Oxysterols and Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome: Implications for an Improved Therapeutic Intervention


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The RSH/Smith-Lemli-Opitz Syndrome (SLOS)
3. AY9944 and the Development of a Rat Model of SLOS
3.1. Retinal Degeneration in the AY9944-Induced SLOS Rat Model
3.2. Sterols and Oxysterols in the Retina in the AY9944-Induced Rat Model of SLOS
4. Perspective and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chevreul, M.E. Recherches chimiques sur les corps gras, et particulièrement sur leurs combinaisons avec les alcalis. Sixième mémoire. Examen des graisses d’homme, de mouton, de boeuf, de jaguar et d’oie. Ann. Chim. Phys. 1816, 2, 339–372. (In French) [Google Scholar]
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef] [PubMed]
- Myant, N.B. The Biology of Cholesterol and Related Sterols, 1st ed.; Butterworth-Heinemann: Oxford, UK, 1981. [Google Scholar]
- Yeagle, P.L. Biology of Cholesterol; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Bittman, R. Cholesterol: Its functions and metabolism in biology and medicine. In Subcellular Biochemistry; Plenum Press: New York, NY, USA, 1997; Volume 28. [Google Scholar]
- Finegold, L. (Ed.) Cholesterol in Membrane Models; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Smith, L.L.; Johnson, B.H. Biological activities of oxysterols. Free Radic Biol Med. 1989, 7, 285–332. [Google Scholar] [CrossRef]
- Luu, B.; Moog, C. Oxysterols: Biological activities and physiochemical studies. Biochemie 1991, 73, 1317–1320. [Google Scholar] [CrossRef]
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis 1999, 142, 1–28. [Google Scholar] [CrossRef]
- Schroepfer, G.J.J. Oxysterols: Modulators of cholesterol metabolism and other processes. Physiol. Rev. 2000, 80, 361–554. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.; Jörnvall, H. Oxysterols. Biochim. Biophys. Res. Commun. 2014, 446, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Mutemberezi, V.; Guillermot-Legris, O.; Muccioloi, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Rossin, D.; Poli, G.; Biasi, F.; Leonarduzzi, G. Implication of oxysterols in chronic inflammatory human diseases. Biochemie 2018, 153, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.W.; Lemmli, L.; Opitz, J.M. A newly recognized syndrome of multiple congenital anomalies. J. Pediatr. 1964, 64, 210–217. [Google Scholar] [CrossRef]
- Tint, G.S.; Batta, A.K.; Xu, G.; Shefer, S.; Honda, A.; Irons, M.; Elias, E.R.; Salen, G. The Smith-Lemli-Opitz syndrome: A potentially fatal birth defect caused by a block in the last enzymatic step in cholesterol biosynthesis. Subcell Biochem. 1997, 28, 117–144. [Google Scholar] [PubMed]
