RNA–Protein Interactions Prevent Long RNA Duplex Formation: Implications for the Design of RNA-Based Therapeutics

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
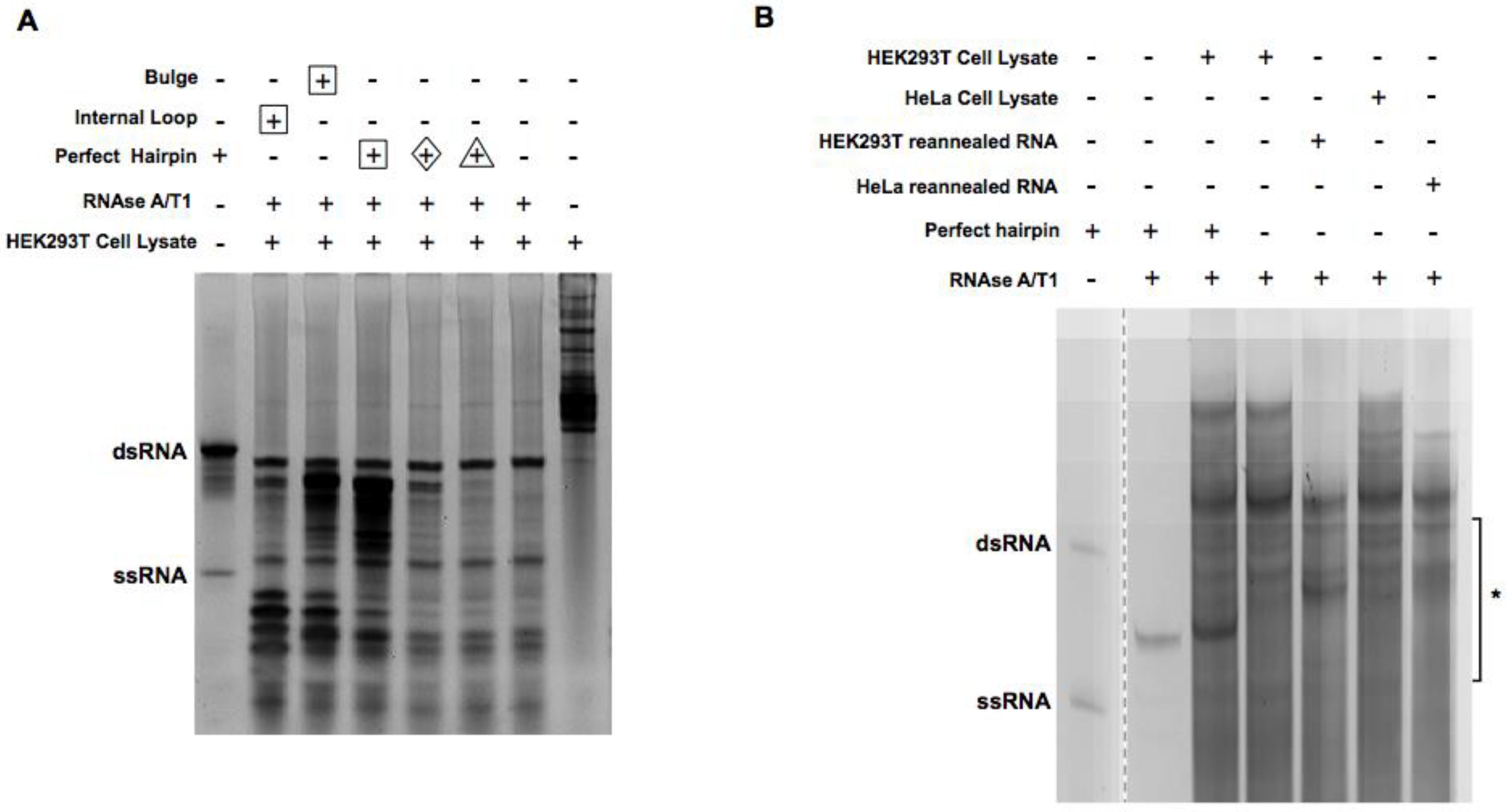
2.1. Digestion of Non-Perfect RNA Duplexes with Endoribonucleases
2.2. Enrichment of the Native Form of RNA Duplexes in Human Cells
2.3. Focus on Pronounced Band
2.4. The Effect of Removing Proteins
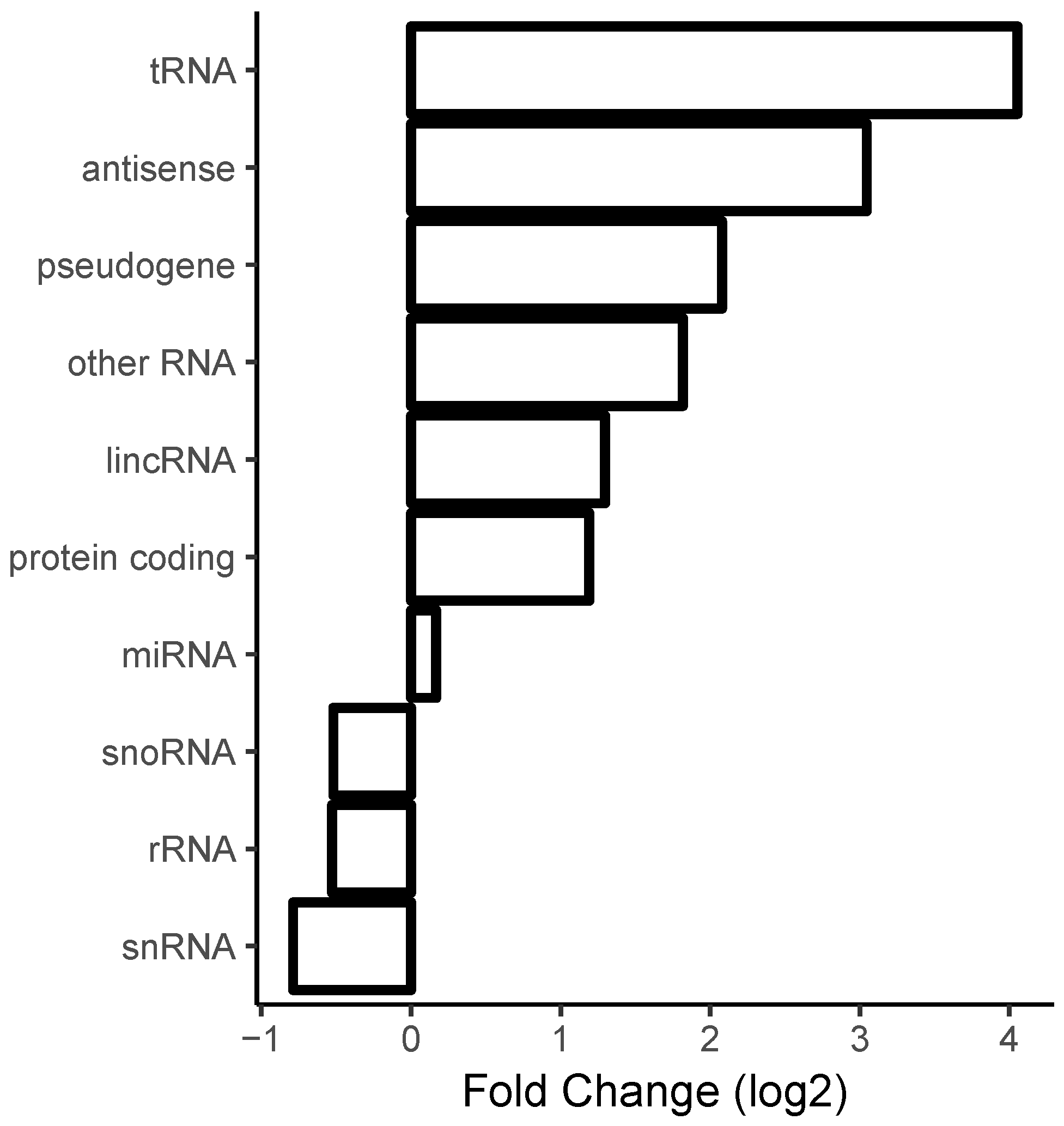
2.5. Correlation with Sense/Antisense Pairing
2.6. Evidence for Endogenous Ligation Events
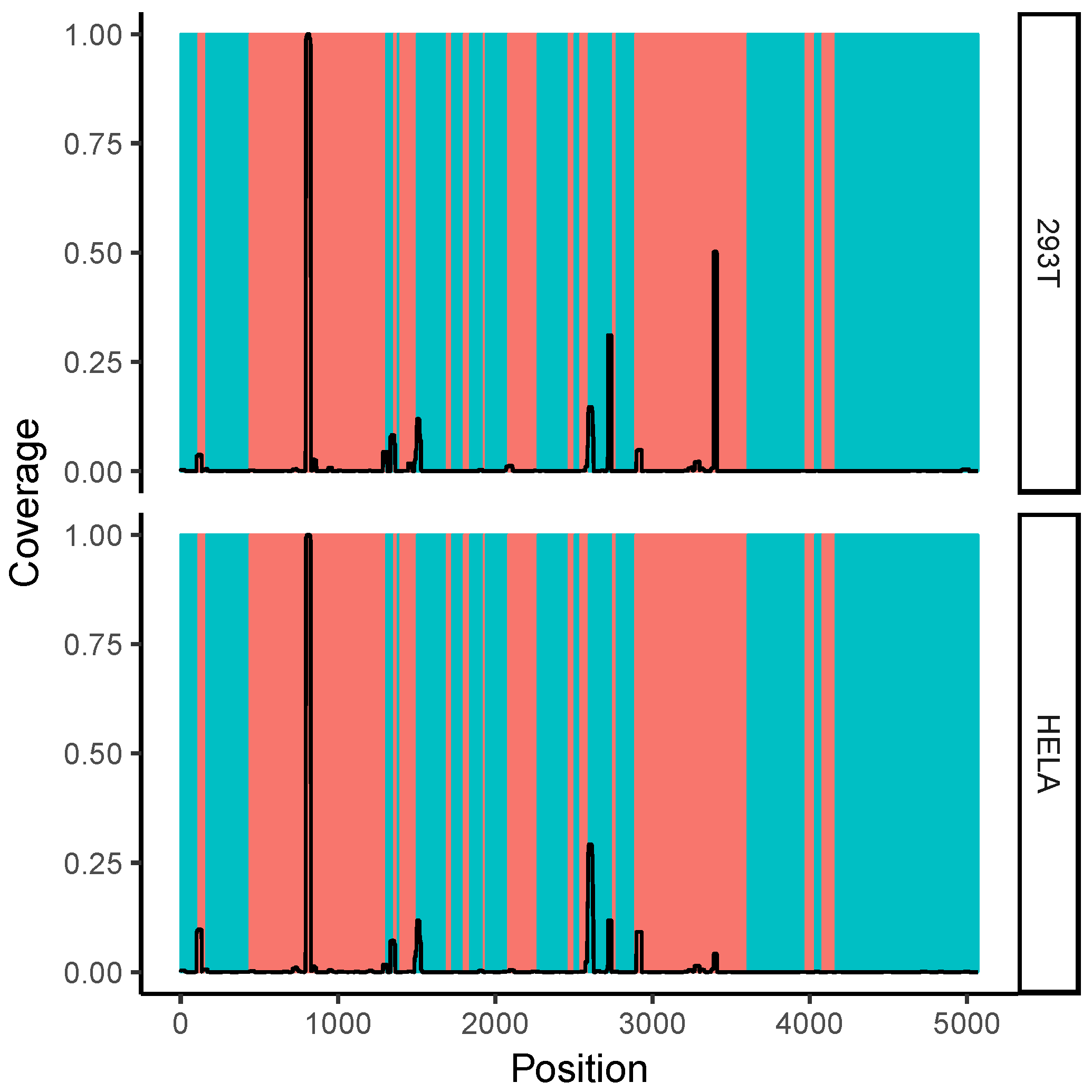
2.7. Correlation with Solvent Accessibility in the Ribosome
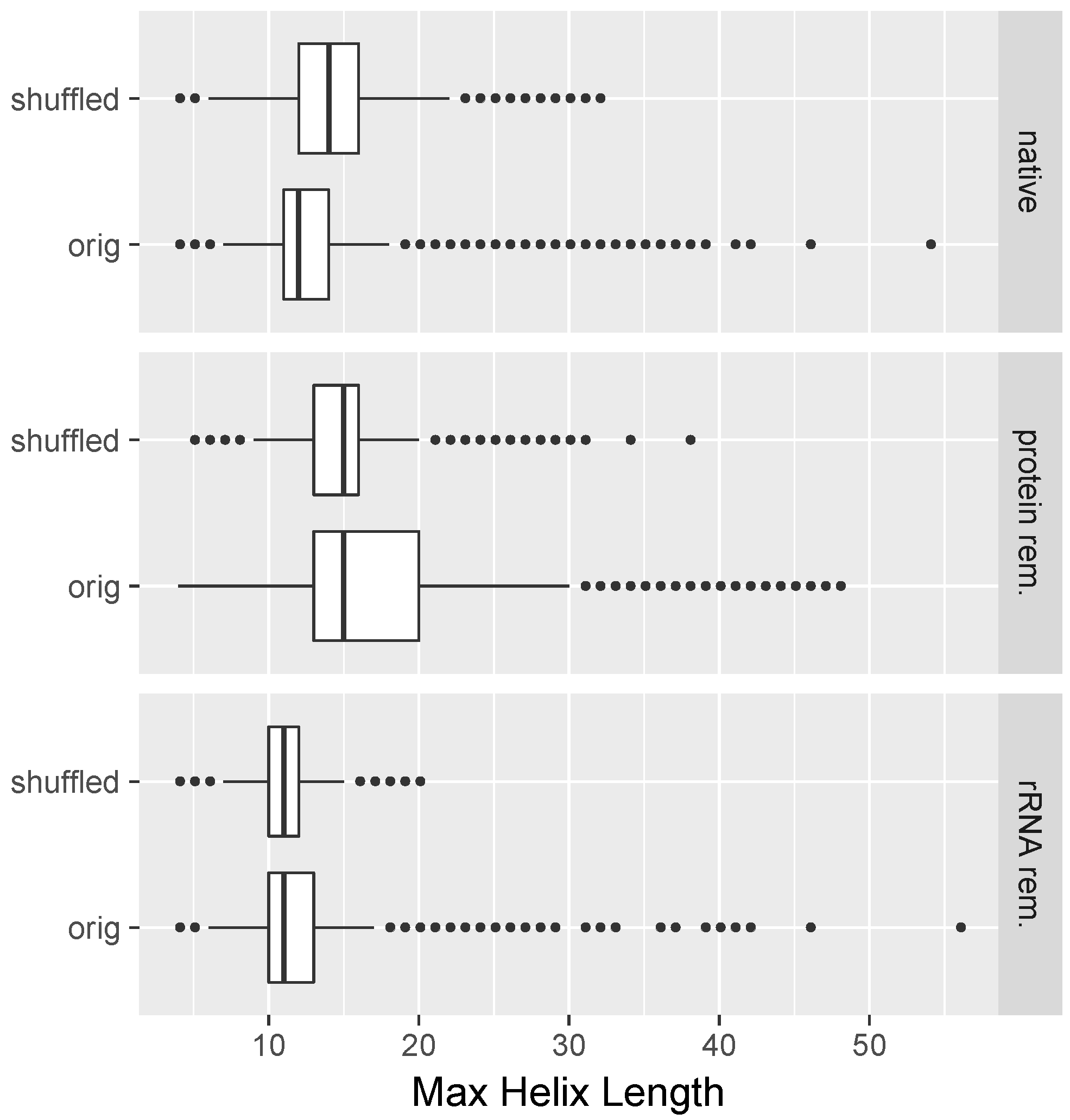
2.8. All-versus-All Search for Duplex Formation
3. Discussion
3.1. Long Perfectly Complementary Regions Are Relatively Uncommon
3.2. Ribosomal RNA Dominates RNA–RNA Interactions
3.3. Implications for the Design of RNA-Based Therapeutics
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Oligos
4.3. RNase A/T1 Digestion
4.4. Cellular RNA Duplex Enrichment
4.5. Cloning of dsRNAs and Deep Sequencing
4.5.1. Gel Extraction
4.5.2. 3′ Adaptor Ligation
4.5.3. 5′ Adaptor Ligation
4.5.4. RT-PCR
4.5.5. Deep Sequencing
4.6. Datasets
4.7. Data Analysis
4.8. Reference Data Set
4.9. Mapping of Reads to Transcripts
4.10. Search for Long Duplexes
4.11. Identifying Enrichment in Coverage
