Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing

 , ,
, ,  and
and 
Abstract
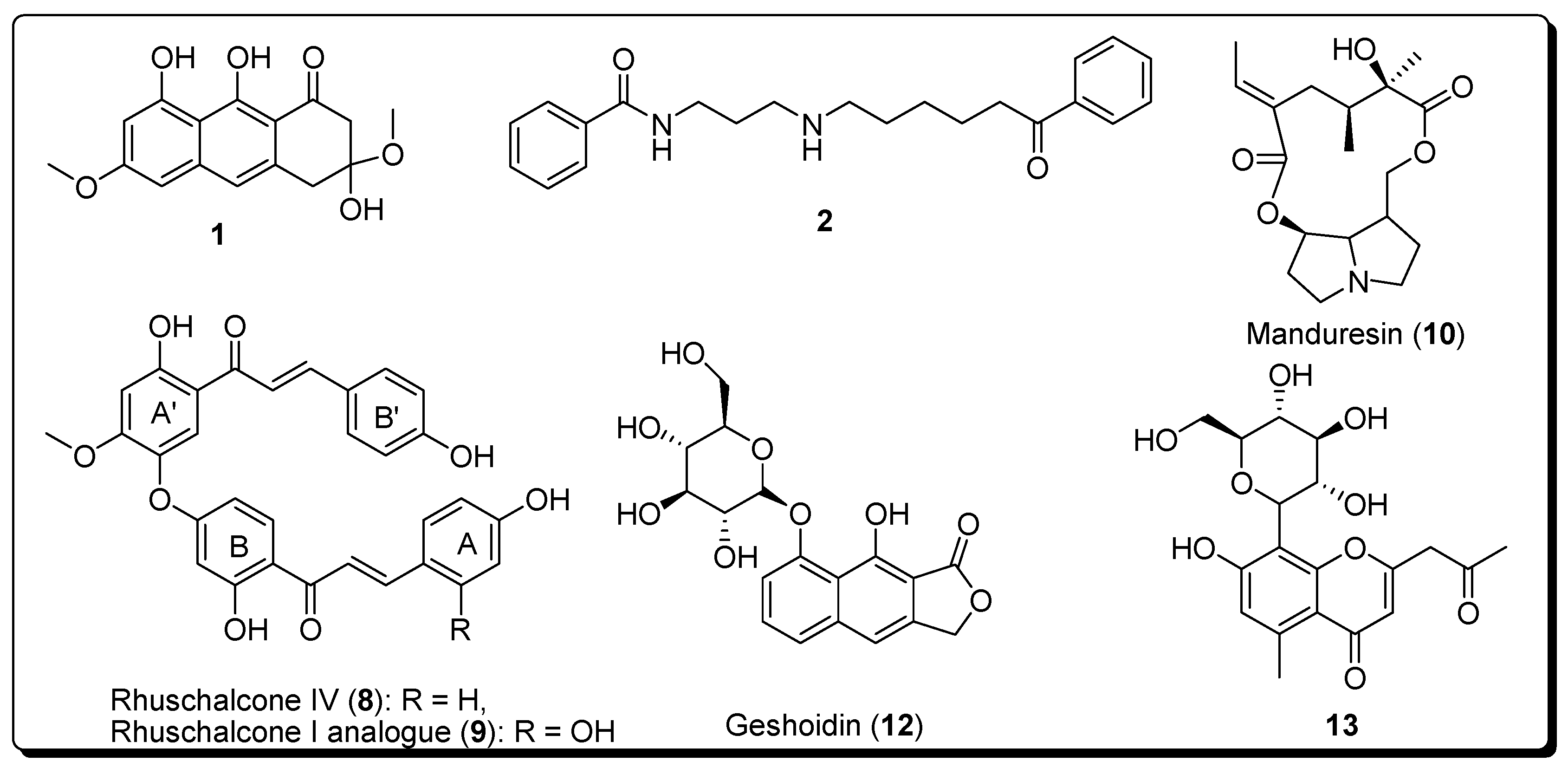
:
1. Introduction
2. Results
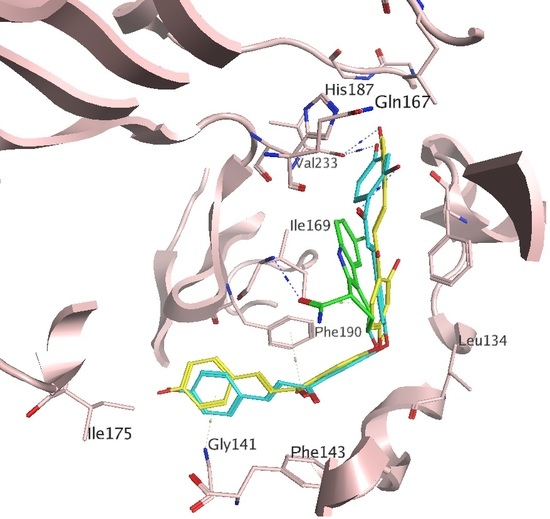
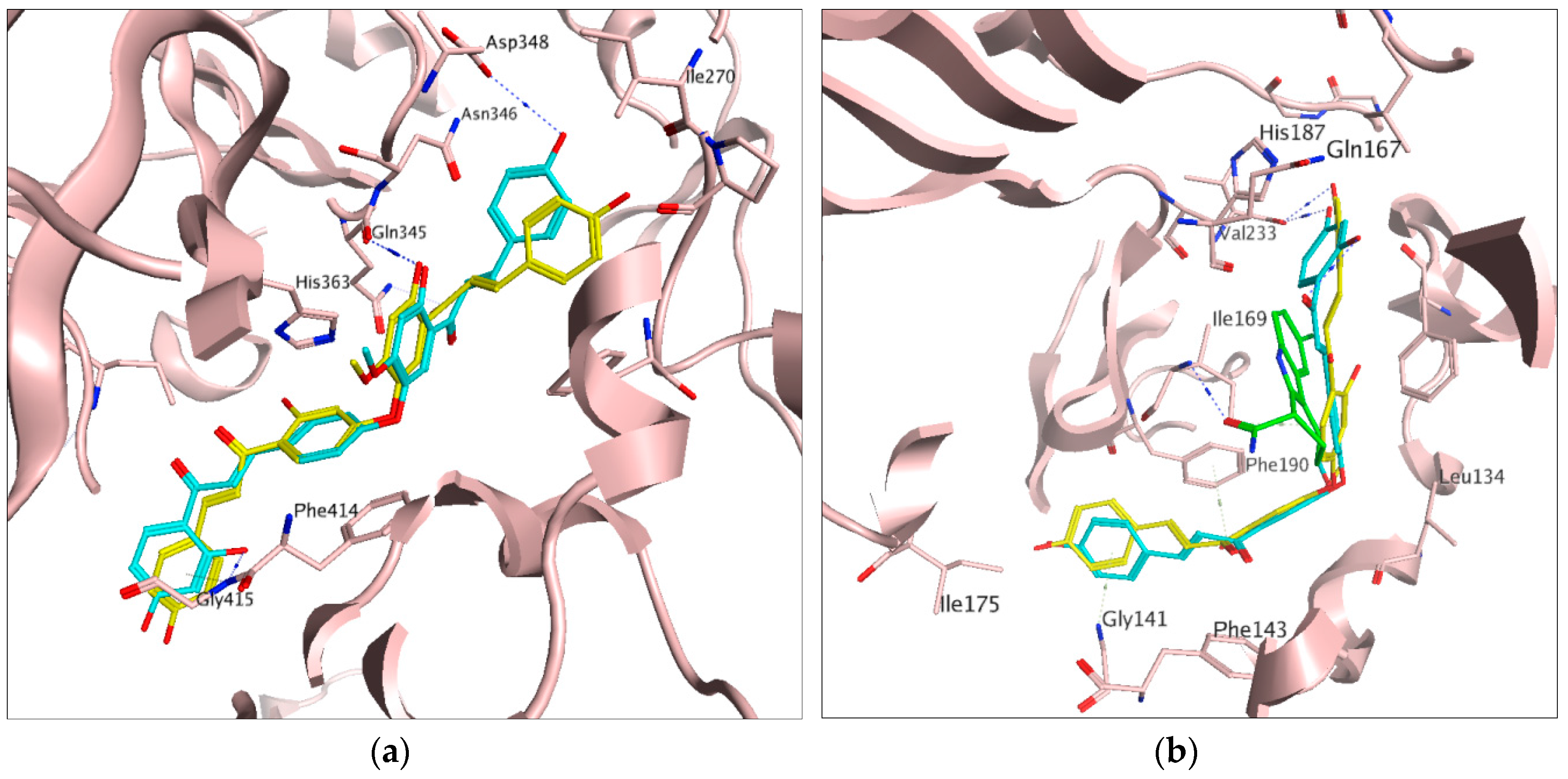
2.1. Docking Results
2.2. In Vitro Activities
3. Discussion
4. Materials and Methods
4.1. Database Preparation
4.2. Protein Preparation
4.3. Docking, Scoring and Hit Selection
4.4. In Vitro Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products as a screening resource. Curr. Opin. Chem. Biol. 2007, 11, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Nwodo, J.N.; Ibezim, A.; Simoben, C.V.; Karaman, B.; Ngwa, V.F.; Sippl, W.; Adikwu, M.U.; Mbaze, L.M. Molecular modeling of potential anticancer agents from African medicinal plants. J. Chem. Inf. Model. 2014, 54, 2433–2450. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Simoben, C.V.; Karaman, B.; Ngwa, V.F.; Judson, P.N.; Sippl, W.; Mbaze, L.M. Pharmacophore modeling and in silico toxicity assessment of potential anticancer agents from African medicinal plants. Drug Des. Dev. Ther. 2016, 10, 2137–2154. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef] [PubMed]
- Zofou, D.; Ntie-Kang, F.; Sippl, W.; Efange, S.M.N. Bioactive natural products derived from the Central African flora against neglected tropical diseases and HIV. Nat. Prod. Rep. 2013, 30, 1098–1120. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Onguéné, P.A.; Fotso, G.W.; Andrae-Marobela, K.; Bezabih, M.; Ndom, J.C.; Ngadjui, B.T.; Ogundaini, A.O.; Abegaz, B.M.; Meva’a, L.M. Virtualizing the p-ANAPL library: A step towards drug discovery from African medicinal plants. PLoS ONE 2014, 9, e90655. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, I.; Ntie-Kang, F.; Mwimanzi, P.; Onguéné, P.A.; Scull, M.A.; Idowu, T.O.; Ogundaini, A.O.; Meva’a, LM.; Abegaz, B.M.; Rice, C.M.; et al. Screening of the pan-African natural product library identifies ixoratannin A-2 and boldine as novel HIV-1 inhibitors. PLoS ONE 2015, 10, e0121099. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B. Finding potent sirt inhibitor in coffee: Isolation, confirmation and synthesis of javamide-II (N-caffeoyltryptophan) as sirt1/2 inhibitor. PLoS ONE 2016, 11, e0150392. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, P.; Kokkola, T.; Suuronen, T.; Poso, A.; Jarho, E.; Lahtela-Kakkonen, M. Virtual screening approach of sirtuin inhibitors results in two new scaffolds. Eur. J. Pharm. Sci. 2015, 76, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Salo, H.S.; Laitinen, T.; Poso, A.; Jarho, E.; Lahtela-Kakkonen, M. Identification of novel Sirt3 inhibitor scaffolds by virtual screening. Bioorg. Med. Chem. Lett. 2013, 23, 2990–2995. [Google Scholar] [CrossRef] [PubMed]
- Sacconnay, L.; Angleviel, M.; Randazzo, G.M.; Queiroz, M.M.; Queiroz, E.F.; Wolfender, J.L.; Carrupt, P.A.; Nurisso, A. Computational studies on sirtuins from Trypanosoma cruzi: Structures, conformations and interactions with phytochemicals. PLoS Negl. Trop. Dis. 2014, 8, e2689. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, H.; Zhu, H.; Leung, S.W. Ligand-based virtual screening and inductive learning for identification of Sirt1 inhibitors in natural products. Sci. Rep. 2016, 6, 19312. [Google Scholar] [CrossRef] [PubMed]
