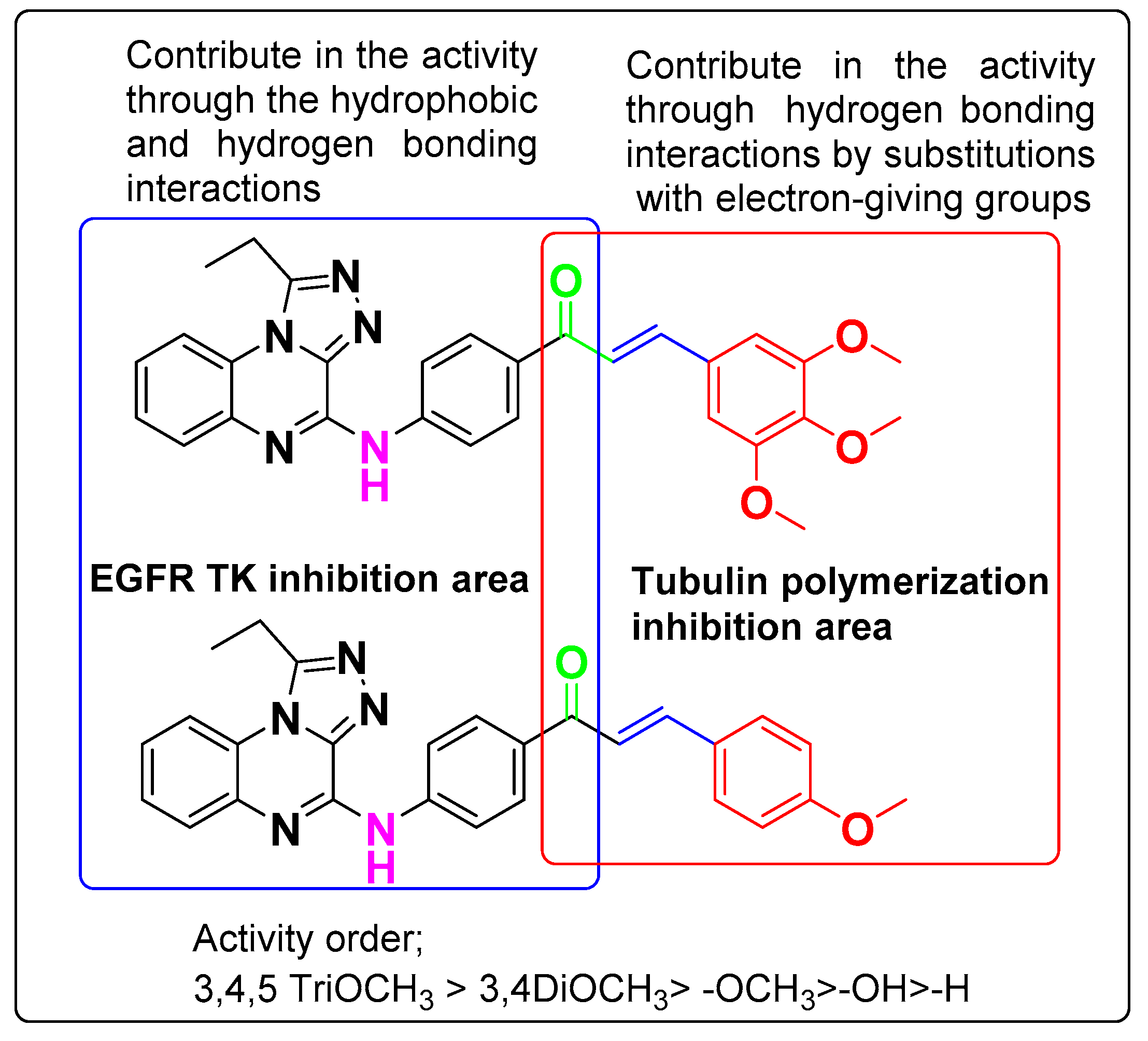
3.6. SAR Analyses
4. Experimental Section
4.1. General Information
4.2. Chemistry
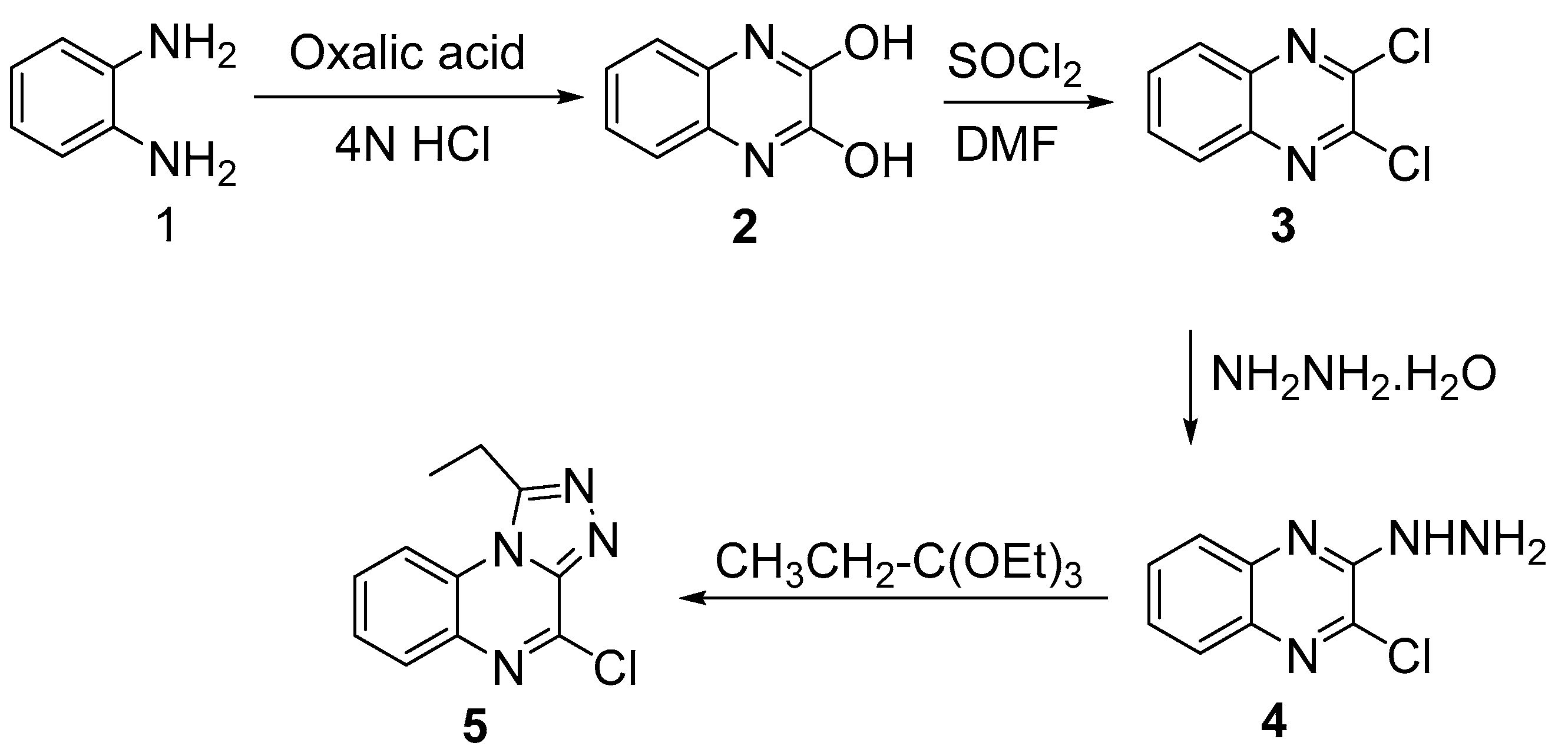
4.2.1. Synthesis of 2,3-(1H,4H)-Quinoxalinedione (2)
4.2.2. Synthesis of 2,3-Dichloroquinoxaline (3)
4.2.3. Synthesis of 2-Chloro-3-hydrazinylquinoxaline (4)
4.2.4. Synthesis of 4-Chloro-1-ethyl-[1,2,4]triazolo[4,3-a]quinoxaline (5)
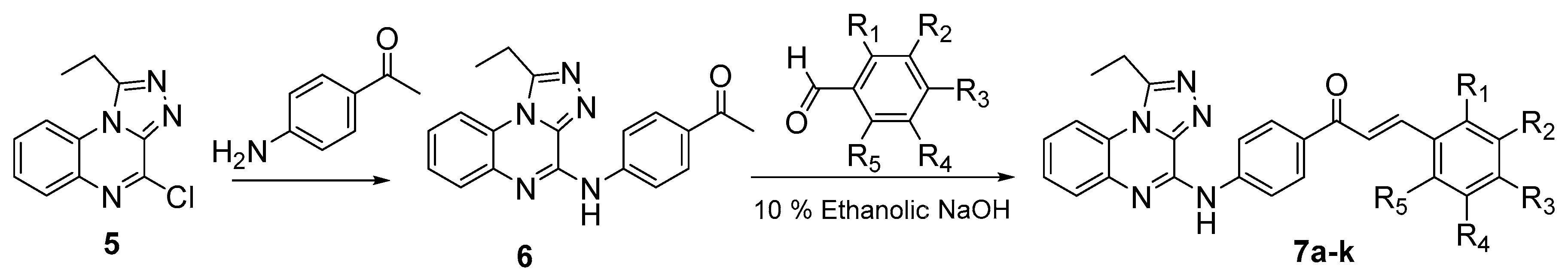
4.2.5. Synthesis of 1-(4-((1-ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)ethan-1-one (6)
4.2.6. General Method for the preparation of (Z)-{4-[(1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino]phenyl}-3-substituted phenylprop-2-en-1-ones 7a–k
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-phenylprop-2-en-1-one (7a)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(2-methoxyphenyl)prop-2-en-1-one (7b)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(2-hydroxyphenyl)prop-2-en-1-one (7c)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(p-tolyl)prop-2-en-1-one (7d)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (7e)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (7f)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (7g)
(E)-1-(4-((1-Ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)-3-(4-nitrophenyl)prop-2-en-1-one (7h)
(E)-3-(4-Chlorophenyl)-1-(4-((1-ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)prop-2-en-1-one (7i)
(E)-3-(2,4-Dichlorophenyl)-1-(4-((1-ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)prop-2-en-1-one (7j)
(E)-3-(2,6-Dichlorophenyl)-1-(4-((1-ethyl-[1,2,4]triazolo[4,3-a]quinoxalin-4-yl)amino)phenyl)prop-2-en-1-one (7k)
4.3. Cancer Cell Antiproliferative Assay
4.4. EGFR Inhibition Assay
4.5. Enzyme-Linked Immunosorbent Assay for Tubulin Beta (TUBb)
4.6. Molecular Docking
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Joshi, M.; Pal, S.K.; Drabick, J.J. Novel approaches in cancer immunotherapy—A light at the end of the tunnel. Dis. Med. 2016, 21, 479–487. [Google Scholar]
- Ali, N.A.; O’Brien, J.M., Jr.; Blum, W.; Byrd, J.C.; Klisovic, R.B.; Marcucci, G.; Phillips, G.; Marsh, C.B.; Lemeshow, S.; Grever, M.R. Hyperglycemia in patients with acute myeloid leukemia is associated with increased hospital mortality. Cancer 2007, 110, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M.; Arion, V.B.; Jakupec, M.A.; Keppler, B.K. Recent developments in the field of tumor-inhibiting metal complexes. Curr. Pharm. Des. 2003, 9, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, W.; Shan, Y.; Liu, F.; Xu, X.; Yang, Y.; Zhang, Q.; Zhang, Y.; Kuang, H.; Wang, Z.; et al. Design, synthesis and anticancer activity of novel nopinone-based thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2360–2363. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T. New challenges and inspired answers for anticancer drug discovery and development. Jpn. J. Clin. Oncol. 2013, 43, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Ingle, R.; Marathe, R.; Magar, D.; Patel, H.M.; Surana, S.J. Sulphonamido-quinoxalines: Search for anticancer agent. Eur. J. Med. Chem. 2013, 65, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cho, S.; Namgoong, K.; Jung, J.K.; Cho, J.; Yang, S.I. Synthesis and in vitro evaluation of 7-dialkylaminomethylbenzo[g]quinoxaline-5,10-diones. Bioorg. Med. Chem. Lett. 2004, 14, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Sridevi, C.H.; Balaji, K.; Naidu, A.; Sudhakaran, R. Synthesis of some phenylpyrazolo benzimidazolo quinoxaline derivatives as potent antihistaminic agents. E J. Chem. 2010, 7, 234–238. [Google Scholar] [CrossRef]
- Patel, N.; Bergman, J.; Graslund, A. 1H-NMR studies of the interaction between a self-complementary deoxyoligonucleotide duplex and indolo[2,3-b]quinoxaline derivatives active against herpes virus. Eur. J. Biochem. 1991, 197, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.E.A.; Ihmaid, S.K.; Omar, A.M.; Shehata, A.M.; Rateb, H.S.; Zayed, M.F.; Ahmed, S.; Elaasser, M.M. Design, synthesis, molecular docking of new lipophilic acetamide derivatives affording potential anticancer and antimicrobial agents. Bioorg. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Refaat, H.M.; Moneer, A.A.; Khalil, O.M. Synthesis and antimicrobial activity of certain novel quinoxalines. Arch. Pharm. Res. 2004, 27, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Tandon, V.K.; Yadav, D.B.; Maurya, H.K.; Chaturvedi, A.K.; Shukla, P.K. Design, synthesis, and biological evaluation of 1,2,3-trisubstituted-1,4-dihydrobenzo[g]quinoxaline-5,10-diones and related compounds as antifungal and antibacterial agents. Bioorg. Med. Chem. 2006, 14, 6120–6126. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Sanna, P.; Gherardini, L.; Usai, D.; Zanetti, S. Novel functionalized pyrido[2,3-g]quinoxalinones as antibacterial, antifungal and anticancer agents. Farmaco 2001, 56, 933–938. [Google Scholar] [CrossRef]
