3.1. Chemistry
3.1.1. General Information
All solvents and reagents were obtained from Sigma Aldrich Chemical Co. (Saint Louis, MO, USA), Iris Biotech GmbH (Marktredwitz, Germany), Alfa Aesar Co. (Karlsruhe, Germany), VWR (Radnor, PA, USA) and FluoroChem UK (Hadfield, UK) and used without further purification unless indicated otherwise. Silica gel chromatography was conducted with 230–400 mesh 60 Å silica gel (Sigma Aldrich Chemical Co.). The progress of reaction was monitored by TLC exposure to UV light (254 nm and 366 nm). Thin layer chromatography plates (Kieselgel 60 F254) were purchased from Merck (Darmstadt, Germany). Microwave assisted organic syntheses were performed on a Biotage Initiator 2.0 microwave system (Uppsala, Sweden).
1H (400 MHz) and
13C-NMR (100 MHz) spectra were obtained on a Brüker AC-400 spectrometer (Billerica, MA, USA). Chemical shifts are given as parts per million (ppm) using residual dimethylsulfoxide signal for protons (
δDMSO = 2.46 ppm) and carbons (
δDMSO = 40.00 ppm). Coupling constants are reported in Hertz (Hz). Spectral splitting partners are designed as follow: singlet (s); doublet (d); triplet (t); quartet (q); multiplet (m). Mass spectral data were obtained on a Waters Micromass Q-Tof (Milford, MA, USA) spectrometer equipped with ESI source (Laboratoires de Mesures Physiques, Plateau technique de l’Institut des Biomolecules Max Mousseron, Université de Montpellier, Montpellier, France). Mass spectra were recorded in positive mode between 50 and 1500 Da, capillary and cone tension were 3000 and 20 V, respectively. The High Resolution Mass Spectroscopy (HRMS) analyses are carried out by direct introduction on a Synapt G2-S mass spectrometer (Waters, SN: UEB205) equipped with ESI source. The mass spectra were recorded in positive mode, between 100 and 1500 Da. The capillary tension is 1000 V and the cone tension is 30 V. The source and desolvation temperature are 120 °C and 250 °C, respectively. NMR
1H and
13C spectra of all compounds are in
Supplementary Materials.
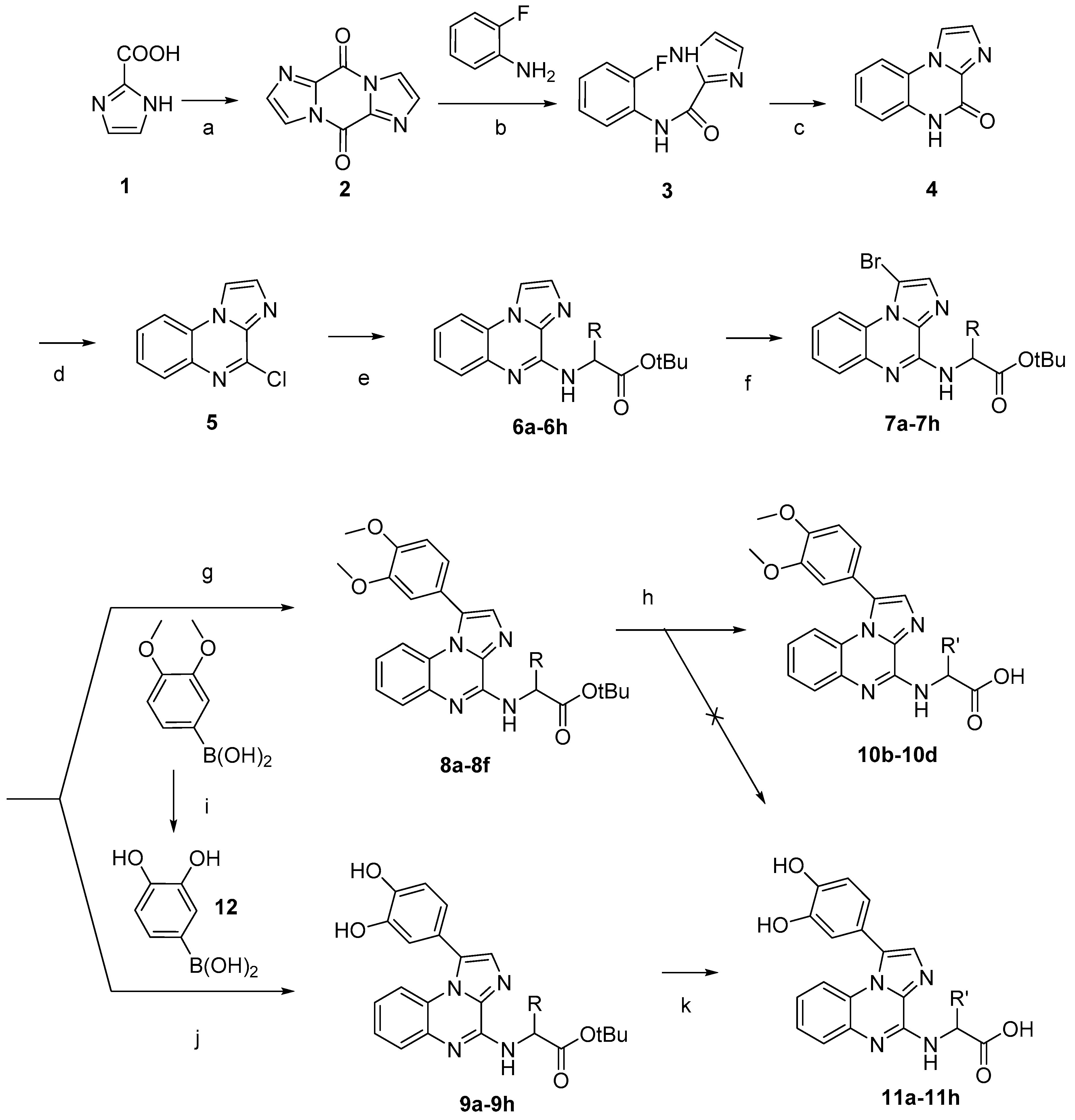
3.1.2. Amino Acids Grafted on 4-Chloroimidazo[1,2-a]quinoxaline
Tert-butyl 2-(imidazo[1,2-a]quinoxalin-4-ylamino)acetate (6a): Glycine tert-butyl ester hydrochloride (0.824 g, 4.9 mmol), N,N-diisopropylethylamine (1.6 mL, 9.8 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) were dissolved in dimethyl-formamide (10 mL) in a microwave adapted vial and sealed. The reaction mixture was irradiated at 150 °C for 30 min. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (100 mL) and successively washed with saturated aqueous ammonium chloride, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (42% yield). C16H18N4O2. MW: 298.34 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.42 (s, 9H, 3 × CH3 OtBu), 4.13 (d, 2H, J = 8 Hz, CH2 α), 7.31–7.35 (m, 1H, CH 7), 7.40–7.44 (m, 1H, CH 8), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.66 (d, 1H, J = 4 Hz, CH 2), 7.96 (t, 1H, J = 8 Hz, NH), 8.13 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.64 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 28.23 (CH3 tBu), 43.30 (CH2 α), 80.85 (Cq tBu), 115.14 (CH 1), 115.91 (CH 6), 123.57 (CH 7), 124.93 (Cq 5a), 126.74 (CH 9), 126.88 (CH 8), 132.52 (CH 2), 132.71 (Cq 3a), 136.81 (Cq 9a), 147.49 (Cq 4), 169.79 (C=O). MS (ESI +, QTof, m/z): 299.0 [M + H]+.
Tert-butyl 2-(imidazo[1,2-a]quinoxalin-4-ylamino)propanoate (6b): Same procedure used for the synthesis of 6a was employed. L-Alanine tert-butyl ester hydrochloride (1.339 g, 7.4 mmol), N,N-diisopropylethylamine (2.4 mL, 14.7 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) were dissolved and reacted in dimethyl-formamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (48% yield). C17H20N4O2. MW: 312.37 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.41 (s, 9H, 3 × CH3 OtBu), 1.51 (d, 3H, J = 8 Hz, CH3 β), 4.56–4.63 (m, 1H, CH α), 7.33–7.36 (m, 1H, CH 7), 7.41–7.43 (m, 1H, CH 8), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.66 (d, 1H, J = 4 Hz, CH 2), 7.75 (d, 1H, J = 4 Hz, NH), 8.13 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.64 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 17.46 (CH3 β), 28.14 (CH3 tBu), 50.22 (CH α), 80.59 (Cq tBu), 115.18 (CH 1), 115.91 (CH 6), 123.64 (CH 7), 124.93 (Cq 5a), 126.73 (CH 9), 126.88 (CH 8), 132.42 (CH 2), 132.58 (Cq 3a), 136.72 (Cq 9a), 147.03 (Cq 4), 172.84 (C=O). MS (ESI +, QTof, m/z): 313.2 [M + H]+.
Tert-Butyl 2-(imidazo[1,2-a]quinoxalin-10-ylamino)-3-methylbutanoate (6c): Using the same procedure as for the synthesis of 6a, L-valine tert-butyl ester hydrochloride (2.060 g, 9.8 mmol), N,N-diisopropylethylamine (2.4 mL, 14.7 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) were mixed in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (51% yield). C19H24N4O2. MW: 340.42 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.03 (d, 3H, J = 8 Hz, CH3 γ), 1.06 (d, 3H, J = 8 Hz, CH3 γ’), 1.43 (s, 9H, 3 × CH3 OtBu), 2.29–2.37 (m, 1H, CH β), 4.52 (t, 1H, J = 16 Hz, CH α), 7.15 (d, 1H, J = 8 Hz, NH), 7.33–7.37 (m, 1H, CH 7), 7.42–7.44 (m, 1H, CH 8), 7.59 (dd, 1H J = 4 Hz, J = 8 Hz, CH 9), 7.68 (d, 1H, J = 4 Hz, CH 2), 8.14 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.66 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 19.42 (CH3 γ, CH3 γ’), 28.17 (CH3 tBu), 30.41 (CH β), 59.76 (CH α), 81.15 (Cq tBu), 115.37 (CH 1), 115.95 (CH 6), 123.88 (CH 7), 125.00 (Cq 5a), 126.84 (CH 9), 126.95 (CH 8), 132.48 (CH 2), 132.58 (Cq 3a), 136.59 (Cq 9a), 147.29 (Cq 4), 171.59 (C=O). MS (ESI +, QTof, m/z): 341.0 [M + H]+.
Tert-butyl 2-(imidazo[1,2-a]quinoxalin-4-ylamino)-4-methylpentanoate (6d): The same as for the synthesis of 6a was used, employing L-leucine tert-butyl ester hydrochloride (2.198 g, 9.8 mmol), N,N-diisopropylethylamine (2.4 mL, 14.7 mmol) and 4-chloro-imidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (60% yield). C20H26N4O2. MW: 354.45 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.93 (d, 3H, J = 8 Hz, CH3 δ), 0.96 (d, 3H, J = 8 Hz, CH3 δ’), 1.40 (s, 9H, 3 × CH3 OtBu), 1.60–1.67 (m, 1H, CH2 β), 1.77–1.79 (m, 1H, CH γ), 1.93–1.95 (m, 1H, CH2 β), 4.63 (t, 1H, J = 4 Hz, CH α), 7.26–7.33 (m, 1H, CH 7), 7.40–7.44 (m, 1H, CH 8), 7.57 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.64 (d, 1H, J = 4 Hz, NH), 7.66 (d, 1H, J = 4 Hz, CH 2), 8.13 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.64 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 22.06 (CH3 δ), 23.31 (CH3 δ’), 25.14 (CH γ), 28.15 (CH3 tBu), 40.01 (CH2 β), 52.93 (CH α), 80.67 (Cq tBu), 115.21 (CH 1), 115.90 (CH 6), 123.64 (CH 7), 124.93 (Cq 5a), 126.76 (CH 8), 126.89 (CH 9), 132.41 (CH 2), 132.58 (Cq 3a), 136.74 (Cq 9a), 147.40 (Cq 4), 172.75 (C=O). MS (ESI +, QTof, m/z): 355.0 [M + H]+.