- Porter, F.D.; Herman, G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2011, 51, 6–34. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, M.J.; Irons, M.B. Smith-Lemli-Opitz syndrome: Phenotype, natural history, and epidemiology. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 250–262. [Google Scholar] [CrossRef] [PubMed]
- DeBarber, A.E.; Eroglu, Y.; Merkens, L.S.; Pappu, A.S.; Steiner, R.D. Smith-Lemli-Opitz syndrome. Expert Rev. Mol. Med. 2011, 13, e24. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Herman, G.E. Inborn errors of sterol biosynthesis. Annu. Rev. Genomics Hum. Genet. 2001, 2, 299–341. [Google Scholar] [CrossRef] [PubMed]
- Tint, G.S.; Irons, M.E.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Tint, G.S.; Seller, M.; Hughes-Benzie, R.; Batta, A.K.; Shefer, S.; Genest, D.; Irons, M.E.; Elias, E.R.; Salen, G. Markedly increased tissue concentrations of 7-dehydrocholesterol combined with low levels of cholesterol are characteristic of the Smith-Lemli-Opitz syndrome. J. Lipid Res. 1995, 36, 89–95. [Google Scholar] [PubMed]
- Herman, G.E.; Kratz, L. Disorders of sterol synthesis: Beyond Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 301–321. [Google Scholar] [CrossRef] [PubMed]
- Fierro, M.; Martinez, A.J.; Harbison, J.W.; Hay, S.H. Smith-Lemli-Opitz syndrome: Neuropathological and ophthalmological observations. Dev. Med. Child. Neurol. 1977, 19, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Elias, E.R.; Hansen, R.M.; Irons, M.; Quinn, N.B.; Fulton, A.B. Rod photoreceptor responses in children with Smith-Lemli-Opitz syndrome. Arch. Ophthalmol. 2003, 121, 1738–1743. [Google Scholar] [CrossRef] [PubMed]
- Garry, D.; Hansen, R.M.; Moskowitz, A.; Elias, E.R.; Irons, M.; Fulton, A.B. Cone ERG responses in patients with Smith-Lemli-Opitz syndrome (SLOS). Doc. Ophthalmol. 2010, 121, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Montigny, M.; Dufour, R. HMG-CoA reductase inhibitors: A look back and a look ahead. Can. J. Cardiol. 1992, 8, 843–864. [Google Scholar] [PubMed]
- Givner, M.L.; Dvornik, D. Agents affecting lipid metabolism—XV. Biochemical studies with the cholesterol biosynthesis inhibitor AY-9944 in young and mature rats. Biochem. Pharmacol. 1965, 14, 611–619. [Google Scholar] [CrossRef]
- Kraml, M.; Marton, A.V.; Dvornik, D. Agents affecting lipid metabolism. XXVIII: A 7-dehydrocholesterol delat-7-reductase inhibitor (AY-9944) as tool in studies of delta-7-sterol metabolism. Biochemistry 1966, 5, 1060–1064. [Google Scholar]