- Vicente, E.; Duchowicz, P.R.; Castro, E.A.; Monge, A. Qsar analysis for quinoxaline-2-carboxylate 1,4-di-N-oxides as anti-mycobacterial agents. J. Mol. Graph. Model. 2009, 28, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new 2-acetyl and 2-benzoyl quinoxaline 1,4-di-N-oxide derivatives as anti-mycobacterium tuberculosis agents. Eur. J. Med. Chem. 2003, 38, 791–800. [Google Scholar] [CrossRef]
- Burguete, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Villar, R.; Vicente, E.; Solano, B.; Ancizu, S.; Perez-Silanes, S.; Aldana, I.; Monge, A. Synthesis and anti-inflammatory/antioxidant activities of some new ring substituted 3-phenyl-1-(1,4-di-N-oxide quinoxalin-2-yl)-2-propen-1-one derivatives and of their 4,5-dihydro-(1H)-pyrazole analogues. Bioorg. Med. Chem. Lett. 2007, 17, 6439–6443. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Hobrath, J.V.; Connelly, M.C.; Kiplin Guy, R.; Reynolds, R.C. Diverse amide analogs of sulindac for cancer treatment and prevention. Bioorg. Med. Chem. Lett. 2017, 27, 4614–4621. [Google Scholar] [CrossRef] [PubMed]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Hearn, M.J.; Cynamon, M.H. Design and synthesis of antituberculars: Preparation and evaluation against mycobacterium tuberculosis of an isoniazid schiff base. J. Antimicrob. Chemother. 2004, 53, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.; Ammar, Y.A.; Ibrahim, M.K.; El-Zahaby, H.S.; Mahmoud, S.S. Synthesis and pharmacological evaluation of novel quinoxalines as potential nonulcerogenic anti-inflammatory and analgesic agents. Arzneimittelforschung 2005, 55, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.; Werfel, E.; Galla, F.; Lehmkuhl, K.; Torres-Gomez, H.; Schepmann, D.; Kogel, B.; Christoph, T.; Strassburger, W.; Englberger, W.; et al. Synthesis and pharmacological evaluation of 5-pyrrolidinylquinoxalines as a novel class of peripherally restricted kappa-opioid receptor agonists. J. Med. Chem. 2014, 57, 6845–6860. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Desrivot, J.; Bories, C.; Loiseau, P.M.; Franck, X.; Hocquemiller, R.; Figadere, B. Synthesis and antiprotozoal activity of some new synthetic substituted quinoxalines. Bioorg. Med. Chem. Lett. 2006, 16, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Sarges, R.; Howard, H.R.; Browne, R.G.; Lebel, L.A.; Seymour, P.A.; Koe, B.K. 4-amino[1,2,4]triazolo[4,3-a]quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants. J. Med. Chem. 1990, 33, 2240–2254. [Google Scholar] [CrossRef] [PubMed]