Tert-butyl 6-((tert-butoxycarbonyl)amino)-2-(imidazo[1,2-a]quinoxalin-4-ylamino)hexanoate (6e): The same procedure used for the synthesis of 6a was employed with N-ε-tert-butyloxycarbonyl-L-lysine tert-butyl ester hydrochloride (2.565 g, 7.6 mmol), N,N-diisopropyl-ethylamine (2.4 mL, 14.7 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (32% yield). C25H35N5O4. MW: 469.58 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.35 (s, 9H, 3 × CH3 OtBu), 1.41 (s, 9H, 3 × CH3 OtBu), 1.43–1.49 (m, 4H, CH2 γ, CH2 δ), 1.86–1.92 (m, 2H, CH2 β), 2.91–2.93 (m, 2H, CH2 ε), 4.52–4.56 (m, 1H, CH α), 6.79 (t, 1H, J = 4 Hz, NH-CH2 ε), 7.26–7.35 (m, 1H, CH 7), 7.39–7.42 (m, 1H, CH 8), 7.52 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.64 (d, 1H, J = 4 Hz, NH-CH α), 7.66 (d, 1H, J = 4 Hz, CH 2), 8.12 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.64 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 23.52 (CH2 γ), 28.19 (CH3 tBu), 28.74 (CH3 tBu), 29.67 (CH2 δ), 30.92 (CH2 β), 40.35 (CH2 ε), 54.64 (CH α), 79.68 (Cq tBu), 80.80 (Cq tBu), 115.25 (CH 1), 115.93 (CH 6), 123.70 (CH 7), 124.98 (Cq 5a), 126.81 (CH 9), 126.92 (CH 8), 132.44 (CH 2), 132.60 (Cq 3a), 136.75 (Cq 9a), 147.35 (Cq 4), 172.41 (C=O). MS (ESI +, QTof, m/z): 470.0 [M + H]+.
Tert-butyl 5-(((benzyloxy)carbonyl)amino)-2-(imidazo[1,2-a]quinoxalin-4-ylamino)-pentanoate (6f): Using the same procedure used for the synthesis of 6a with N-δ-carbobenzoxy-L-ornithine α-tert-butyl ester hydrochloride (3.702 g, 10.3 mmol), N,N-diisopropylethylamine (2.4 mL, 14.7 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (38% yield). C27H31N5O4. MW: 489.57 g/mol. 1H-NMR δ (ppm, 400 MHz, DMSO-d6): 1H-NMR δ (ppm, DMSO-d6): 1.29 (s, 9H, 3 × CH3 OtBu), 1.46–1.50 (m, 2H, CH2 γ), 1.79–1.84 (m, 2H, CH2 β), 2.94–2.98 (m, 2H, CH2 δ), 4.44–4.46 (m, 1H, CH α), 4.90 (s, 2H, CH2–Phenyl), 7.17–7.21 (m, 1H, CH 7), 7.23 (s, 1H, NH-CH2 δ), 7.24–7.27 (m, 5H, CH Phenyl), 7.29–7.33 (m, 1H, CH 8), 7.46 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.52 (d, 1H, J = 4 Hz, NH-CH α), 7.56 (d, 1H, J = 4 Hz, CH 2), 8.02 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.53 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 26.63 (CH2 γ), 28.13 (CH3 tBu), 28.66 (CH2 β), 40.71 (CH2 δ), 54.50 (CH α), 65.67 (CH2-Phenyl), 80.91 (Cq tBu), 115.30 (CH 1), 115.99 (CH 6), 123.77 (CH 7), 125.03 (Cq 5a), 126.84 (CH 9), 126.97 (CH 8), 128.86 (CH Phenyl), 132.50 (CH 2), 132.65 (Cq 3a), 136.77 (Cq 9a), 147.36 (Cq 4), 156.65 (Cq Phenyl), 172.28 (C=O). MS (ESI +, QTof, m/z): 490.0 [M + H]+.
Tert-Butyl 2-(imidazo[1,2-a]quinoxalin-4-ylamino)-3-phenylpropanoate (6g):The same procedure as for the synthesis of 6a was used with L-phenylalanine tert-butyl ester hydrochloride (1.9 g, 7.4 mmol), N,N-diisopropylethylamine (2.4 mL, 14.7 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (37% yield). C23H24N4O2. MW: 388.46 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.33 (s, 9H, 3 × CH3 OtBu), 3.19–3.23 (m, 1H, CH2 β), 3.32–3.36 (m, 1H, CH2 β), 4.82–4.84 (m, 1H, CH α), 7.21 (t, 1H, J = 8 Hz, CH Phenyl), 7.29 (t, 2H, J = 8 Hz, 2 × CH Phenyl), 7.32–7.34 (m, 3H, CH 7, 2 × CH Phenyl), 7.41–7.44 (m, 1H, CH 8), 7.57 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.61 (d, 1H, J = 8 Hz, NH), 7.66 (d, 1H, J = 4 Hz, CH 2), 8.13 (dd, 1H, J = 8, 4 Hz, CH 6), 8.64 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 26.97 (CH3 tBu), 35.91 (CH2 β), 54.93 (CH α), 79.88 (Cq tBu), 114.18 (CH 1), 114.85 (CH 6), 122.74 (CH 7), 123.89 (Cq 5a), 125.74 (CH 9), 125.84 (CH Phenyl), 125.86 (CH 8), 127.59 (CH Phenyl), 128.69 (CH Phenyl), 131.41 (CH 2), 135.54 (Cq 3a), 137.05 (Cq 9a), 146.02 (Cq 4), 170.53 (C=O). MS (ESI +, QTof, m/z): 389.0 [M + H]+.
Tert-butyl 3-(4-(tert-butoxy)phenyl)-2-(imidazo[1,2-a]quinoxalin-4-ylamino)propanoate (6h): The same procedure as for the synthesis of 6a was used with O-tert-butyl-L-tyrosine tert-butyl ester hydrochloride (1.6 g, 4.9 mmol), N,N-diisopropylethylamine (2.4 mL, 14.7 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (26% yield). C27H32N4O3. MW: 460.57 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.23 (s, 9H, 3 × CH3 OtBu), 1.31 (s, 9H, 3 × CH3 OtBu), 3.11–3.17 (m, 1H, CH2 β), 3.25–3.31 (m, 1H, CH2 β), 4.78–4.83 (m, 1H, CH α), 6.86 (dd, 2H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.22 (dd, 2H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.32–7.36 (m, 1H, CH 7), 7.40–7.44 (m, 1H, CH 8), 7.57 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.61 (d, 1H, J = 4 Hz, NH), 7.66 (d, 1H, J = 4 Hz, CH 2), 8.13 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.64 (d, 1H, J = 4 Hz, CH 1). 13C-NMR δ (ppm, DMSO-d6): 28.02 (CH3 tBu), 28.93 (CH3 tBu), 36.63 (CH2 β), 56.09 (CH α), 78.11 (Cq tBu), 80.87 (Cq tBu), 115.22 (CH 1), 115.90 (CH 6), 123.80 (CH 7), 123.98 (2 × CH Phenyl), 124.96 (Cq 5a), 126.79 (CH 9), 126.90 (CH 8), 130.31 (2 × CH Phenyl), 132.47 (CH 2), 132.61 (Cq 3a), 136.60 (Cq 9a), 147.04 (Cq 4), 154.03 (2 × Cq Phenyl), 171.72 (C=O). MS (ESI +, QTof, m/z): 461.2 [M + H]+.
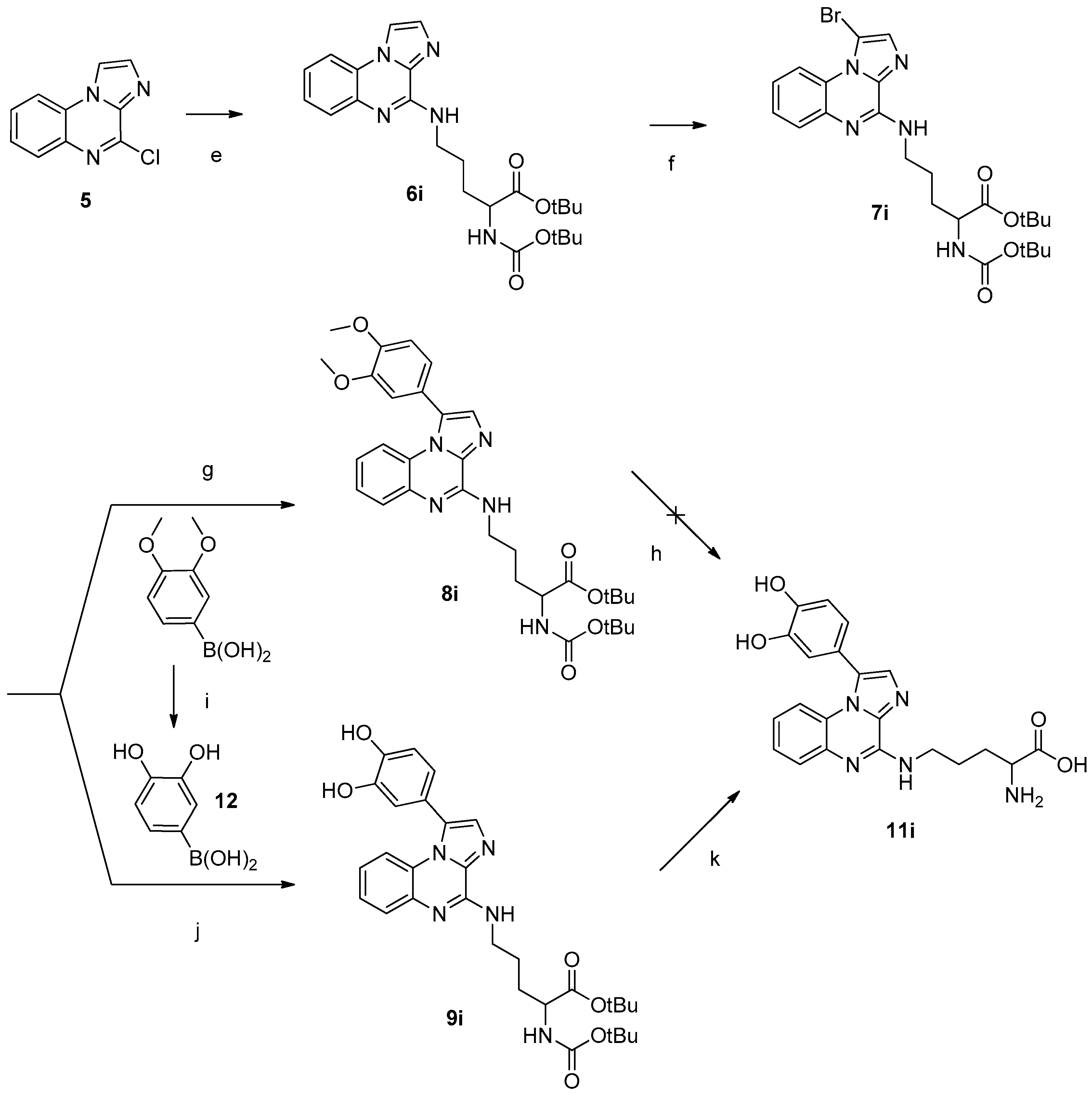
Tert-butyl 6-((tert-butoxycarbonyl)amino)-2-(imidazo[1,2-a]quinoxalin-4-ylamino)hexanoate (6i): Same procedure used for the synthesis of 6a was employed with N-α-tert-butyloxycarbonyl-L-ornithine tert-butyl ester hydrochloride (1.596 g, 4.9 mmol), N,N-diisopropyl-ethylamine (1.6 mL, 9.8 mmol) and 4-chloroimidazo[1,2-a]quinoxaline 5 (0.5 g, 2.5 mmol) in dimethylformamide (10 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a beige solid (48% yield). C24H33N5O4. MW: 455.56 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.35 (s, 9H, CH-COOtBu), 1.41 (s, 9H, NH-COOtBu), 1.85–1.96 (m, 2H, CH2 β), 2.91–2.95 (m, 2H, CH2 γ), 3.35–3.39 (m, 2H, CH2 δ), 4.51–4.56 (m, 1H, CH α), 6.79 (d, 1H, J = 4 Hz, NH-CH α), 7.32–7.35 (m, 1H, CH 7), 7.39–7.42 (m, 1H, CH 8), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.60 (d, 1H, J = 4 Hz, NH-CH2 δ), 7.66 (s, 1H, CH 2), 8.13 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 8.64 (d, 1H, CH 1). 13C-NMR δ (ppm, DMSO-d6): 25.87 (CH2 β), 28.04 (CH3 tBu), 28.67 (CH3 tBu), 28.43 (CH2 γ), 39.79 (CH2 δ), 54.75 (CH α), 78.45 (Cq tBu), 80.53 (Cq tBu), 115.02 (CH 1), 115.79 (CH 6), 122.99 (CH 7), 124.65 (Cq 5a), 126.50 (CH 9), 126.73 (CH 8), 132.20 (CH 2), 132.87 (Cq 3a), 147.78 (Cq 4), 156.00 (Cq 9a), 172.33 (C=O). MS (ESI +, QTof, m/z): 456.0.1 [M + H]+.