- Roux, C.; Horvath, C.; Dupuis, R. Teratogenic action and embryo lethality of AY 9944R: Prevention by a hypercholesterolemia-provoking diet. Teratology 1979, 19, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Dupuis, R.; Horvath, C.; Talbot, J.N. Teratogenic effect of an inhibitor of cholesterol synthesis (AY 9944) in rats: Correlation with maternal cholesterolemia. J. Nutrition. 1980, 110, 2310–2312. [Google Scholar] [CrossRef] [PubMed]
- Kolf Clauw, M.; Chevy, F.; Wolf, C.; Siliart, B.; Citadelle, D.; Roux, C. Inhibition of 7-dehydrocholesterol reductase by the teratogen AY9944: A rat model for Smith-Lemli-Opitz syndrome. Teratology 1996, 54, 115–125. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Richards, M.J.; Miller, C.-Y.; Peachey, N.S. Marked alteration of sterol metabolism and composition without compromising retinal development or function. Invest. Ophthalmol. Vis. Sci. 1999, 40, 1792–1801. [Google Scholar] [PubMed]
- Fliesler, S.J.; Peachey, N.S.; Richards, M.J.; Nagel, B.A.; Vaughan, D.K. Retinal degeneration in a rodent model of Smith-Lemli-Opitz syndrome: Electrophysiological, biochemical, and morphological features. Arch. Ophthalmol. 2004, 122, 1190–2000. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.R.; Fliesler, S.J. Photodamage generates 7-keto- and 7-hydroxycholesterol in the rat retina via a free radical-mediated mechanism. Photochem. Photobiol. 2009, 85, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Rózanowska, M.; Rózanowski, B. Retinal photodamage. J. Photochem. Photobiol. B 2001, 64, 144–161. [Google Scholar] [CrossRef]
- Organisciak, D.T.; Vaughan, D.K. Retinal light damage: mechanisms and protection. Prog. Retin. Eye Res. 2010, 29, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.J.; Morgan, J.I.; Merigan, W.H.; Sliney, D.H.; Sparrow, J.R.; Williams, D.R. The susceptibility of the retina to photochemical damage from visible light. Prog. Retin. Eye Res. 2012, 31, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Davis, T.A.; Porter, N.A. Rate constants for peroxidation of polyunsaturated fatty acids and sterols in solution and in liposomes. J. Am. Chem. Soc. 2009, 131, 13037–13044. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Li, J.; Sheflin, L.G.; Pfeffer, B.A.; Behringer, M.; Fliesler, S.J.; Qu, J. Ion-current-based proteomic profiling of the retina in a rat model of Smith-Lemli-Opitz syndrome. Mol. Cell Proteomics 2013, 12, 3583–3598. [Google Scholar] [CrossRef] [PubMed]
- Serfis, A.B.; Brancato, S.; Fliesler, S.J. Comparative behavior of sterols in phosphatidylcholine-sterol monolayer films. Biochim. Biophys. Acta 2001, 1511, 341–348. [Google Scholar] [CrossRef]