- Alswah, M.; Ghiaty, A.; El-Morsy, A.; El-Gamal, K. Synthesis and biological evaluation of some [1,2,4]triazole[4,3-a]quinoxaline derivatives as novel anticonvulsant agents. ISRN Org. Chem. 2013, 2013, 587054. [Google Scholar] [CrossRef] [PubMed]
- Mielcke, T.R.; Mascarello, A.; Filippi-Chiela, E.; Zanin, R.F.; Lenz, G.; Leal, P.C.; Chiaradia, L.D.; Yunes, R.A.; Nunes, R.J.; Battastini, A.M.; et al. Activity of novel quinoxaline-derived chalcones on in vitro glioma cell proliferation. Eur. J. Med. Chem. 2012, 48, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururaja, T.L.; Yung, S.; Ding, R.; Huang, J.; Zhou, X.; McLaughlin, J.; Daniel-Issakani, S.; Singh, R.; Cooper, R.D.; Payan, D.G.; et al. A class of small molecules that inhibit tnfalpha-induced survival and death pathways via prevention of interactions between tnfalphari, tradd, and rip1. Chem. Biol. 2007, 14, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Matwijczuk, A.; Janik, E.; Luchowski, R.; Niewiadomy, A.; Gruszecki, W.I.; Gagoś, M. Spectroscopic studies of the molecular organization of 4-([1,2,4] triazolo [4,3-a] pyridin-3-yl)-6-methylbenzene-1,3-diol in selected solvents. J. Lumin. 2018, 194, 208–218. [Google Scholar] [CrossRef]
- Yuan, C.; Li, S.; Wu, Y.; Lu, L.; Zhu, M. Zn(ii)-selective and sensitive fluorescent chemosensor based on steric constrains and inhibition of esipt. Sens. Actuators B Chem. 2017, 242, 1035–1042. [Google Scholar] [CrossRef]
- Padalkar, V.S.; Chemate, S.B.; Lanke, S.K.; Sekar, N. N-2-aryl-1,2,3-triazoles: A novel class of blue–green emitting fluorophores-synthesis, photophysical properties study and dft computations. J. Lumin. 2015, 168, 114–123. [Google Scholar] [CrossRef]
- Habib, N.S.; El-Hawash, S.A. Synthesis and antimicrobial testing of thiazolinyl-, thiazolidinonyl-quinoxalines and 1,2,4-triazolo[4,3-a]quinoxalines. Die Pharm. 1997, 52, 594–598. [Google Scholar]
- El-Hawash, S.A.; Habib, N.S.; Fanaki, N.H. Quinoxaline derivatives. Part ii: Synthesis and antimicrobial testing of 1,2,4-triazolo[4,3-a]quinoxalines, 1,2,4-triazino[4,3-a]quinoxalines and 2-pyrazolylquinoxalines. Pharmazie 1999, 54, 808–813. [Google Scholar] [PubMed]
- Suresh, M.; Lavanya, P.; Sudhakar, D.; Vasu, K.; Rao, C.V. Synthesis and biological activity of 8-chloro-[1,2,4]triazolo[4,3-a]quinoxalines. J. Chem. Pharm. Res. 2010, 2, 497–504. [Google Scholar]