3.1.3. Bromination
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)acetate (7a): A solution of 6a (0.26 g, 0.87 mmol) and N-bromosuccinimide (0.19 g, 1.0 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine (50 mL). The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (97% yield) and used without purification. C16H17BrN4O2. MW: 377.24 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.42 (s, 9H, 3 × CH3 OtBu), 4.12 (d, 2H, J = 8 Hz, CH2 α), 7.34–7.37 (m, 1H, CH 7), 7.45–7.50 (m, 1H, CH 8), 7.59 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.76 (s, 1H, CH 2), 8.02 (t, 1H, J = 8 Hz, NH), 8.96 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 28.22 (CH3 tBu), 43.26 (CH2 α), 80.93 (Cq tBu), 99.61 (Cq 1), 115.12 (CH 6), 123.08 (CH 7), 126.08 (Cq 5a), 127.35 (CH 9), 127.78 (CH 8), 134.01 (Cq 3a), 134.79 (CH 2), 137.63 (Cq 9a), 147.05 (Cq 4), 169.64 (C=O). MS (ESI +, QTof, m/z): 377.0 [M + H]+.
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)propanoate (7b): A solution of 6b (0.3 g, 0.96 mmol) and N-bromosuccinimide (0.2 g, 1.1 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (98% yield) and used without purification. C17H19BrN4O2. MW: 391.26 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.41 (s, 9H, 3 × CH3 OtBu), 1.51 (d, 3H, J = 8 Hz, CH3 β), 4.55–4.59 (m, 1H, CH α), 7.34–7.38 (m, 1H, CH 7), 7.46–7.48 (m, 1H, CH 8), 7.60 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.76 (s, 1H, CH 2), 7.82 (d, 1H, J = 4 Hz, NH), 8.97 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 17.40 (CH3 β), 28.03 (CH3 tBu), 50.24 (CH α), 80.70 (Cq tBu), 99.68 (CH 1), 115.06 (CH 6), 123.17 (CH 7), 126.06 (Cq 5a), 127.30 (CH 9), 127.45 (CH 8), 133.87 (Cq 3a), 134.73 (CH 2), 137.47 (Cq 9a), 146.58 (Cq 4), 172.66 (C=O). MS (ESI +, QTof, m/z): 391.1 [M + H]+.
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)-3-methylbutanoate (7c): A solution of 6c (0.38 g, 1.1 mmol) and N-bromosuccinimide (0.24 g, 1.4 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (96% yield) and used without purification. C19H23BrN4O2. MW: 419.31 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.02 (d, 3H, J = 8 Hz, CH3 γ), 1.05 (d, 3H, J = 8 Hz, CH3 γ’), 1.43 (s, 9H, 3 × CH3 OtBu), 2.31–2.36 (m, 1H, CH β), 4.49 (t, 1H, J = 16 Hz, CH α), 7.20 (d, 1H, J = 8 Hz, NH), 7.35–7.40 (m, 1H, CH 7), 7.47–7.49 (m, 1H, CH 8), 7.62 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.76 (s, 1H, CH 2), 8.97 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 19.42 (CH3 γ, CH3 γ’), 28.17 (CH3 tBu), 30.40 (CH β), 59.76 (CH α), 81.25 (Cq tBu), 99.86 (Cq 1), 115.08 (CH 6), 123.38 (CH 7), 126.16 (Cq 5a), 127.44 (CH 9), 127.47 (CH 8), 133.81 (Cq 3a), 134.74 (CH 2), 137.40 (Cq 9a), 146.84 (Cq 4), 171.44 (C=O). MS (ESI +, QTof, m/z): 419.1 [M + H]+.
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)-4-methylpentanoate (7d): A solution of 6d (0.35 g, 1.0 mmol) and N-bromosuccinimide (0.2 g, 1.2 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (95% yield) and used without purification. C20H25BrN4O2. MW: 433.34 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.91 (d, 3H, J = 8 Hz, CH3 δ), 0.95 (d, 3H, J = 8 Hz, CH3 δ’), 1.40 (s, 9H, 3 × CH3 OtBu), 1.60–1.62 (m, 1H, CH2 β), 1.77–1.79 (m, 1H, CH γ), 1.94–1.96 (m, 1H, CH2 β), 4.60 (t, 1H, J = 4 Hz, CH α), 7.35–7.38 (m, 1H, CH 7), 7.46–7.50 (m, 1H, CH 8), 7.60 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.71 (d, 1H, J = 4 Hz, NH), 7.76 (s, 1H, CH 2), 8.97 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 22.03 (CH3 δ), 23.31 (CH3 δ’), 25.12 (CH γ), 28.14 (CH3 tBu), 40.10 (CH2 β), 52.95 (CH α), 80.76 (Cq tBu), 99.21 (Cq 1), 115.07 (CH 6), 123.17 (CH 7), 126.08 (Cq 5a), 127.37 (CH 9), 127.46 (CH 8), 133.88 (Cq 3a), 134.71 (CH 2), 137.55 (Cq 9a), 146.97 (Cq 4), 172.59 (C=O). MS (ESI +, QTof, m/z): 433.1 [M + H]+.
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)-6-((Tert-butoxycarbonyl)amino)-hexanoate (7e): A solution of 6e (1.2 g, 2.5 mmol) and N-bromosuccinimide (0.54 g, 3.1 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (78% yield) and used without purification. C25H34BrN5O4. MW: 548.47 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.22–1.24 (m, 2H, CH2 γ), 1.34 (s, 9H, 3 × CH3 OtBu), 1.40 (s, 9H, 3 × CH3 OtBu), 1.54–1.56 (m, 2H, CH2 β), 1.89–1.91 (m, 2H, CH2 δ), 2.83–2.89 (m, 2H, CH2 ε), 4.13–4.19 (m, 1H, CH α), 6.78 (t, 1H, J = 4 Hz, NH-CH2 ε), 7.34–7.39 (m, 1H, CH 7), 7.46–7.48 (m, 1H, CH 8), 7.65 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 8.03 (s, 1H, CH 2), 8.34 (d, 1H, J = 4 Hz, NH-CH α), 8.97 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 22.90 (CH2 γ), 28.15 (CH3 tBu), 28.73 (CH3 tBu), 29.46 (CH2 δ), 31.34 (CH2 β), 39.36 (CH2 ε), 54.61 (CH α), 80.85 (Cq tBu), 81.14 (Cq tBu), 99.70 (Cq 1), 115.06 (CH 6), 123.19 (CH 7), 126.09 (Cq 5a), 127.39 (CH 9), 127.46 (CH 8), 133.87 (CH 2), 134.70 (Cq 3a), 137.54 (Cq 9a), 146.88 (Cq 4), 171.39 (C=O), 172.21 (C=O). MS (ESI +, QTof, m/z): 548.1 [M + H]+.
Tert-butyl 5-(((benzyloxy)carbonyl)amino)-2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)-amino)pentanoate (7f): A solution of 6f (0.45 g, 0.9 mmol) and N-bromosuccinimide (0.20 g, 1.1 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (94% yield) and used without purification. C27H30BrN5O4. MW: 568.46 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.40 (s, 9H, 3 × CH3 OtBu), 1.51–1.59 (m, 2H, CH2 γ), 1.89–1.92 (m, 2H, CH2 β), 3.03–3.08 (m, 2H, CH2 δ), 4.52–4.54 (m, 1H, CH α), 5.00 (s, 2H, CH2-Phenyl), 7.26 (s, 1H, NH-CH2 δ), 7.31–7.35 (m, 5H, CH Phenyl), 7.36–7.39 (m, 1H, CH 7), 7.46–7.50 (m, 1H, CH 8), 7.59 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.67 (d, 1H, J = 4 Hz, NH-CH α), 7.76 (s, 1H, CH 2), 8.99 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 26.53 (CH2 γ), 28.13 (CH3 tBu), 28.50 (CH2 β), 40.42 (CH2 δ), 54.43 (CH α), 65.59 (CH2-Phenyl), 80.92 (Cq tBu), 99.69 (Cq 1), 115.07 (CH 6), 123.20 (CH 7), 126.10 (Cq 5a), 127.39 (CH 9), 127.45 (CH 8), 128.18 (CH Phenyl), 128.78 (CH Phenyl), 133.87 (Cq 3a), 134.70 (CH 2), 137.72 (Cq 9a), 146.85 (Cq 4), 156.56 (Cq Phenyl), 172.05 (C=O). MS (ESI +, QTof, m/z): 568.3 [M + H]+.
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)-3-phenylpropanoate (7g): A solution of 6g (0.34 g, 0.9 mmol) and N-bromosuccinimide (0.18 g, 1.1 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (83% yield) and used without purification. C23H23BrN4O2. MW: 467.36 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.33 (s, 9H, 3 × CH3 OtBu), 3.18–3.22 (m, 1H, CH2 β), 3.31–3.34 (m, 1H, CH2 β), 4.79–4.81 (m, 1H, CH α), 7.21 (t, 1H, J = 8 Hz, CH Phenyl), 7.27–7.30 (m, 2H, 2 × CH Phenyl), 7.32–7.34 (m, 2H, 2xCH Phenyl), 7.35–7.38 (m, 1H, CH 7), 7.46–7.49 (m, 1H, CH 8), 7.60 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.65 (d, 1H, J = 8 Hz, NH), 7.75 (s, 1H, CH 2), 8.97 (dd, 1H, J = 2 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 28.04 (CH3 tBu), 36.91 (CH2 β), 56.01 (CH α), 81.06 (Cq tBu), 99.76 (Cq 1), 115.07 (CH 6), 123.33 (CH 7), 126.13 (Cq 5a), 126.96 (CH Phenyl), 127.45 (CH 9), 127.47 (CH 8), 128.68 (CH Phenyl), 129.77 (CH Phenyl), 133.80 (Cq Phenyl), 134.76 (CH 2), 137.45 (Cq 3a), 138.05 (Cq 9a), 146.66 (Cq 4), 171.45 (C=O). MS (ESI +, QTof, m/z): 467.1 [M + H]+.
Tert-butyl 2-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)-3-(4-(Tert-butoxy)phenyl)-propanoate (7h): A solution of 6h (0.34 g, 0.9 mmol) and N-bromosuccinimide (0.18 g, 1.1 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (91% yield) and used without purification. C23H23BrN4O2. MW: 467.36 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.22 (s, 9H, 3 × CH3 OtBu), 1.31 (s, 9H, 3 × CH3 OtBu), 3.11–3.16 (m, 1H, CH2 β), 3.24–3.26 (m, 1H, CH2 β), 4.76–4.81 (m, 1H, CH α), 6.85 (dd, 2H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.20 (dd, 2H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.34–7.38 (m, 1H, CH 7), 7.46–7.50 (m, 1H, CH 8), 7.59 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.65 (d, 1H, J = 8 Hz, NH), 7.75 (d, 1H, J = 4 Hz, CH 2), 8.98 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 28.04 (CH3 tBu), 28.93 (CH3 tBu), 36.61 (CH2 β), 56.03 (CH α), 78.10 (Cq tBu), 81.01 (Cq tBu), 99.71 (Cq 1), 115.08 (CH 6), 123.32 (CH 7), 123.96 (2 × CH Phenyl), 126.13 (Cq 5a), 127.42 (CH 9), 127.47 (CH 8), 130.33 (2 × CH Phenyl), 132.54 (Cq 3a), 134.78 (CH 2), 137.43 (Cq 9a), 146.61 (Cq 4), 154.06 (2 × Cq Phenyl), 171.53 (C=O). MS (ESI +, QTof, m/z): 467.1 [M + H]+.