4.12. Search for Evidence of Endo-Ligases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Loughrey, D.; Watters, K.E.; Settle, A.H.; Lucks, J.B. SHAPE-Seq 2.0: Systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Kudla, G.; Granneman, S.; Hahn, D.; Beggs, J.D.; Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 10010–10015. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.; Qiu, R.; Shendure, J. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 2015, 33, 980–984. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Cook, K.B.; Hughes, T.R.; Morris, Q.D. High-throughput characterization of protein-RNA interactions. Briefgs. Funct. Genom. 2015, 14, 74–89. [Google Scholar] [CrossRef]
- Berglund, A.C.; Sjolund, E.; Ostlund, G.; Sonnhammer, E.L. InParanoid 6: Eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 2008, 36, 263–266. [Google Scholar] [CrossRef]
- Ray, D.; Kazan, H.; Cook, K.B.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Dieterich, C.; Wurmus, R.; Rajewsky, N.; Landthaler, M.; Akalin, A. DoRiNA 2.0—Upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2015, 43, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Khorshid, M.; Rodak, C.; Zavolan, M. CLIPZ: A database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 2011, 39, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Van Treeck, B.; Protter, D.S.W.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 2008, 381, 351–360. [Google Scholar] [CrossRef]
- Marchal, J.A.; Lopez, G.J.; Peran, M.; Comino, A.; Delgado, J.R.; Garcia-Garcia, J.A.; Conde, V.; Aranda, F.M.; Rivas, C.; Esteban, M.; et al. The impact of PKR activation: From neurodegeneration to cancer. FASEB J. 2014, 28, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Liao, C.Y.; Yang, Q.; Xie, X.Q.; Shu, H.B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med. 2017, 214, 973–989. [Google Scholar] [CrossRef]
- Afonin, K.A.; Kireeva, M.; Grabow, W.W.; Kashlev, M.; Jaeger, L.; Shapiro, B.A. Co-transcriptional assembly of chemically modified RNA nanoparticles functionalized with siRNAs. Nano. Lett. 2012, 12, 5192–5195. [Google Scholar] [CrossRef]
- Guo, P.; Zhang, C.; Chen, C.; Garver, K.; Trottier, M. Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol. Cell 1998, 2, 149–155. [Google Scholar] [CrossRef]
- Afonin, K.A.; Kasprzak, W.; Bindewald, E.; Puppala, P.S.; Diehl, A.R.; Hall, K.T.; Kim, T.J.; Zimmermann, M.T.; Jernigan, R.L.; Jaeger, L.; et al. Computational and experimental characterization of RNA cubic nanoscaffolds. Methods 2014, 67, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Bindewald, E.; Afonin, K.A.; Viard, M.; Zakrevsky, P.; Kim, T.; Shapiro, B.A. Multistrand Structure Prediction of Nucleic Acid Assemblies and Design of RNA Switches. Nano. Lett. 2016, 16, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Eyras, E.; Wu, J.; Khanna, A.; Josiah, S.; Rederstorff, M.; Zhang, M.Q.; Stamm, S. Direct cloning of double-stranded RNAs from RNase protection analysis reveals processing patterns of C/D box snoRNAs and provides evidence for widespread antisense transcript expression. Nucleic Acids Res. 2011, 39, 9720–9730. [Google Scholar] [CrossRef] [PubMed]