- Johnson, L.B.; Kauffman, C.A. Voriconazole: A new triazole antifungal agent. Clin. Infect. Dis. 2003, 36, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Torres, H.A.; Hachem, R.Y.; Chemaly, R.F.; Kontoyiannis, D.P.; Raad, I.I. Posaconazole: A broad-spectrum triazole antifungal. Lancet Infect. Dis. 2005, 5, 775–785. [Google Scholar] [CrossRef]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzyme Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Issa, D.A.E.; Habib, N.S.; Abdel Wahab, A.E. Design, synthesis and biological evaluation of novel 1,2,4-triazolo and 1,2,4-triazino[4,3-a]quinoxalines as potential anticancer and antimicrobial agents. Med. Chem. Comm. 2015, 6, 202–211. [Google Scholar] [CrossRef]
- Ali, I.; Lee, J.; Go, A.; Choi, G.; Lee, K. Discovery of novel [1,2,4]triazolo[4,3-a]quinoxaline aminophenyl derivatives as bet inhibitors for cancer treatment. Bioorg. Med. Chem. Lett. 2017, 27, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed]
- El-Hawash, S.A.M.; Habib, N.S.; Kassem, M.A. Synthesis of some new quinoxalines and 1,2,4-triazolo[4,3-a]-quinoxalines for evaluation of in vitro antitumor and antimicrobial activities. Arch. Der Pharm. 2006, 339, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Dell, A.; Williams, D.H.; Morris, H.R.; Smith, G.A.; Feeney, J.; Roberts, G.C. Structure revision of the antibiotic echinomycin. J. Am. Chem. Soc. 1975, 97, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Wakelin, S.P.; Waring, M.J. The binding of echinomycin to deoxyribonucleic acid. Biochem. J. 1976, 157, 721–740. [Google Scholar] [CrossRef] [PubMed]
- Lieva, D.B.; Todorova, I.T. Trends in utilization of the pharmacological potential of chalcones. Curr. Clin. Pharmacol. 2010, 5, 1–29. [Google Scholar]
- Mielcke, T.R.; Muradas, T.C.; Filippi-Chiela, E.C.; Amaral, M.E.A.; Kist, L.W.; Bogo, M.R.; Mascarello, A.; Neuenfeldt, P.D.; Nunes, R.J.; Campos, M.M. Mechanisms underlying the antiproliferative effects of a series of quinoxaline-derived chalcones. Sci. Rep. 2017, 7, 15850. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Wang, K.; Edler, M.C.; Hamel, E.; Mooberry, S.L.; Paige, M.A.; Brown, M.L. A boronic acid chalcone analog of combretastatin a-4 as a potent anti-proliferation agent. Bioorg. Med. Chem. 2010, 18, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Qiu, K.M.; Lu, X.; Liu, K.; Fu, J.; Zhu, H.L. Synthesis, biological evaluation, and molecular modeling of cinnamic acyl sulfonamide derivatives as novel antitubulin agents. Bioorg. Med. Chem. 2011, 19, 4730–4738. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kaur, C.; Budhiraja, A.; Nepali, K.; Gupta, M.K.; Saxena, A.K.; Bedi, P.M.S. Chalcone based azacarboline analogues as novel antitubulin agents: Design, synthesis, biological evaluation and molecular modelling studies. Eur. J. Med. Chem. 2014, 85, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Dyrager, C.; Wickstrom, M.; Friden-Saxin, M.; Friberg, A.; Dahlen, K.; Wallen, E.A.; Gullbo, J.; Grotli, M.; Luthman, K. Inhibitors and promoters of tubulin polymerization: Synthesis and biological evaluation of chalcones and related dienones as potential anticancer agents. Bioorg. Med. Chem. 2011, 19, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Kumar, G.B.; Vishnuvardhan, M.V.; Shaik, A.B.; Reddy, V.S.; Mahesh, R.; Sayeeda, I.B.; Kapure, J.S. Synthesis of phenstatin/isocombretastatin-chalcone conjugates as potent tubulin polymerization inhibitors and mitochondrial apoptotic inducers. Org. Biomol. Chem. 2015, 13, 3963–3981. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.; McGown, A.T.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; Boccard, J.; Carrupt, P.A.; Nicolle, E.; Blanc, M.; Geze, A.; Choisnard, L.; Wouessidjewe, D.; Matera, E.L.; Dumontet, C. Antimitotic and antiproliferative activities of chalcones: Forward structure-activity relationship. J. Med. Chem. 2008, 51, 2307–2310. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Caputo, V.; Karadimitris, A. Inhibition of bromodomain and extra-terminal proteins (bet) as a potential therapeutic approach in haematological malignancies: Emerging preclinical and clinical evidence. Ther. Adv. Hematol. 2015, 6, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.V.; Siddiqui, S.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Lokwani, D.K.; Nikalje, A.P.G. Microwave-assisted facile synthesis, anticancer evaluation and docking study of n-((5-(substituted methylene amino)-1,3,4-thiadiazol-2-yl)methyl) benzamide derivatives. Molecules 2017, 22, 995. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Romer, D.R. Synthesis of 2,3-dichloroquinoxalines via vilsmeier reagent chlorination. J. Heterocycl. Chem. 2009, 46, 317–319. [Google Scholar] [CrossRef]
- Klancnik, A.; Piskernik, S.; Jersek, B.; Mozina, S.S. Evaluation of diffusion and dilution methods to determine the antibacterial activity of plant extracts. J. Microbiol. Methods 2010, 81, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Gangadevi, V.J.M. Preliminary studies on cytotoxic effect of fungal taxol on cancer cell lines. Afr. J. Biotechnol. 2007, 6, 1382–1386. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2012; Available online: http://www.chemcomp.com (accessed on 30 February 2013).