Tert-butyl 5-((1-bromoimidazo[1,2-a]quinoxalin-4-yl)amino)-2-((Tert-butoxycarbonyl)amino)-pentanoate (7i): A solution of 6i (0.49 g, 1.1 mmol) and N-bromosuccinimide (0.23 g, 1.3 mmol) in CHCl3 (50 mL) was heated under reflux for 2 h. The solvent was removed under reduced pressure. The crude mixture was dissolved in ethyl acetate (50 mL) and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The compound was obtained as a beige oil (92% yield) and used without purification. C24H32BrN5O4. MW: 534.45 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.44 (s, 9H, CH-COOtBu), 1.46 (s, 9H, NH-COOtBu), 1.79–1.83 (m, 2H, CH2 β), 1.91–1.94 (m, 2H, CH2 γ), 3.73–3.78 (m, 2H, CH2 δ), 4.25–4.29 (m, 1H, CH α), 5.39 (d, 1H, J = 4 Hz, NH-CH α), 6.33 (d, 1H, J = 4 Hz, NH-CH2 δ), 7.27–7.31 (m, 1H, CH 7), 7.41–7.45 (m, 1H, CH 8), 7.48 (s, 1H, CH 2), 7.81 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 8.98 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6). 13C-NMR δ (ppm, DMSO-d6): 25.74 (CH2 β), 28.07 (CH3 tBu), 28.45 (CH3 tBu), 30.17 (CH2 γ), 40.42 (CH2 δ), 54.00 (CH α), 79.75 (Cq tBu), 81.98 (Cq tBu), 99.05 (Cq 1), 115.02 (CH 6), 122.77 (CH 7), 124.54 (Cq 5a), 127.12 (CH 8), 127.98 (CH 9), 134.22 (CH 2), 135.98 (Cq 3a), 146.86 (Cq 4), 155.59 (Cq 9a), 171.92 (C=O). MS (ESI +, QTof, m/z): 534.1 [M + H]+.
3.1.4. Suzuki-Miyaura Cross-Coupling Reactions
Tert-butyl 2-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)acetate (8a): To a mixture of 7a (0.270 g, 0.71 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.143 g, 0.79 mmol), tetrakis(triphenylphosphine) palladium (0.042 g, 0.036 mmol) and sodium carbonate (0.152 g, 1.43 mmol) in a microwave-adapted vial. The reaction was submitted to microwave irradiations during 20 min at 150 °C and then filtered on a Celite pad. The filtrate was concentrated under reduced pressure and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (19% yield). C24H26N4O4. MW: 434.49 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.44 (s, 9H, 3 × CH3 OtBu), 3.74 (s, 3H, OCH3 Phenyl), 3.87 (s, 3H, OCH3 Phenyl), 4.14 (d, 2H, J = 8 Hz, CH2 α), 7.01–7.05 (m, 1H, CH 7), 7.11 (d, 1H, J = 4 Hz, CH Phenyl), 7.14 (s, 1H, J = 4 Hz, CH Phenyl), 7.18 (d, 1H, J = 4 Hz, CH Phenyl), 7.31 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.33–7.35 (m, 1H, CH 8), 7.54 (s, 1H, CH 2), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.96 (t, 1H, J = 8 Hz, NH). 13C-NMR δ (ppm, DMSO-d6): 28.26 (CH3 tBu), 43.33 (CH2 α), 80.86 (Cq tBu), 112.31 (CH Phenyl), 114.25 (CH Phenyl), 115.93 (CH 6), 122.75 (CH 7), 123.30 (CH Phenyl), 126.03 (Cq 5a), 126.58 (CH 8), 127.20 (CH 9), 132.66 (CH 2), 133.141 (Cq 3a), 130.93 (Cq 1), 137.71 (Cq 9a), 149.27 (Cq Phenyl), 150.14 (Cq Phenyl), 147.74 (Cq 4), 169.85 (C=O). MS (ESI +, QTof, m/z): 435.1 [M + H]+.
Tert-butyl 2-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)propanoate (8b): Following the same procedure used for the synthesis of 8a, to a mixture of 7b (0.440 g, 1.12 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.225 g, 1.24 mmol), tetrakis(triphenylphosphine) palladium (0.065 g, 0.056 mmol) and sodium carbonate (0.237 g, 2.24 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (42% yield). C25H28N4O4. MW: 448.51 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.32 (s, 9H, 3 × CH3 OtBu), 1.42 (d, 3H, J = 8 Hz, CH3 β), 3.64 (s, 3H, OCH3 Phenyl), 3.76 (s, 3H, OCH3 Phenyl), 4.48–4.52 (m, 1H, CH α), 6.90–6.95 (m, 1H, CH 7), 7.01 (d, 1H, J = 4 Hz, CH Phenyl), 7.03 (s, 1H, CH Phenyl), 7.08 (d, 1H, J = 4 Hz, CH Phenyl), 7.20 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.22–7.25 (m, 1H, CH 8), 7.42 (s, 1H, CH 2), 7.46 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.82 (d, 1H, J = 4 Hz, NH). 13C-NMR δ (ppm, DMSO-d6): 17.59 (CH3 β), 28.25 (CH3 tBu), 50.30 (CH α), 80.72 (Cq tBu), 112.40 (CH Phenyl), 114.33 (CH Phenyl), 116.00 (CH 6), 122.77 (CH 7), 122.90 (Cq 1), 123.37 (CH Phenyl), 126.10 (CH 8), 126.65 (Cq 5a), 127.28 (CH 9), 131.05 (Cq 3a), 132.63 (CH 2), 137.68 (Cq 9a), 147.33 (Cq 4), 149.36 (Cq Phenyl), 150.22 (Cq Phenyl), 172.96 (C=O). MS (ESI +, QTof, m/z): 449.2 [M + H]+. HRMS calculated for C25H29N4O4 449.2189, found 449.2186.
Tert-butyl 2-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-methylbutanoate (8c): Followigng the same procedure used for the synthesis of 8a, to a mixture of 7c (0.370 g, 0.88 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.176 g, 0.97 mmol), tetrakis(triphenylphosphine) palladium (0.051 g, 0.044 mmol) and sodium carbonate (0.186 g, 1.76 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (69% yield). C27H32N4O4. MW: 476.57 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.95 (d, 3H, J = 8 Hz, CH3 γ), 0.98 (d, 3H, J = 8 Hz, CH3 γ’), 1.34 (s, 9H, 3 × CH3 OtBu), 2.20–2.28 (m, 1H, CH β), 3.63 (s, 3H, OCH3 Phenyl), 3.77 (s, 3H, OCH3 Phenyl), 4.44 (t, 1H, J = 16 Hz, CH α), 6.92–6.97 (m, 1H, CH 7), 7.00 (s, 1H, CH Phenyl), 7.02 (d, 1H, J = 4 Hz, NH), 7.06 (d, 1H, J = 4 Hz, CH Phenyl), 7.08 (d, 1H, J = 4 Hz, CH Phenyl), 7.22 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.23–7.26 (m, 1H, CH 8), 7.42 (s, 1H, CH 2), 7.49 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9). 13C-NMR δ (ppm, DMSO-d6): 19.43 (CH3 γ’), 19.56 (CH3 γ), 28.28 (CH3 tBu), 30.57 (CH β), 56.15 (OCH3 Phenyl), 56.24 (OCH3 Phenyl), 59.73 (CH α), 81.31 (Cq tBu), 112.41 (CH Phenyl), 114.35 (CH Phenyl), 116.02 (CH 6), 122.67 (Cq 1), 123.14 (CH 7), 123.39 (CH Phenyl), 126.19 (Cq 5a), 126.71 (CH 8), 127.39 (CH 9), 131.25 (Cq Phenyl), 132.68 (CH 2), 133.00 (Cq 3a), 137.55 (Cq 9a), 147.56 (Cq 4), 149.36 (Cq Phenyl), 150.26 (Cq Phenyl), 171.70 (C=O). MS (ESI +, QTof, m/z): 477.1 [M + H]+. HRMS calculated for C27H33N4O4 477.2502, found 477.2505.
Tert-butyl 2-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-4-methyl-pentanoate: (8d): Following the same procedure used for the synthesis of 8a, to a mixture of 7d (0.360 g, 0.83 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.166 g, 0.91 mmol), tetrakis(triphenylphosphine) palladium (0.048 g, 0.041 mmol) and sodium carbonate (0.176 g, 1.66 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (61% yield). C28H34N4O4. MW: 490.59 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.83 (d, 3H, J = 8 Hz, CH3 δ), 0.86 (d, 3H, J = 8 Hz, CH3 δ’), 1.31 (s, 9H, 3 × CH3 OtBu), 1.55–1.58 (m, 1H, CH2 β), 1.67–1.70 (m, 1H, CH γ), 1.84–1.88 (m, 1H, CH2 β), 3.63 (s, 3H, OCH3 Phenyl), 3.76 (s, 3H, OCH3 Phenyl), 4.55 (t, 1H, J = 4 Hz, CH α), 6.90–6.94 (m, 1H, CH 7), 7.01 (d, 1H, J = 4 Hz, CH Phenyl), 7.05 (s, 1H, CH Phenyl), 7.08 (d, 1H, J = 4 Hz, CH Phenyl), 7.20 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.22–7.24 (m, 1H, CH 8), 7.41 (s, 1H, CH 2), 7.45 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.50 (d, 1H, J = 4 Hz, NH). 13C-NMR δ (ppm, DMSO-d6): 22.14 (CH3 δ), 23.41 (CH3 δ’), 25.25 (CH γ), 28.25 (CH3 tBu), 40.29 (CH2 β), 52.96 (CH α), 56.14 (OCH3 Phenyl), 56.23 (OCH3 Phenyl), 80.79 (Cq tBu), 112.40 (CH Phenyl), 114.34 (CH Phenyl), 115.99 (CH 6), 122.78 (Cq 1), 122.88 (CH 7), 123.36 (CH Phenyl), 126.11 (Cq 5a), 126.69 (CH 8), 127.29 (CH 9), 131.09 (Cq 3a), 132.61 (CH 2), 137.70 (Cq 9a), 147.66 (Cq 4), 149.36 (Cq Phenyl), 150.23 (Cq Phenyl), 172.86 (C=O). MS (ESI +, QTof, m/z): 491.0 [M + H]+. HRMS calculated for C28H35N4O4 491.2658, found 491.2654.