- Panek, J.; Kolar, M.; Vohradsky, J.; Shivaya Valasek, L. An evolutionary conserved pattern of 18S rRNA sequence complementarity to mRNA 5′ UTRs and its implications for eukaryotic gene translation regulation. Nucleic Acids Res. 2013, 41, 7625–7634. [Google Scholar] [CrossRef] [PubMed]
- Anger, A.M.; Armache, J.P.; Berninghausen, O.; Habeck, M.; Subklewe, M.; Wilson, D.N.; Beckmann, R. Structures of the human and Drosophila 80S ribosome. Nature 2013, 497, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.S.; Balasubramaniam, A.; Sallee, F.R.; Parker, S.L. The Expansion Segments of 28S Ribosomal RNA Extensively Match Human Messenger RNAs. Front. Genet. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.S.; Sallee, F.R.; Park, E.A.; Parker, S.L. Homoiterons and expansion in ribosomal RNAs. FEBS Open Biol. 2015, 5, 864–876. [Google Scholar] [CrossRef]
- He, Y.; Vogelstein, B.; Velculescu, V.E.; Papadopoulos, N.; Kinzler, K.W. The antisense transcriptomes of human cells. Science 2008, 322, 1855–1857. [Google Scholar] [CrossRef]
- Frellsen, J.; Moltke, I.; Thiim, M.; Mardia, K.V.; Ferkinghoff-Borg, J.; Hamelryck, T. A probabilistic model of RNA conformational space. PLoS Comput. Biol. 2009, 5, e1000406. [Google Scholar] [CrossRef]