Sample Availability: Samples of the compounds are currently not available from the authors. |

| Compound | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|
| 7a | H | H | H | H | H |
| 7b | OCH3 | H | H | H | H |
| 7c | OH | H | H | H | H |
| 7d | H | H | CH3 | H | H |
| 7e | H | H | OCH3 | H | H |
| 7f | H | H | OH | H | H |
| 7g | H | OCH3 | OCH3 | OCH3 | H |
| 7h | H | H | NO2 | H | H |
| 7i | H | H | Cl | H | H |
| 7j | Cl | H | Cl | H | H |
| 7k | Cl | H | H | H | Cl |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) * | ||
|---|---|---|---|
| HCT-116 | MCF-7 | HepG-2 | |
| 5 | 236 ± 0.10 | 246 ± 0.13 | 322 ± 0.15 |
| 6 | 411 ± 0.21 | 410 ± 0.25 | 446 ± 0.20 |
| 7a | 57.49 ± 0.29 | 58.92 ± 0.32 | 105.21 ± 0.50 |
| 7b | 25.60 ± 0.03 | 24.04 ± 0.12 | 30.72 ± 0.12 |
| 7c | 19.76 ± 0.23 | 22.17 ± 0.11 | 17.32 ± 0.12 |
| 7d | >100 | 93.40 ± 0.14 | >100 |
| 7e | 9.57 ± 0.11 | 8.23 ± 0.25 | 34.28 ± 0.14 |
| 7f | 18.10 ± 0.15 | >100 | >100 |
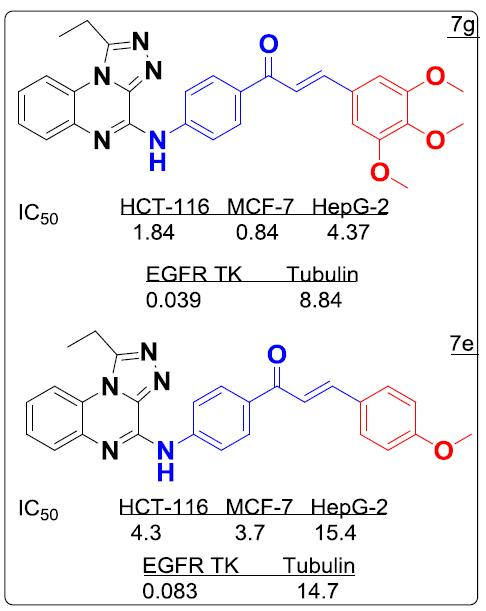
| 7g | 3.61 ± 0.18 | 1.65 ± 0.13 | 8.58 ± 0.06 |
| 7h | >100 | >100 | >100 |
| 7i | >100 | >100 | >100 |
| 7j | >100 | >100 | >100 |
| 7k | 19.66 ± 0.25 | >100 | >100 |
| Doxorubicin | 1.55 ± 0.03 | 0.27 ± 0.08 | 0.22 ± 0.01 |
| Compounds | EGFR Inhibition (IC50 µM) a,b | Tubulin Polymerization (IC50 µM) c |
|---|---|---|
| 7a | 661.0 ± 0.11 | 77.1 ± 0.21 |
| 7b | 127.0 ± 0.32 | 18.1 ± 0.30 |
| 7c | 136.0 ± 0.05 | 43.4 ± 0.01 |
| 7d | >100 | ND |
| 7e | 0.083 ± 0.04 | 14.7 ± 0.11 |
| 7f | >50 | >100 |
| 7g | 0.039 ± 0.16 | 8.84 ± 0.02 |