Tert-butyl 6-((Tert-butoxycarbonyl)amino)-2-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]-quinoxalin-4-yl)amino)hexanoate (8e): Following the same procedure used for the synthesis of 8a, to a mixture of 7e (1.090 g, 1.99 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.397 g, 2.18 mmol), tetrakis(triphenylphosphine) palladium (0.114 g, 0.099 mmol) and sodium carbonate (0.421 g, 3.97 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (7% yield). C33H43N5O6. MW: 605.72 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.36 (s, 9H, 3 × CH3 OtBu), 1.41–1.46 (m, 13H, CH2 γ, 3 × CH3 OtBu, CH2 δ), 1.88–1.91 (m, 2H, CH2 β), 2.93–2.95 (m, 2H, CH2 ε), 3.73 (s, 3H, OCH3 Phenyl), 3.87 (s, 3H, OCH3 Phenyl), 4.54–4.58 (m, 1H, CH α), 6.80 (t, 1H, J = 4 Hz, NH-CH2 ε), 7.01–7.05 (m, 1H, CH 7), 7.11 (d, 1H, J = 4 Hz, CH Phenyl), 7.15 (s, 1H, CH Phenyl), 7.18 (d, 1H, J = 4 Hz, CH Phenyl), 7.31 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.31–7.35 (m, 1H, CH 8), 7.52 (s, 1H, CH 2), 7.55 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.59 (d, 1H, J = 4 Hz, NH-CH α). 13C-NMR δ (ppm, DMSO-d6): 23.49 (CH2 γ), 28.18 (CH3 tBu), 28.72 (CH3 tBu), 29.64 (CH2 δ), 30.94 (CH2 β), 40.40 (CH2 ε), 54.56 (CH α), 56.05 (OCH3 Phenyl), 56.14 (OCH3 Phenyl), 77.78 (Cq tBu), 80.80 (Cq tBu), 112.30 (CH Phenyl), 114.22 (CH Phenyl), 115.91 (CH 6), 122.67 (CH 7), 123.28 (CH Phenyl), 126.03 (Cq 5a), 126.56 (CH 8), 127.23 (CH 9), 131.01 (Cq 1), 132.53 (CH 2), 132.98 (Cq 3a), 137.61 (Cq 9a), 147.51 (Cq 4), 149.27 (Cq Phenyl), 150.14 (Cq Phenyl), 156.04 (Cq Phenyl), 172.41 (C=O). MS (ESI +, QTof, m/z): 606.2 [M + H]+. HRMS calculated for C33H44N5O6 606.3292, found 606.3291.
Tert-butyl 5-(((benzyloxy)carbonyl)amino)-2-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]-quinoxalin-4-yl)amino)pentanoate (8f): Following the same procedure used for the synthesis of 8a, to a mixture of 7f (0.470 g, 0.89 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.165 g, 0.91 mmol), tetrakis(triphenylphosphine) palladium (0.048 g, 0.041 mmol) and sodium carbonate (0.175 g, 1.65 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (42% yield). C35H39N5O6. MW: 625.71 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1H-NMR δ (ppm, 400 MHz, DMSO-d6): 1.52 (s, 9H, 3 × CH3 OtBu), 1.77–1.81 (m, 2H, CH2 γ), 1.99–2.03 (m, 1H, CH2 δ), 2.10–2.13 (m, 1H, CH2 δ), 3.34–3.39 (m, 2H, CH2 β), 3.88 (s, 3H, OCH3 Phenyl), 4.02 (s, 3H, OCH3 Phenyl), 5.12 (s, 2H, CH2-Phenyl), 5.37–5.39 (m, 1H, CH α), 6.99 (d, 2H, J = 4 Hz, CH Phenyl), 7.03 (s, 1H, CH Phenyl), 7.10–7.12 (m, 1H, CH 7), 7.28 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.32–7.35 (m, 7H, CH 8, CH2, CH Phenyl), 7.36 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.47 (d, 1H, J = 4 Hz, NH-CH2 δ), 7.76 (dd, 1H, J = 4 Hz, J = 8 Hz, NH-CH α). 13C-NMR δ (ppm, DMSO-d6): 25.62 (CH2 γ), 28.09 (CH3 tBu), 30.04 (CH2 β), 40.61 (CH2 δ), 54.42 (CH α), 66.58 (CH2-Phenyl), 82.42 (Cq tBu), 111.30 (CH Phenyl), 113.24 (CH Phenyl), 114.69 (Cq 1), 115.99 (CH 6), 123.16 (CH 7), 126.40 (Cq 5a), 126.82 (CH 9), 126.87 (CH 8), 128.01 (CH Phenyl), 128.08 (CH Phenyl), 129.49 (CH Phenyl), 131.85 (CH 2), 132.87 (Cq 3a), 136.68 (Cq 9a), 149.16 (Cq 4), 150.12 (Cq Phenyl), 152.37 (Cq Phenyl), 156.51 (Cq Phenyl), 171.59 (C=O). MS (ESI +, QTof, m/z): 626.0 [M + H]+. HRMS calculated for C35H40N5O6 626.2979, found 626.2982.
Tert-butyl 2-((Tert-butoxycarbonyl)amino)-5-((1-(3,4-dimethoxyphenyl)imidazo[1,2-a]-quinoxalin-4-yl)amino)pentanoate (8i): Following the same procedure used for the synthesis of 8a, to a mixture of 7i (0.515 g, 0.96 mmol) in DME/H2O (2/1, 15 mL) were added 3,4-dimethoxyphenylboronic acid (0.193 g, 1.06 mmol), tetrakis(triphenylphosphine) palladium (0.056 g, 0.048 mmol) and sodium carbonate (0.204 g, 1.93 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (45% yield). C32H41N5O6. MW: 591.70 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.34 (s, 9H, CH-COOtBu), 1.39 (s, 9H, NH-COOtBu), 1.73–1.74 (m, 2H, CH2 β), 1.77–1.78 (m, 2H, CH2 γ), 3.56–3.59 (m, 2H, CH2 δ), 3.74 (s, 3H, OCH3 Phenyl), 3.87 (s, 3H, OCH3 Phenyl), 3.89–3.92 (m, 1H, CH α), 6.95–7.00 (m, 1H, CH 7), 7.09 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.14 (s, 1H, CH Phenyl), 7.15 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.19 (d, 1H, J = 4 Hz, NH-CH α), 7.28 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.30–7.32 (m, 1H, CH 8), 7.47 (s, 1H, CH 2), 7.58 (dd, 1H, J = 4 Hz, J = 8 Hz CH 9), 7.72 (t, 1H, J = 4 Hz, NH-CH2 δ). 13C-NMR δ (ppm, DMSO-d6): 25.97 (CH2 β), 28.06 (CH3 tBu), 28.25 (CH3 tBu), 28.67 (CH2 γ), 39.79 (CH2 δ), 54.77 (CH α), 56.05 (OCH3 Phenyl), 56.15 (OCH3 Phenyl), 78.46 (Cq tBu), 80.54 (Cq tBu), 112.32 (CH Phenyl), 114.22 (CH Phenyl), 115.83 (CH 6), 122.12 (CH 7), 122.82 (CH Phenyl), 123.26 (Cq 1), 125.75 (Cq 5a), 126.41 (CH 8), 127.07 (CH 9), 132.32 (CH 2), 138.24 (Cq 3a), 148.04 (Cq 4), 149.26 (Cq Phenyl), 150.10 (Cq Phenyl), 156.01 (Cq 9a), 172.36 (C=O). MS (ESI +, QTof, m/z): 592.1 [M + H]+. HRMS calculated for C32H42N5O6 592.3135, found 592.3137.
Tert-butyl 2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)acetate (9a): To a mixture of 7a (0.320 g, 0.85 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.392 g, 2.54 mmol), tetrakis(triphenylphosphine) palladium (0.049 g, 0.040 mmol) and sodium carbonate (0.179 g, 1.70 mmol) in a microwave-adapted vial. The reaction was submitted to microwave irradiations during 20 min at 150 °C and then filtered on a Celite pad. The filtrate was concentrated under reduced pressure and successively washed with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate, distilled water and finally brine. The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (16% yield). C22H22N4O4. MW: 406.43 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1H-NMR δ (ppm, DMSO-d6): 1.42 (s, 9H, 3 × CH3 OtBu), 4.15 (d, 2H, J = 8 Hz, CH2 α), 6.82 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.89 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.93 (s, 1H, CH Phenyl), 7.02–7.06 (m, 1H, CH 7), 7.33 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.35–7.38 (m, 1H, CH 8), 7.46 (s, 1H, CH 2), 7.54 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.96–8.00 (m, 1H, NH), 9.32 (s, 1H, C-OH Phenyl), 9.41 (s, 1H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 28.23 (CH3 tBu), 43.30 (CH2 α), 80.85 (Cq tBu), 115.37 (Cq 1), 115.92 (CH 6), 116.48 (CH 7), 117.89 (CH Phenyl), 121.13 (CH Phenyl), 122.06 (CH 8), 122.81 (CH Phenyl), 125.98 (Cq 5a), 126.54 (CH 9), 127.77 (Cq Phenyl), 131.43 (Cq 3a), 132.36 (CH 2), 132.89 (Cq 9a), 146.04 (Cq Phenyl), 147.13 (Cq Phenyl), 147.65 (Cq 4), 169.76 (C=O). MS (ESI +, QTof, m/z): 407.2 [M + H]+. HRMS calculated for C22H23N4O4 407.1706, found 407.1710.
Tert-butyl 2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)propanoate (9b): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7b (0.290 g, 0.74 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.342 g, 2.22 mmol), tetrakis- (triphenylphosphine) palladium (0.043 g, 0.037 mmol) and sodium carbonate (0.157 g, 1.48 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (6% yield). C23H24N4O4. MW: 420.46 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.42 (s, 9H, 3 × CH3 OtBu), 1.51 (d, 3H, J = 8 Hz, CH3 β), 4.59–4.63 (m, 1H, CH α), 6.81 (d, 1H, J = 4 Hz, CH Phenyl), 6.90 (d, 1H, J = 4 Hz, CH Phenyl), 6.93 (s, 1H, CH Phenyl), 7.01–7.05 (m, 1H, CH 7), 7.30 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.33–7.37 (m, 1H, CH 8), 7.45 (s, 1H, CH 2), 7.53 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.66 (d, 1H, J = 4 Hz, NH), 9.36 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 17.57 (CH3 β), 28.16 (CH3 tBu), 50.17 (CH α), 80.66 (Cq tBu), 115.87 (CH 8), 116.47 (CH Phenyl), 117.88 (CH Phenyl), 121.16 (Cq 1), 122.05 (CH Phenyl), 122.78 (CH 7), 126.02 (Cq 5a), 127.16 (CH 9), 131.35 (Cq 3a), 132.19 (CH 2), 132.80 (Cq 9a), 137.57 (Cq 4), 146.03 (Cq Phenyl), 147.10 (Cq Phenyl), 147.23 (Cq Phenyl), 172.87 (C=O). MS (ESI +, QTof, m/z): 421.2 [M + H]+. HRMS calculated for C23H25N4O4 421.1876, found 421.1875.
Tert-butyl 2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-methyl-butanoate (9c): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7c (0.380 g, 0.91 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.340 g, 2.72 mmol), tetrakis- (triphenylphosphine) palladium (0.052 g, 0.045 mmol) and sodium carbonate (0.192 g, 1.81 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (32% yield). C25H28N4O4. MW: 448.51 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.03 (d, 3H, J = 8 Hz, CH3 γ), 1.06 (d, 3H, J = 8 Hz, CH3 γ’), 1.44 (s, 9H, 3 × CH3 OtBu), 2.31–2.35 (m, 1H, CH β), 4.53 (t, 1H, J = 8 Hz, CH α), 6.81 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.90 (d, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.93 (d, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.03–7.05 (m, 1H, CH 7), 7.07 (d, 1H, J = 4 Hz, NH), 7.34–7.35 (m, 1H, CH 8), 7.37 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.46 (s, 1H, CH 2), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 9.32 (s, 1H, C-OH Phenyl), 9.41 (s, 1H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 19.30 (CH3 γ’), 19.47 (CH3 γ), 28.19 (CH3 tBu), 30.54 (CH β), 59.60 (CH α), 81.25 (Cq tBu), 115.90 (CH 6), 116.47 (CH Phenyl), 117.89 (CH Phenyl), 121.05 (Cq 1), 122.07 (CH Phenyl), 123.07 (CH 7), 126.11 (Cq 5a), 126.56 (CH 8), 127.28 (CH 9), 131.55 (Cq Phenyl), 132.24 (CH 2), 132.72 (Cq 3a), 137.43 (Cq 9a), 146.03 (Cq 4), 147.14 (Cq Phenyl), 147.47 (Cq Phenyl), 171.62 (C=O). MS (ESI +, QTof, m/z): 449.3 [M + H]+. HRMS calculated for C25H29N4O4 449.2189, found 449.2188.