- Umu, S.U.; Poole, A.M.; Dobson, R.C.; Gardner, P.P. Avoidance of stochastic RNA interactions can be harnessed to control protein expression levels in bacteria and archaea. eLife 2016, 5, e13479. [Google Scholar] [CrossRef]
- Song, R.; Hennig, G.W.; Wu, Q.; Jose, C.; Zheng, H.; Yan, W. Male germ cells express abundant endogenous siRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 13159–13164. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Regulation of mRNA transport, localization and translation in the nervous system of mammals (Review). Int. J. Mol. Med. 2014, 33, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2014, 505, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Zeng, J.M.; Huang, S.F.; Wang, X.Z.; Zhao, S.Q.; Bai, W.J.; Cao, W.X.; Huang, Z.G.; Feng, W.L. Selective leukemia cell death by activation of the double-stranded RNA-dependent protein kinase PKR. Int. J. Mol. Med. 2011, 28, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.R.; Ghanbariniaki, A.; Myong, S. RNA stem structure governs coupling of dicing and gene silencing in RNA interference. Proc. Natl. Acad. Sci. USA 2017, 114, E10349–E10358. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Cheng, H.; Ke, X.; Sun, L.; Zhang, Q.C.; Wang, H.W. Cryo-EM Structure of Human Dicer and Its Complexes with a Pre-miRNA Substrate. Cell 2018, 173, 1191–1203. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, 710–716. [Google Scholar] [CrossRef]
- Volders, P.J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, 4363–4364. [Google Scholar] [CrossRef]