- Lintker, K.B.; Kpere-Daibo, P.; Fliesler, S.J.; Serfis, A.B. A comparison of the packing behavior of egg phosphatidylcholine with cholesterol and biogenically related sterols in Langmuir monolayer films. Chem. Phys. Lipids 2009, 161, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.K.; Arnold, T.P.; Fliesler, S.J. Formation of 7-dehydrocholesterol-containing membrane rafts in vitro and in vivo, with relevance to the Smith-Lemli-Opitz syndrome. J. Lipid Res. 2004, 45, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Bittman, R.; Duportail, G.; Heissler, D.; Vilcheze, C.; London, E. Effect of the structure of natural sterols and sphingolipids on the formation of ordered sphingolipid/sterol domains (rafts): Comparison of cholesterol to plant, fungal, and disease-associated sterols and comparison of sphingomyelin, cerebrosides, and ceramide. J. Biol. Chem. 2001, 276, 33540–33546. [Google Scholar] [PubMed]
- Xu, L.; Korade, Z.; Porter, N.A. Oxysterols from free radical chain oxidation of 7-dehydrocholesterol: Product and mechanistic studies. J. Am. Chem. Soc. 2010, 132, 2222–2232. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Porter, N.A. Free radical oxidation of cholesterol and its precursors: Implications in cholesterol biosynthesis disorders. Free Radic. Res. 2015, 49, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Korade, Z.; Rosado, D.A., Jr.; Liu, W.; Lamberson, C.R.; Porter, N.A. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2011, 52, 1222–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Korade, Z.; Rosdado, D.A., Jr.; Mirnics, K.; Porter, N.A. Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. J. Lipid Res. 2013, 54, 1135–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Liu, W.; Sheflin, L.G.; Fliesler, S.J.; Porter, N.A. Novel oxysterols observed in tissues and fluids of AY9944-treated rats: a model for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2011, 52, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Shinkyo, R.; Xu, L.; Tallman, K.A.; Cheng, Q.; Porter, N.A.; Guengerich, F.P. Conversion of 7-dehydrocholesterol to 7-ketocholesterol is catalyzed by human cytochrome P450 7A1 and occurs by direct oxidation without an epoxide intermediate. J. Biol. Chem. 2011, 286, 33021–33028. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Mirnics, K.; Bowman, A.B.; Liu, W.; Da, J.; Porter, N.A.; Korade, Z. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol. Dis. 2012, 45, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Panini, S.R.; Sinensky, M.S. Mechanisms of oxysterol-induced apoptosis. Curr. Opin. Lipidol. 2001, 12, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Vejux, A.; Malvitte, L.; Lizard, G. Side effects of oxysterols: Cytotoxicity, oxidation, inflammation, and phospholipidosis. Braz. J. Med. Biol. Res. 2008, 41, 545–556. [Google Scholar] [CrossRef]