Tert-butyl 2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-4-methyl-pentanoate (9d): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7d (0.460 g, 1.06 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.400 g, 2.6 mmol), tetrakis- (triphenylphosphine) palladium (0.062 g, 0.053 mmol) and sodium carbonate (0.224 g, 2.11 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (16% yield). C26H30N4O4. MW: 462.54 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.93 (d, 3H, J = 8 Hz, CH3 δ), 0.97 (d, 3H, J = 8 Hz, CH3 δ’), 1.42 (s, 9H, 3 × CH3 OtBu), 1.56–1.60 (m, 1H, CH2 β), 1.79–1.83 (m, 1H, CH γ), 1.91–1.95 (m, 1H, CH2 β), 4.65 (t, 1H, J = 4 Hz, CH α), 6.81 (d, 1H, J = 4 Hz, CH Phenyl), 6.86 (s, 1H, CH Phenyl), 6.90 (d, 1H, J = 4 Hz, CH Phenyl), 7.01–7.05 (m, 1H, CH 7), 7.28–7.32 (m, 1H, CH 8), 7.34 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.46 (s, 1H, CH 2), 7.55 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.59 (d, 1H, J = 4 Hz, NH), 9.38 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 22.09 (CH3 δ), 23.32 (CH3 δ’), 25.17 (CH γ), 28.18 (CH3 tBu), 39.60 (CH2 β), 52.88 (CH α), 80.76 (Cq tBu), 115.87 (CH 6), 116.48 (CH Phenyl), 117.90 (CH Phenyl), 121.15 (Cq 1), 122.18 (CH Phenyl), 122.79 (CH 7), 126.11 (Cq 5a), 125.80 (CH 8), 126.04 (CH 9), 131.42 (Cq 3a), 132.18 (CH 2), 137.58 (Cq 9a), 146.06 (Cq 4), 147.14 (Cq Phenyl), 147.57 (Cq Phenyl), 172.77 (C=O). MS (ESI +, QTof, m/z): 463.2 [M + H]+. HRMS calculated for C26H31N4O4 463.2345, found 463.2343.
Tert-butyl 6-((Tert-butoxycarbonyl)amino)-2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]-quinoxalin-4-yl)amino)hexanoate (9e): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7e (0.242 g, 0.44 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.170 g, 1.10 mmol), tetrakis- (triphenylphosphine) palladium (0.025 g, 0.022 mmol) and sodium carbonate (0.093 g, 0.88 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (40% yield). C31H39N5O6. MW: 577.67 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.35 (s, 9H, 3 × CH3 OtBu), 1.40–1.42 (m, 2H, CH2 γ), 1.43 (s, 9H, 3 × CH3 OtBu), 1.45–1.47 (m, 2H, CH2 δ), 1.88–2.00 (m, 2H, CH2 β), 2.92–2.94 (m, 2H, CH2 ε), 4.55–4.56 (m, 1H, CH α), 6.79 (t, 1H, J = 4 Hz, NH-CH2 ε), 6.83 (d, 1H, J = 4 Hz, CH Phenyl), 6.89 (s, 1H, CH Phenyl), 6.93 (d, 1H, J = 4 Hz, CH Phenyl), 7.02–7.05 (m, 1H, CH 7), 7.31–7.33 (m, 1H, CH 8), 7.36 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.45 (s, 1H, CH 2), 7.54 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.57 (d, 1H, J = 4 Hz, NH-CH α), 9.33 (s, 1H, C-OH Phenyl), 9.41 (s, 1H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 22.40 (CH2 γ), 27.11 (CH3 tBu), 27.65 (CH3 tBu), 28.56 (CH2 δ), 29.91 (CH2 β), 39.25 (CH2 ε), 53.44 (CH α), 76.70 (Cq tBu), 79.75 (Cq tBu), 114.79 (CH 6), 115.40 (CH Phenyl), 116.80 (CH Phenyl), 120.07 (CH Phenyl), 121.73 (CH 7), 124.97 (Cq 5a), 125.42 (CH 8), 126.12 (CH 9), 130.32 (Cq 1), 131.09 (CH 2), 131.70 (Cq 3a), 136.49 (Cq 9a), 144.96 (Cq 4), 146.04 (Cq Phenyl), 146.43 (Cq Phenyl), 154.96 (Cq Phenyl), 172.41 (C=O). MS (ESI +, QTof, m/z): 578.3 [M + H]+. HRMS calculated for C31H40N5O6 578.2979, found 578.2980.
Tert-butyl 5-(((benzyloxy)carbonyl)amino)-2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]-quinoxalin-4-yl)amino)pentanoate (9f): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7f (0.260 g, 0.46 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.211 g, 1.37 mmol), tetrakis- (triphenylphosphine) palladium (0.026 g, 0.023 mmol) and sodium carbonate (0.097 g, 0.91 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (26% yield). C33H35N5O6. MW: 597.66 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1H-NMR δ (ppm, 400 MHz, DMSO-d6): 1.40 (s, 9H, 3 × CH3 OtBu), 1.58–1.60 (m, 2H, CH2 γ), 1.91–1.95 (m, 2H, CH2 β), 3.05–3.08 (m, 2H, CH2 δ), 4.55–4.59 (m, 1H, CH α), 5.01 (s, 2H, CH2-Phenyl), 6.81 (d, 1H, J = 4 Hz, CH Phenyl), 6.89 (s, 1H, CH Phenyl), 6.93 (d, 1H, J = 4 Hz, CH Phenyl), 7.01–7.06 (m, 1H, CH 7), 7.27 (d, 1H, J = 4 Hz, NH-CH2 δ), 7.31–7.34 (m, 5H, CH Phenyl), 7.37 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.45 (s, 1H, CH 2), 7.54 (d, 1H, J = 4 Hz, NH-CH α), 7.56–7.60 (m, 1H, CH 8), 7.63 (dd, 1H, J = J = 4 Hz, J = 8 Hz, CH 9). 13C-NMR δ (ppm, DMSO-d6): 26.54 (CH2 γ), 28.09 (CH2 β), 28.17 (CH3 tBu), 40.45 (CH2 δ), 54.34 (CH α), 65.60 (CH2-Phenyl), 80.92 (Cq tBu), 115.88 (CH 6), 116.48 (CH Phenyl), 117.90 (CH Phenyl), 121.16 (Cq 1), 122.84 (CH 7), 126.06 (Cq 5a), 126.50 (CH 9), 127.20 (CH 8), 128.18 (CH Phenyl), 128.79 (CH Phenyl), 129.16 (CH Phenyl), 131.90 (CH 2), 132.79 (Cq 3a), 137.55 (Cq 9a), 146.04 (Cq 4), 147.12 (Cq Phenyl), 147.47 (Cq Phenyl), 156.59 (Cq Phenyl), 172.23 (C=O). MS (ESI +, QTof, m/z): 598.3 [M + H]+. HRMS calculated for C33H36N5O6 598.2666, found 598.2670.
Tert-butyl 2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-phenyl-propanoate (9g): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7g (0.360 g, 0.77 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.356 g, 2.31 mmol), tetrakis- (triphenylphosphine) palladium (0.044 g, 0.038 mmol) and sodium carbonate (0.163 g, 1.54 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (16% yield). C29H28N4O4. MW: 496.56 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.36 (s, 9H, 3 × CH3 OtBu), 3.23–3.27 (m, 1H, CH2 β), 3.33–3.38 (m, 1H, CH2 β), 4.91–4.93 (m, 1H, CH α), 6.81 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.89 (d, 1H, J = 4 Hz, CH Phenyl), 6.92 (d, 1H, J = 8 Hz, CH Phenyl), 7.08 (t, 1H, J = 8 Hz, CH Phenyl), 7.20 (t, 1H, J = 8 Hz, CH Phenyl), 7.29 (t, 2H, J = 8 Hz, CH Phenyl), 7.34 (d, 2H, J = 8 Hz, CH 6, CH Phenyl), 7.34–7.37 (m, 1H, CH 7), 7.38–7.40 (m, 1H, CH 8), 7.52 (s, 1H, CH 2), 7.60 (d, 1H, J = 8 Hz, CH 9), 7.92–7.93 (m, 1H, NH), 9.38 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 26.98 (CH3 tBu), 35.93 (CH2 β), 55.04 (CH α), 80.23 (Cq tBu), 114.96 (CH 6), 115.42 (CH Phenyl), 116.75 (CH Phenyl), 119.67 (Cq 1), 120.96 (CH Phenyl), 122.32 (CH 7), 124.69 (Cq 5a), 125.73 (CH Phenyl, CH 9), 125.94 (CH Phenyl), 127.90 (CH Phenyl), 128.71 (CH 8), 130.76 (Cq Phenyl), 131.11 (CH 2), 131.21 (Cq Phenyl), 136.87 (Cq 3a), 138.12 (Cq 9a), 146.17 (Cq 4), 170.13 (C=O). MS (ESI +, QTof, m/z): 497.1 [M + H]+. HRMS calculated for C29H29N4O4 497.2189, found 497.2198.
Tert-butyl 3-(4-(Tert-butoxy)phenyl)-2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)propanoate (9h): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7h (0.260 g, 0.57 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.170 g, 1.72 mmol), tetrakis- (triphenylphosphine) palladium (0.028 g, 0.028 mmol) and sodium carbonate (0.101 g, 1.15 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (22% yield). C33H36N4O5. MW: 568.66 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.24 (s, 9H, 3 × CH3 OtBu), 1.33 (s, 9H, 3 × CH3 OtBu), 3.12–3.18 (m, 1H, CH2 β), 3.26–3.32 (m, 1H, CH2 β), 4.80–4.84 (m, 1H, CH α), 6.80 (d, 1H, J = 8 Hz, CH Phenyl), 6.88 (s, 1H, CH Phenyl), 6.90 (dd, 2H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.92 (d, 1H, J = 8 Hz, CH Phenyl), 7.01–7.05 (m, 1H, CH 7), 7.25 (d, 2H, J = 8 Hz, CH Phenyl), 7.30–7.32 (m, 1H, CH 8), 7.34 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.45 (s, 1H, CH 2), 7.54 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.57 (d, 1H, J = 8 Hz, NH), 9.31 (s, 1H, C-OH Phenyl), 9.40 (s, 1H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 28.05 (CH3 tBu), 28.95 (CH3 tBu), 36.64 (CH2 β), 55.87 (CH α), 78.12 (Cq tBu), 80.91 (Cq tBu), 115.90 (CH 6), 116.46 (CH Phenyl), 117.87 (CH Phenyl), 121.08 (Cq 1), 122.05 (CH Phenyl), 122.91 (CH 7), 124.00 (2 × CH Phenyl), 126.07 (Cq 5a), 126.49 (CH 8), 127.21 (CH 9), 130.33 (2 × CH Phenyl), 132.26 (CH 2), 132.62 (Cq 3a), 137.46 (Cq 9a), 146.02 (Cq 4), 147.11 (Cq Phenyl), 147.22 (Cq Phenyl), 154.05 (2 × Cq Phenyl), 171.75 (C=O). MS (ESI +, QTof, m/z): 569.3 [M + H]+. HRMS calculated for C33H37N4O5 569.2764, found 569.2773.
Tert-butyl 2-((Tert-butoxycarbonyl)amino)-5-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]-quinoxalin-4-yl)amino)pentanoate (9i): Following the same procedure used for the synthesis of 9a (see 4.1.4.8.), to a mixture of 7i (0.460 g, 0.86 mmol) in DME/H2O (2/1, 15 mL) were added compound 12 (0.330 g, 2.15 mmol), tetrakis- (triphenylphosphine) palladium (0.050 g, 0.043 mmol) and sodium carbonate (0.182 g, 1.72 mmol) in a microwave-adapted vial. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 80/20 to 50/50. The compound is obtained as a white solid (30% yield). C30H37N5O6. MW: 563.64 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.33 (s, 9H, CH-COOtBu), 1.39 (s, 9H, NH-COOtBu), 1.66–1.72 (m, 4H, CH2 β, CH2 γ), 3.56–3.57 (m, 2H, CH2 δ), 3.89–3.93 (m, 1H, CH α), 6.79 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.87 (d, 1H, J = 4 Hz, CH Phenyl), 6.90 (d, 1H, J = 8 Hz, CH Phenyl), 6.95–7.00 (m, 1H, CH 7), 7.17 (d, 1H, J = 8 Hz, NH-CH α), 7.27–7.29 (m, 1H, CH 8), 7.31 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.40 (s, 1H, CH 2), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.69 (t, 1H, J = 4 Hz, NH-CH2 δ), 9.35 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 26.80 (CH2 β), 28.04 (CH3 tBu), 28.67 (CH3 tBu), 30.42 (CH2 γ), 39.79 (CH2 δ), 54.78 (CH α), 78.46 (Cq tBu), 80.53 (Cq tBu), 115.79 (CH 6), 116.45 (CH Phenyl), 117.86 (CH Phenyl), 121.29 (Cq 1), 122.01 (CH Phenyl), 122.06 (CH 7), 125.76 (Cq 5a), 126.29 (CH 8), 127.03 (CH 9), 131.91 (CH 2), 138.20 (Cq 3a), 146.02 (Cq 4), 147.05 (Cq Phenyl), 148.03 (Cq Phenyl), 156.02 (Cq 9a), 172.34 (C=O). MS (ESI +, QTof, m/z): 564.3 [M + H]+. HRMS calculated for C30H38N5O6 564.2822, found 564.2827.