| 7h | >100 | >100 |
| 7i | >100 | >100 |
| 7j | >100 | >100 |
| 7k | >100 | >100 |
| Colchicine | - | 26.8 ± 0.12 |
| Staurosporine | 0.054 ± 0.10 | - |
| Compound | Target | Binding Energy (kcal/mol) | Interacting Moieties | Amino Acids |
|---|---|---|---|---|
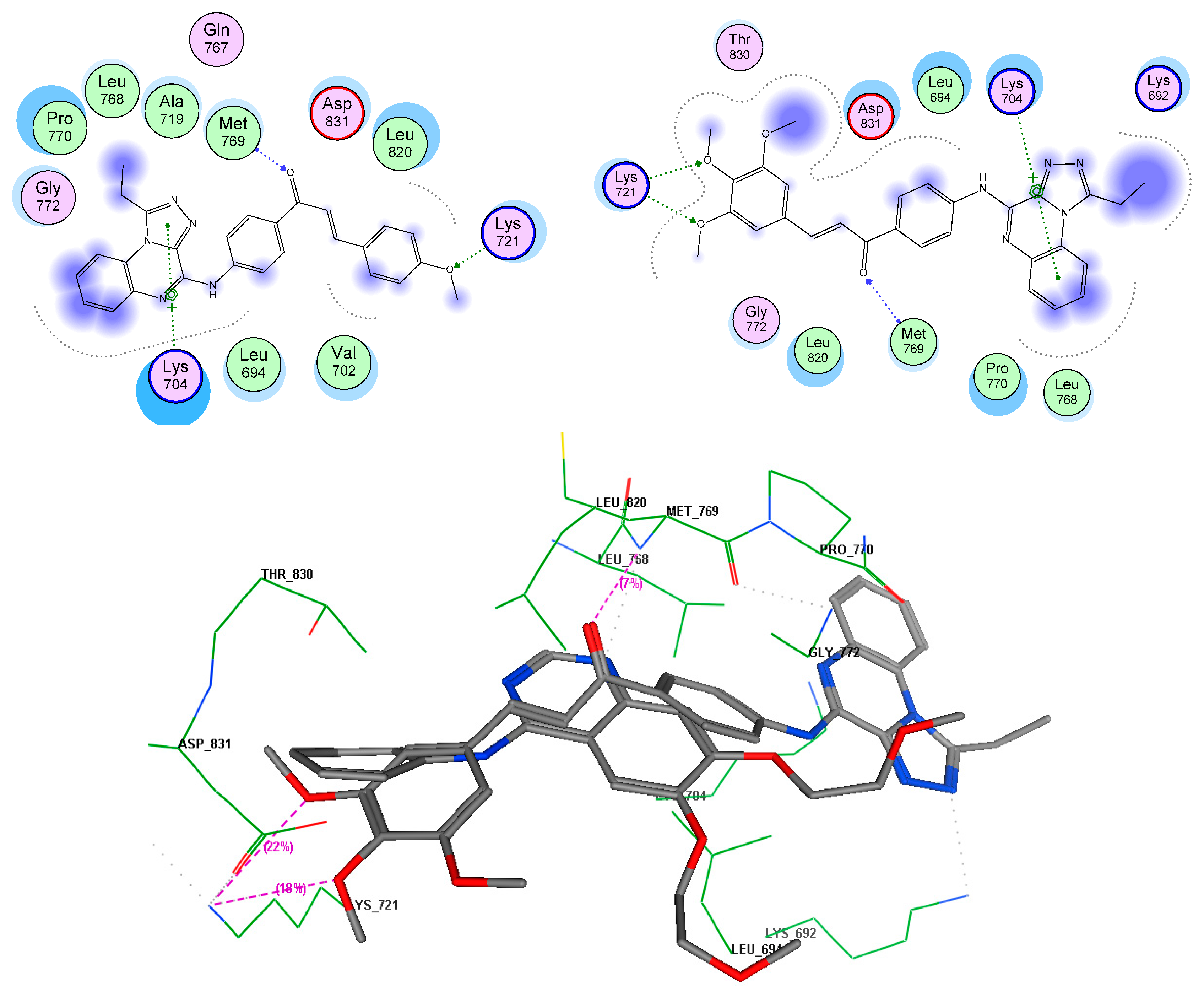
| 7e | EGFR TK (1M17) | −13.51 | 3,4 diO-CH3 | Lys721 |
| C=O iminone linker | Met769 | |||
| Quinoxaline | Lys704 | |||
| 4-aminophenylcarbonyl | Leu694 | |||
| 7g | −15.5 | 3,4 diO-CH3 | Lys721 | |
| C=O iminone linker | Met769 | |||
| Quinoxaline | Lys704, Pro770, Leu768 | |||
| 4-aminophenylcarbonyl | Leu694 | |||
| Tubulin (3E22) | −12.8 | 3,4 diO-CH3 | Cys241, Thr240 | |
| Quinoxaline | Thr179 | |||
| Chalcone | Leu248, Ala315, Ile378, Leu255 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2018, 23, 48. https://doi.org/10.3390/molecules23010048
Alswah M, Bayoumi AH, Elgamal K, Elmorsy A, Ihmaid S, Ahmed HEA. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules. 2018; 23(1):48. https://doi.org/10.3390/molecules23010048
Chicago/Turabian StyleAlswah, Mohamed, Ashraf H. Bayoumi, Kamal Elgamal, Ahmed Elmorsy, Saleh Ihmaid, and Hany E. A. Ahmed. 2018. "Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects" Molecules 23, no. 1: 48. https://doi.org/10.3390/molecules23010048
APA StyleAlswah, M., Bayoumi, A. H., Elgamal, K., Elmorsy, A., Ihmaid, S., & Ahmed, H. E. A. (2018). Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules, 23(1), 48. https://doi.org/10.3390/molecules23010048