- Lordan, S.; Mackrill, J.J.; O'Brien, N.M. Oxysterols and mechanisms of apoptotic signaling: implications in the pathology of degenerative diseases. J. Nutr. Biochem. 2009, 20, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Cilla, A.; Alegría, A.; Attanzio, A.; Garcia-Llatas, G.; Tesoriere, L.; Livrea, M.A. Dietary phytochemicals in the protection against oxysterol-induced damage. Chem. Phys. Lipids. 2017, 207 (Pt B), 192–205. [Google Scholar] [CrossRef]
- Goyal, S.; Xiao, Y.; Porter, N.A.; Xu, L.; Guengerich, F.P. Oxidation of 7-dehydrocholesterol and desmosterol by human cytochrome P450 46A1. J. Lipid Res. 2014, 55, 1933–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Sheflin, L.G.; Porter, N.A.; Fliesler, S.J. 7-Dehydrocholesterol-derived oxysterols and retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Biochim. Biophys. Acta 2012, 1821, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Xu, L.; Shelton, R.; Porter, N.A. Biological activities of 7-dehydrocholesterol-derived oxysterols: Implications for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2010, 51, 3259–3269. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, B.A.; Xu, L.; Porter, N.A.; Rao, S.R.; Fliesler, S.J. Differential cytotoxic effects of 7-dehydrocholesterol-derived oxysterols on cultured retina-derived cells: Dependence on sterol structure, cell type, and density. Exp. Eye Res. 2016, 145, 297–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, M.J.; Nagel, B.A.; Fliesler, S.J. Lipid hydroperoxide formation in the retina: Correlation with retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Exp. Eye Res. 2006, 82, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.K.; Peachey, N.S.; Richards, M.J.; Buchan, B.; Fliesler, S.J. Light-induced exacerbation of retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Exp. Eye Res. 2006, 82, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J. Antioxidants: The missing key to improved therapeutic intervention in Smith-Lemli-Opitz syndrome? Hereditary Genet. 2013, 2, 119. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J. Retinal Degeneration and Cholesterol Deficiency. In Handbook of Nutrition, Diet and the Eye, 1st ed.; Preedy, V.R., Ed.; Elsevier: London, UK, 2014; pp. 287–297. ISBN 9780124017177. [Google Scholar]