3.1.5. Cleavage of the Protective Groups
2-((1-(3,4-Dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)propanoic acid (10b): To a cooled (0 °C) solution of 8b (0.170 g, 0.36 mmol) in anhydrous CH2Cl2 (20 mL) was added boron tribromide (2.1 mL, 2.1 mmol). The resulting solution was allowed to warm to room temperature and stirred until complete consumption of the starting material (1–3 h, monitored by TLC). The solution was neutralized by addition of saturated aqueous sodium bicarbonate (20 mL). The crude mixture was extracted with CH2Cl2 (3 × 20 mL). The organic phase was dried on sodium sulfate, filtered and concentrated under reduced pressure. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (71% yield). C21H20N4O4. MW: 392.41 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.24 (s, 1H, COOH), 1.53 (d, 3H, J = 8 Hz, CH3 β), 3.75 (s, 3H, OCH3 Phenyl), 3.87 (s, 3H, OCH3 Phenyl), 4.61–4.63 (m, 1H, CH α), 7.00–7.04 (m, 1H, CH 7), 7.11 (d, 1H, J = 4 Hz, CH Phenyl), 7.13 (s, 1H, CH Phenyl), 7.17 (d, 1H, J = 4 Hz, CH Phenyl), 7.31 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.33–7.35 (m, 1H, CH 8), 7.50 (s, 1H, CH 2), 7.52 (d, 1H, J = 4 Hz, NH), 7.60 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9). 13C-NMR δ (ppm, DMSO-d6): 18.71 (CH3 β), 50.30 (CH α), 56.07 (OCH3 Phenyl), 56.16 (OCH3 Phenyl), 112.31 (CH Phenyl), 114.22 (CH Phenyl), 115.92 (CH 6), 122.54 (CH 7), 122.70 (Cq 1), 123.28 (CH Phenyl), 126.54 (CH 8), 125.90 (Cq 5a), 127.21 (CH 9), 130.95 (Cq 3a), 132.63 (CH 2), 137.99 (Cq 9a), 147.05 (Cq 4), 149.26 (Cq Phenyl), 150.132 (Cq Phenyl), 172.96 (C=O). MS (ESI +, QTof, m/z): 393.0 [M + H]+. HRMS calculated for C21H21N4O4 393.1563, found 393.1558.
2-((1-(3,4-Dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-methylbutanoic acid (10c): Following the same procedure used for the synthesis of 10b, to a cooled solution of 8c (0.290 g, 0.61 mmol) in anhydrous CH2Cl2 (20 mL) was added boron tribromide (3.6 mL, 3.6 mmol). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (80% yield). C23H24N4O4. MW: 420.46 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.99 (d, 3H, J = 8 Hz, CH3 γ), 1.01 (d, 3H, J = 8 Hz, CH3 γ’), 1.23 (s, 1H, COOH), 2.36–2.41 (m, 1H, CH β), 3.74 (s, 3H, OCH3 Phenyl), 3.87 (s, 3H, OCH3 Phenyl), 4.55 (t, 1H, J = 16 Hz, CH α), 6.97–7.01 (m, 1H, CH 7), 7.10 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.14 (s, 1H, CH Phenyl), 7.16 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.19 (d, 1H, NH-CH α), 7.28 (d, 1H, J = 4 Hz, CH 6), 7.29–7.32 (m, 1H, CH 8), 7.48 (s, 1H, CH 2), 7.54 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9). 13C-NMR δ (ppm, DMSO-d6): 19.02 (CH3 γ’), 19.96 (CH3 γ), 31.19 (CH β), 56.06 (OCH3 Phenyl), 56.16 (OCH3 Phenyl), 59.54 (CH α), 112.31 (CH Phenyl), 114.26 (CH Phenyl), 115.87 (CH 6), 122.35 (CH 7), 122.75 (Cq 1), 123.29 (CH Phenyl), 125.89 (Cq 5a), 126.49 (CH 8), 127.18 (CH 9), 130.95 (Cq Phenyl), 132.44 (CH 2), 133.40 (Cq 3a), 138.10 (Cq 9a), 147.60 (Cq 4), 149.25 (Cq Phenyl), 150.12 (Cq Phenyl), 170.00 (C=O). MS (ESI +, QTof, m/z): 420.9 [M + H]+. HRMS calculated for C23H25N4O4 421.1876, found 421.1873.
2-((1-(3,4-Dimethoxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-4-methylpentanoic acid (10d): Following the same procedure used for the synthesis of 10b, to a cooled solution of 8d (0.210 g, 0.43 mmol) in anhydrous CH2Cl2 (20 mL) was added boron tribromide (2.5 mL, 2.53.6 mmol). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (76% yield). C24H26N4O4. MW: 434.49 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.95 (d, 3H, J = 8 Hz, CH3 δ), 0.98 (d, 3H, J = 8 Hz, CH3 δ’), 1.76–1.80 (m, 3H, CH2 β, CH γ), 3.74 (s, 3H, OCH3 Phenyl), 3.87 (s, 3H, OCH3 Phenyl), 4.66 (t, 1H, J = 4 Hz, CH α), 6.98–7.02 (m, 1H, CH 7), 7.11 (d, 1H, J = 4 Hz, CH Phenyl), 7.13 (s, 1H, CH Phenyl), 7.17 (d, 1H, J = 4 Hz, CH Phenyl), 7.29 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.31–7.33 (m, 1H, CH 8), 7.44 (d, 1H, J = 4 Hz, NH), 7.49 (s, 1H, CH 2), 7.56 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9). 13C-NMR δ (ppm, DMSO-d6): 22.68 (CH3 δ), 23.69 (CH3 δ’), 25.24 (CH γ), 40.42 (CH2 β), 53.00 (CH α), 56.05 (OCH3 Phenyl), 56.15 (OCH3 Phenyl), 112.30 (CH Phenyl), 114.24 (CH Phenyl), 115.88 (CH 6), 122.29 (CH 7), 122.77 (Cq 1), 123.27 (CH Phenyl), 125.86 (Cq 5a), 126.49 (CH 8), 127.13 (CH 9), 130.90 (Cq 3a), 132.42 (CH 2), 138.12 (Cq 9a), 147.44 (Cq 4), 149.25 (Cq Phenyl), 150.10 (Cq Phenyl), 172.31 (C=O). MS (ESI +, QTof, m/z): 434.9 [M + H]+. HRMS calculated for C24H27N4O4 435.2032, found 435.2032.
2-((1-(3,4-Dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)acetic acid (11a): To a cooled (0 °C) solution of 9a (0.055, 0.14 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The resulting solution was allowed to warm to room temperature and stirred until complete consumption of the starting material (1–2 h, monitored by TLC). The solvent was removed under reduced pressure. The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (92% yield). C18H14N4O4. MW: 350.33 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.23 (s, 1H, COOH), 4.24 (d, 2H, J = 8 Hz, CH2 α), 6.82 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.90 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.93 (s, 1H, CH Phenyl), 7.03–7.07 (m, 1H, CH 7), 7.31–7.34 (m, 1H, CH 8), 7.35 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.48 (s, 1H, CH 2), 7.58 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 8.02–8.05 (m, 1H, NH), 9.39–9.43 (m, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 42.37 (CH2 α), 115.97 (CH 6), 116.49 (CH Phenyl), 117.86 (CH Phenyl), 120.69 (Cq 1), 122.06 (CH Phenyl), 122.96 (CH 7), 126.13 (Cq 5a), 126.51 (CH 8), 126.62 (CH 9), 132.36 (CH 2), 146.01 (Cq 3a), 147.17 (Cq 9a), 147.56 (Cq 4), 152.74 (Cq Phenyl), 172.09 (C=O). MS (ESI +, QTof, m/z): 351.2 [M + H]+. HRMS calculated for C18H15N4O4 351.1093, found 351.1093.
2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)propanoic acid (11b): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9b (0.040, 0.09 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (88% yield). C19H16N4O4. MW: 364.35 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.23 (s, 1H, COOH), 1.56 (d, 3H, J = 8 Hz, CH3 β), 4.82–4.84 (m, 1H, CH α), 6.82 (d, 1H, J = 4 Hz, CH Phenyl), 6.90 (d, 1H, J = 4 Hz, CH Phenyl), 6.93 (s, 1H, CH Phenyl), 7.05–7.08 (m, 1H, CH 7), 7.33 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.35–7.37 (m, 1H, CH 8), 7.50 (s, 1H, CH 2), 7.59 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.95 (d, 1H, J = 4 Hz, NH), 9.37 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 17.79 (CH3 β), 49.27 (CH α), 116.01 (CH 6), 116.51 (CH Phenyl), 117.87 (CH Phenyl), 120.42 (Cq 1), 120.95 (Cq 5a), 122.06 (CH Phenyl), 123.17 (CH 7), 125.80 (CH 9), 126.69 (CH 8), 131.82 (Cq 3a), 132.42 (CH 2), 132.64 (Cq 9a), 137.42 (Cq 4), 146.06 (Cq Phenyl), 146.95 (Cq Phenyl), 147.22 (Cq Phenyl), 174.64 (C=O). MS (ESI +, QTof, m/z): 365.1 [M + H]+. HRMS calculated for C19H17N4O4 365.1250, found 365.1240.
2-((1-(3,4-Dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-methylbutanoic acid (11c): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9c (0.035, 0.08 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (91% yield). C21H20N4O4. MW: 392.41 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.04 (d, 3H, J = 8 Hz, CH3 γ), 1.07 (d, 3H, J = 8 Hz, CH3 γ’), 1.18 (s, 1H, COOH), 2.36–2.41 (m, 1H, CH β), 4.74–4.76 (m, 1H, CH α), 6.82 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.90 (d, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.94 (d, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 7.06–7.10 (m, 1H, CH 7), 7.17 (d, 1H, J = 4 Hz, NH), 7.33–7.36 (m, 1H, CH 8), 7.38 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.51 (s, 1H, CH 2), 7.60 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 9.40 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 18.86 (CH3 γ’), 19.63 (CH3 γ), 30.50 (CH β), 58.70 (CH α), 115.98 (CH 6), 116.49 (CH Phenyl), 117.86 (CH Phenyl), 120.83 (Cq 1), 122.07 (CH Phenyl), 123.29 (CH 7), 126.05 (Cq 5a), 126.72 (CH 8), 126.94 (CH 9), 132.12 (CH 2), 132.52 (Cq Phenyl), 132.82 (Cq 3a), 137.42 (Cq 9a), 146.06 (Cq 4), 147.23 (Cq Phenyl), 147.47 (Cq Phenyl), 171.62 (C=O). MS (ESI +, QTof, m/z): 393.2 [M + H]+. HRMS calculated for C21H21N4O4 393.1563, found 393.1561.