- Juhling, F.; Morl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Putz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, 159–162. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ FIles. (Version 1.33) [Software]. Available online: https://github.com/najoshi/sickle (accessed on 10 November 2018).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bindewald, E.; Dai, L.; Kasprzak, W.K.; Kim, T.; Gu, S.; Shapiro, B.A. RNA–Protein Interactions Prevent Long RNA Duplex Formation: Implications for the Design of RNA-Based Therapeutics. Molecules 2018, 23, 3329. https://doi.org/10.3390/molecules23123329
Bindewald E, Dai L, Kasprzak WK, Kim T, Gu S, Shapiro BA. RNA–Protein Interactions Prevent Long RNA Duplex Formation: Implications for the Design of RNA-Based Therapeutics. Molecules. 2018; 23(12):3329. https://doi.org/10.3390/molecules23123329
Chicago/Turabian StyleBindewald, Eckart, Lisheng Dai, Wojciech K. Kasprzak, Taejin Kim, Shuo Gu, and Bruce A. Shapiro. 2018. "RNA–Protein Interactions Prevent Long RNA Duplex Formation: Implications for the Design of RNA-Based Therapeutics" Molecules 23, no. 12: 3329. https://doi.org/10.3390/molecules23123329
APA StyleBindewald, E., Dai, L., Kasprzak, W. K., Kim, T., Gu, S., & Shapiro, B. A. (2018). RNA–Protein Interactions Prevent Long RNA Duplex Formation: Implications for the Design of RNA-Based Therapeutics. Molecules, 23(12), 3329. https://doi.org/10.3390/molecules23123329