- Fliesler, S.J.; Peachey, N.S.; Herron, J.; Hines, K.M.; Weinstock, N.I.; Ramachandra Rao, S.; Xu, L. Prevention of retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Sci Rep. 2018, 8, 1286. [Google Scholar] [CrossRef] [PubMed]
- Windsor, K.; Genaro-Mattos, T.C.; Kim, H.Y.; Liu, W.; Tallman, K.A.; Miyamoto, S.; Korade, Z.; Porter, N.A. Probing lipid-protein adduction with alkynyl surrogates: Application to Smith-Lemli-Opitz syndrome. J. Lipid Res. 2013, 54, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Kapphahn, R.J.; Richards, M.J.; Ferrington, D.A.; Fliesler, S.J. Lipid-derived and other oxidative modifications of retinal proteins in a rat model of Smith-Lemli-Opitz syndrome. Exp Eye Res. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Kretzer, F.L.; Hittner, H.M.; Mehta, R.S. Ocular manifestations of the Smith-Lemli-Opitz syndrome. Arch. Ophthalmol. 1981, 99, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Wassif, C.A.; Zhu, P.; Kratz, L.; Krakowiak, P.A.; Battaile, K.P.; Weight, F.F.; Grinberg, A.; Steiner, R.D.; Nwokoro, N.A.; Kelley, R.I.; et al. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith-Lemli-Opitz syndrome. Hum. Mol. Genet. 2001, 10, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzky, B.U.; Moebius, F.F.; Asaoka, H.; Waage-Baudet, H.; Xu, L.; Xu, G.; Maeda, N.; Kluckman, K.; Hiller, S.; Yu, H.; et al. 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J. Clin. Investig. 2001, 108, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Correa-Cerro, L.S.; Wassif, C.A.; Kratz, L.; Miller, G.F.; Munasinghe, J.P.; Grinberg, A.; Fliesler, S.J.; Porter, F.D. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum. Mol. Genet. 2006, 15, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]


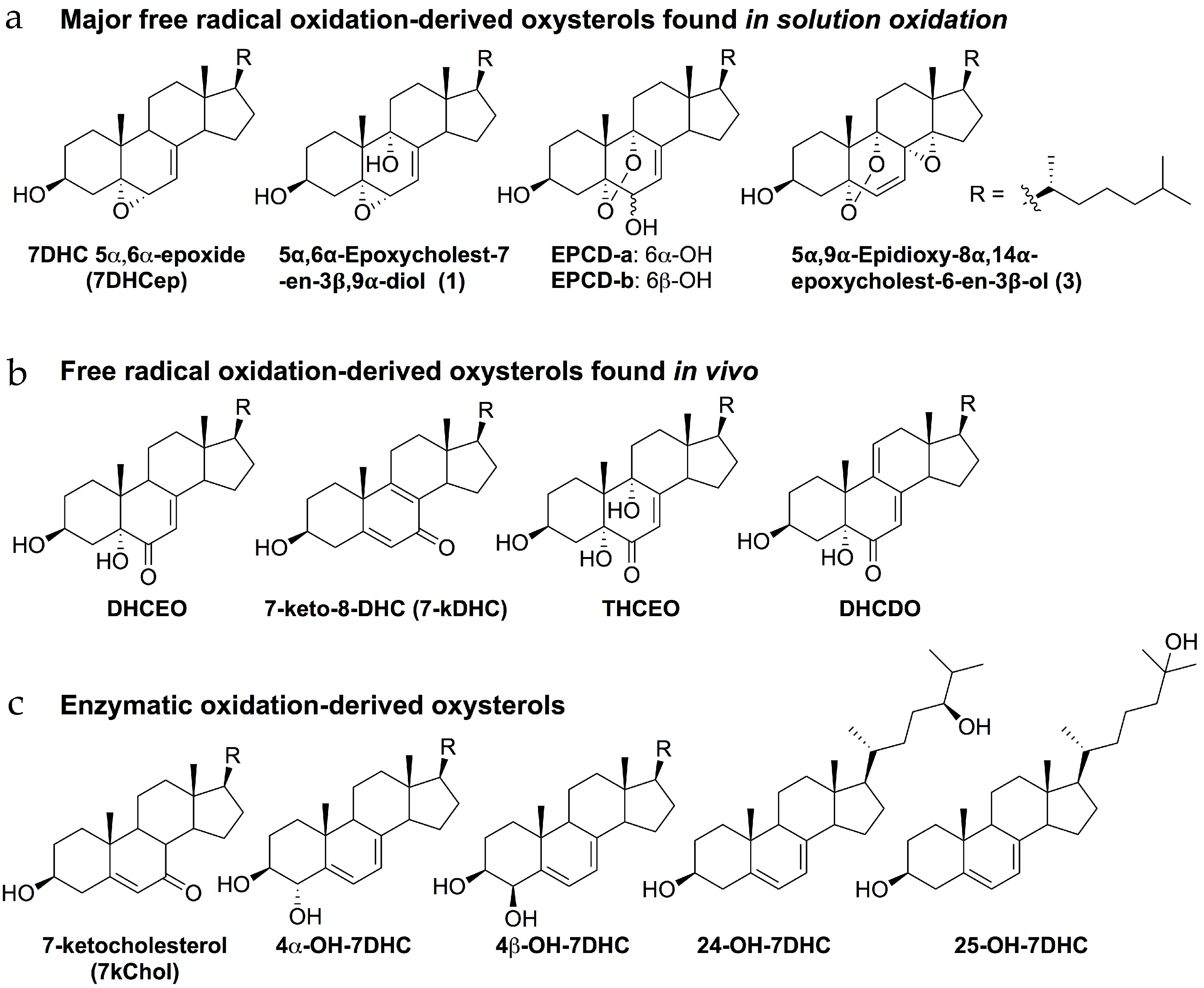

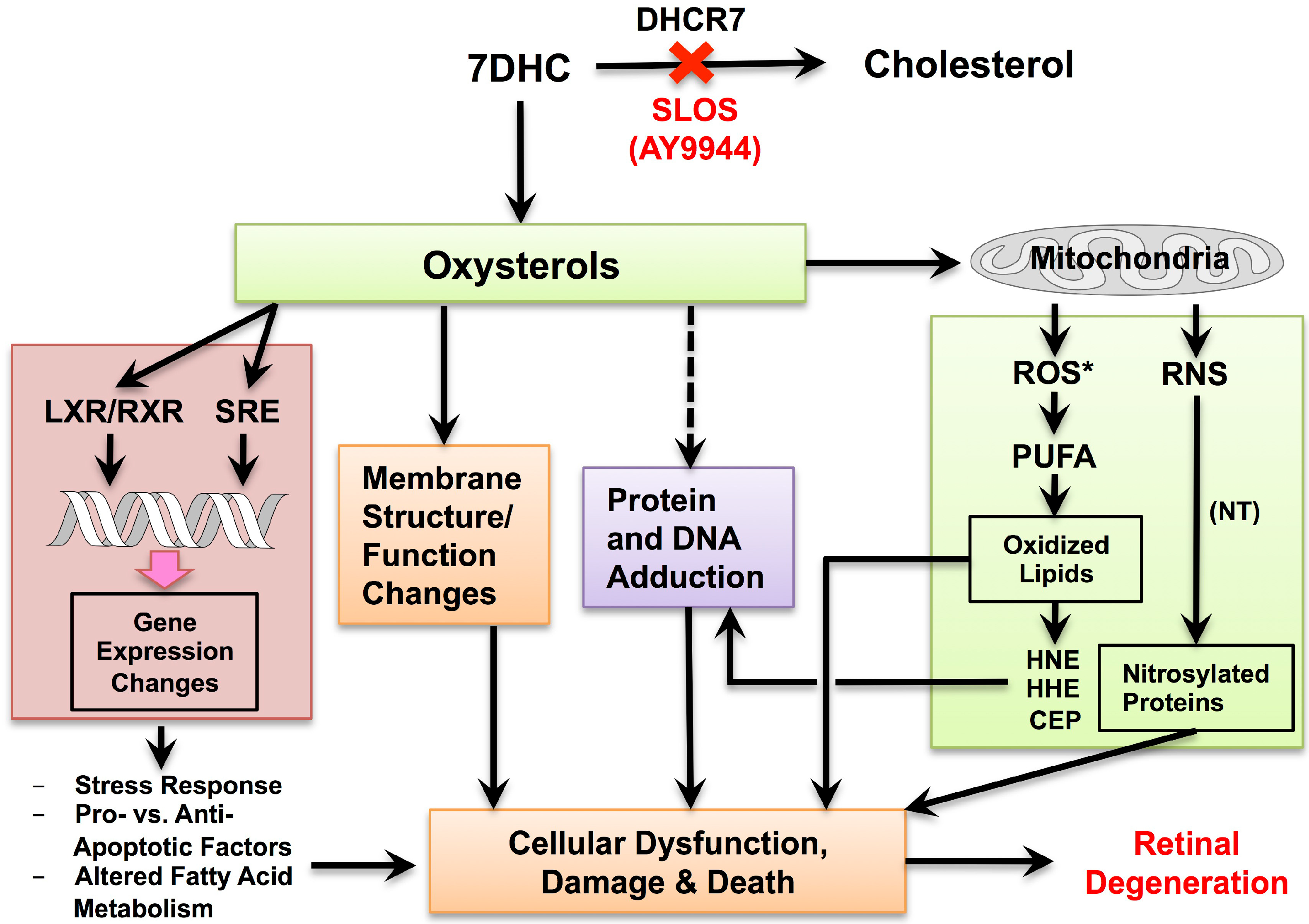
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fliesler, S.J.; Xu, L. Oxysterols and Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome: Implications for an Improved Therapeutic Intervention. Molecules 2018, 23, 2720. https://doi.org/10.3390/molecules23102720
Fliesler SJ, Xu L. Oxysterols and Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome: Implications for an Improved Therapeutic Intervention. Molecules. 2018; 23(10):2720. https://doi.org/10.3390/molecules23102720
Chicago/Turabian StyleFliesler, Steven J., and Libin Xu. 2018. "Oxysterols and Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome: Implications for an Improved Therapeutic Intervention" Molecules 23, no. 10: 2720. https://doi.org/10.3390/molecules23102720