2-((1-(3,4-Dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-4-methylpentanoic acid (11d): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9d (0.050, 0.11 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (78% yield). C22H22N4O4. MW: 406.43 g/mol. 1H-NMR δ (ppm, DMSO-d6): 0.93 (d, 3H, J = 8 Hz, CH3 δ), 0.98 (d, 3H, J = 8 Hz, CH3 δ’), 1.42 (s, 1H, COOH), 1.69–1.72 (m, 1H, CH2 β), 1.76–1.79 (m, 1H, CH γ), 1.99–2.02 (m, 1H, CH2 β), 4.85 (t, 1H, J = 4 Hz, CH α), 6.82 (d, 1H, J = 4 Hz, CH Phenyl), 6.90 (s, 1H, CH Phenyl), 6.93 (d, 1H, J = 4 Hz, CH Phenyl), 7.05–7.08 (m, 1H, CH 7), 7.32–7.35 (m, 1H, CH 8), 7.36 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.50 (s, 1H, CH 2), 7.59 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.90 (d, 1H, J = 4 Hz, NH), 9.38 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 20.74 (CH3 δ), 22.28 (CH3 δ’), 24.07 (CH γ), 39.08 (CH2 β), 50.92 (CH α), 114.92 (CH 6), 115.42 (CH Phenyl), 116.77 (CH Phenyl), 119.82 (Cq 1), 120.98 (CH Phenyl), 122.10 (CH 7), 124.67 (Cq 5a), 125.64 (CH 8), 125.72 (CH 9), 130.79 (Cq 3a), 131.28 (CH 2), 144.99 (Cq 9a), 146.14 (Cq 4), 146.28 (Cq Phenyl), 157.42 (Cq Phenyl), 173.51 (C=O). MS (ESI +, QTof, m/z): 407.1 [M + H]+. HRMS calculated for C22H23N4O4 407.1719, found 407.1712.
6-Amino-2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)hexanoic acid (11e): Following the same procedure for the synthesis of 11a. To a cooled (0 °C) solution of 9e (0.035, 0.06 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (84% yield). C22H23N5O4. MW: 421.45 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.23 (s, 1H, COOH), 1.47–1.51 (m, 2H, CH2 γ), 1.58–1.64 (m, 2H, CH2 δ), 1.98–2.04 (m, 2H, CH2 β), 2.79–2.83 (m, 2H, CH2 ε), 4.76–4.78 (m, 1H, CH α), 6.81 (d, 1H, J = 4 Hz, CH Phenyl), 6.90 (s, 1H, CH Phenyl), 6.93 (d, 1H, J = 4 Hz, CH Phenyl), 7.03–7.07 (m, 1H, CH 7), 7.31–7.35 (m, 1H, CH 8), 7.37 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.47 (s, 1H, CH 2), 7.58 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.64 (d, 1H, J = 4 Hz, NH-CH α), 7.68–7.70 (m, 2H, NH2), 9.40–9.46 (m, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 22.04 (CH2 γ), 26.08 (CH2 δ), 29.65 (CH2 β), 38.04 (CH2 ε), 52.17 (CH α), 114.84 (CH 6), 115.40 (CH Phenyl), 116.79 (CH Phenyl), 120.96 (CH Phenyl), 121.87 (CH 7), 124.88 (Cq 5a), 125.49 (CH 8), 126.01 (CH 9), 130.49 (Cq 1), 131.14 (CH 2), 131.69 (Cq 3a), 136.24 (Cq 9a), 145.00 (Cq 4), 146.10 (Cq Phenyl), 146.51 (Cq Phenyl), 157.59 (Cq Phenyl), 173.29 (C=O). MS (ESI +, QTof, m/z): 422.1 [M + H]+. HRMS calculated for C22H24N5O4 422.1828, found 422.1834.
5-Amino-2-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)pentanoic acid (11f): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9f (0.055, 0.09 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (10% yield). C21H21N5O4. MW: 407.42 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1H-NMR δ (ppm, 400 MHz, DMSO-d6): 1.70–1.74 (m, 2H, CH2 γ), 1.89–1.93 (m, 1H, CH2 β), 2.07–2.11 (m, 1H, CH2 β), 2.66–2.70 (m, 1H, CH2 δ), 2.78–2.82 (m, 1H, CH2 δ), 4.33–4.34 (m, 1H, CH α), 6.69 (d, 1H, J = 8 Hz, CH Phenyl), 6.80 (d, 1H, J = 8 Hz, CH Phenyl), 6.89 (d, 1H, J = 4 Hz, CH Phenyl), 6.97–7.01 (m, 1H, CH 7), 7.29-.33 (m, 1H, CH 8), 7.36 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.40 (s, 1H, J = 4 Hz, CH 2), 7.46 (d, 1H, J = 4 Hz, NH-CH α), 7.57 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 8.69 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 23.61 (CH2 γ), 29.41 (CH2 β), 40.41 (CH2 δ), 54.57 (CH α), 115.88 (CH 6), 116.50 (CH Phenyl), 117.95 (CH Phenyl), 121.23 (Cq 1), 122.04 (CH 7), 122.18 (CH Phenyl), 125.85 (Cq 5a), 126.39 (CH 8), 127.17 (CH 9), 132.08 (CH 2), 133.25 (Cq 3a), 138.31 (Cq 9a), 146.07 (Cq 4), 146.84 (Cq Phenyl), 147.12 (Cq Phenyl), 174.19 (C=O). MS (ESI +, QTof, m/z): 408.2 [M + H]+. HRMS calculated for C21H22N5O4 408.1672, found 408.1668.
2-((1-(3,4-Dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-phenylpropanoic acid (11g): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9g (0.045, 0.09 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (87% yield). C25H20N4O4. MW: 440.45 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.23 (s, 1H, COOH), 3.33–3.39 (m, 2H, CH2 β), 5.04–5.07 (m, 1H, CH α), 6.81 (d, 1H, J = 8 Hz, CH Phenyl), 6.89 (d, 1H, J = 4 Hz, CH Phenyl), 6.91 (d, 1H, J = 8 Hz, CH Phenyl), 7.05–7.08 (m, 1H, CH 7), 7.17 (t, 1H, J = 8 Hz, CH Phenyl), 7.26 (t, 2H, J = 8 Hz, CH Phenyl), 7.31–7.333 (m, 1H, CH 8), 7.34 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.36–7.37 (m, 2H, CH Phenyl), 7.48 (s, 1H, CH 2), 7.61 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.76–7.77 (m, 1H, NH), 9.39 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 35.61 (CH2 β), 5.31 (CH α), 114.91 (CH 6), 115.40 (CH Phenyl), 116.75 (CH Phenyl), 119.75 (Cq 1), 120.96 (CH Phenyl), 122.23 (CH 7), 124.71 (Cq 5a), 125.64 (CH 9), 125.88 (CH 8), 127.67 (CH Phenyl), 128.58 (CH Phenyl), 130.78 (Cq Phenyl), 131.35 (CH 2), 137.26 (Cq 3a), 138.42 (Cq 9a), 144.98 (Cq Phenyl), 146.14 (Cq 4), 157.48 (Cq Phenyl), 157.77 (Cq Phenyl), 172.35 (C=O). MS (ESI +, QTof, m/z): 441.2 [M + H]+. HRMS calculated for C25H21N4O4 441.1563, found 441.1569.
2-((1-(3,4-Dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)-3-(4-hydroxyphenyl)-propanoic acid (11h): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9h (0.030, 0.06 mmol) in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (89% yield). C25H20N4O5. MW: 456.45 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.29 (s, 1H, COOH), 3.21–3.23 (m, 2H, CH2 β), 4.94–4.98 (m, 1H, CH α), 6.71 (d, 2H, J = 8 Hz, CH Phenyl), 6.86 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.94 (d, 1H, CH Phenyl), 6.98 (d, 1H, J = 8 Hz, CH Phenyl), 7.04–7.08 (m, 1H, CH 7), 7.12 (d, 2H, J = 8 Hz, CH Phenyl), 7.31–7.32 (m, 1H, CH 8), 7.35 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.47 (s, 1H, CH 2), 7.59–7.60 (m, 1H, NH), 7.64 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 9.32–9.35 (m, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 36.01 (CH2 β), 55.24 (CH α), 115.56 (2 × CH Phenyl), 115.96 (CH 6), 116.47 (CH Phenyl), 117.82 (CH Phenyl), 120.86 (Cq 1), 122.05 (CH Phenyl), 123.19 (CH 7), 125.81 (Cq 5a), 126.68 (CH 9), 126.70 (CH 8), 130.59 (2 × CH Phenyl), 132.35 (Cq 3a), 132.48 (CH 2), 137.32 (Cq 9a), 146.03 (Cq 4), 147.05 (Cq Phenyl), 147.19 (Cq Phenyl), 156.44 (Cq Phenyl), 173.55 (C=O). MS (ESI +, QTof, m/z): 457.1 [M + H]+. HRMS calculated for C25H21N4O5 457.1512, found 457.1514.
2-Amino-5-((1-(3,4-dihydroxyphenyl)imidazo[1,2-a]quinoxalin-4-yl)amino)pentanoic acid (11i): Following the same procedure for the synthesis of 11a, to a cooled (0 °C) solution of 9i in anhydrous CH2Cl2 (10 mL) was added TFA (5 mL). The product was purified by flash chromatography eluted with cyclohexane/ethyl acetate 90/20 to 40/50. The compound is obtained as a white solid (79% yield). C21H21N5O4. MW: 407.42 g/mol. 1H-NMR δ (ppm, DMSO-d6): 1.23 (s, 1H, COOH), 1.85–1.89 (m, 4H, CH2 γ, CH2 β), 3.64–3.66 (m, 2H, CH2 δ), 3.99–4.01 (m, 1H, CH α), 6.80 (dd, 1H, J = 4 Hz, J = 8 Hz, CH Phenyl), 6.89 (d, 1H, J = 4 Hz, CH Phenyl), 6.92 (d, 1H, J = 8 Hz, CH Phenyl), 7.07–7.11 (m, 1H, CH 7), 7.35 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 6), 7.37–7.39 (m, 1H, CH 8), 7.51 (s, 1H, CH 2), 7.55 (dd, 1H, J = 4 Hz, J = 8 Hz, CH 9), 7.66 (t, 1H, J = 4 Hz, NH-CH2 δ), 8.24 (s, 2H, NH2), 9.40 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 24.80 (CH2 γ), 28.04 (CH2 β), 39.99 (CH2 δ), 52.30 (CH α), 116.12 (CH 6), 116.51 (CH Phenyl), 117.77 (CH Phenyl), 120.74 (Cq 1), 121.96 (CH Phenyl), 123.27 (CH 7), 125.76 (Cq 5a), 126.83 (CH 8), 129.28 (CH 9), 132.68 (CH 2), 138.42 (Cq 3a), 146.12 (Cq 4), 147.29 (Cq Phenyl), 158.41 (Cq Phenyl), 158.75 (Cq 9a), 171.54 (C=O). MS (ESI +, QTof, m/z): 408.2 [M + H]+. HRMS calculated for C21H22N5O4 408.1672, found 408.1668.
3.1.6. 3,4-Dihydroxyphenylboronic Acid (12)
To a cooled (0 °C) solution of 3,4-dimethoxyphenylboronic acid (0.800 g, 4.39 mmol) in anhydrous CH2Cl2 (50 mL) was added boron tribromide (10 mL, 10 mmol). The resulting solution was allowed to warm to room temperature and stirred until complete consumption of the starting material (1–2h, monitored by TLC). The solution was neutralized by addition of methanol (50 mL). The crude mixture was concentrated under reduced pressure. The compound was obtained as a white solid (84% yield) and used without purification. C6H7BO4. MW: 153.93 g/mol. 1H-NMR δ (ppm, DMSO-d6): 2.08 (s, 2H, B-OH), 6.47 (d, 1H, CH Phenyl), 6.60 (d, 1H, CH Phenyl), 6.71 (d, 1H, CH Phenyl), 8.00 (s, 2H, C-OH Phenyl). 13C-NMR δ (ppm, DMSO-d6): 108.10 (CH Phenyl), 116.14 (CH Phenyl), 119.73(CH Phenyl), 145.73 (C-OH). MS (ESI +, QTof, m/z): 153.2 [M−H]−. HRMS calculated for C6H6O4B 153.0359, found 153.0358.

 and
and 




{kind=link}
{kind=link}
{kind=link}
{kind=link}