- Sacconnay, L.; Ryckewaert, L.; Randazzo, G.M.; Petit, C.; Passos Cdos, S.; Jachno, J.; Michailovienė, V.; Zubrienė, A.; Matulis, D.; Carrupt, P.A.; et al. 5-Benzylidene-hydantoin is a new scaffold for Sirt inhibition: From virtual screening to activity assays. Eur. J. Pharm. Sci. 2016, 85, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ji, S.; Yu, Z.Y.; Wang, H.L.; Cheng, X.; Li, W.J.; Jing, L.; Yu, Y.; Chen, Q.; Yang, L.L.; et al. Structure-based discovery of new selective small-molecule sirtuin 5 inhibitors. Chem. Biol. Drug Des. 2017, 91, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Ramu, M.; Mathur, S.; Unni, S.; Thiyagarajan, S. Identification of new inhibitors for human sirt1: An in-silico approach. Med. Chem. 2016, 12, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Pulla, V.K.; Sriram, D.S.; Viswanadha, S.; Sriram, D.; Yogeeswari, P. Energy-based pharmacophore and three-dimensional quantitative structure-activity relationship (3D-QSAR) modeling combined with virtual screening to identify novel small-molecule inhibitors of silent mating-type information regulation 2 homologue 1 (sirt1). J. Chem. Inf. Model. 2016, 56, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific Sirt1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Gallí, M.; van Gool, F.; Leo, O. Sirtuins and inflammation: Friends or foes? Biochem. Pharmacol. 2011, 81, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins link inflammation and metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. Sirt2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Sirt1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.M.; Huber, F.M.; Hoelz, A. Structural and functional analysis of human Sirt1. J. Mol. Biol. 2014, 426, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; He, B.; Zhang, X.; Lin, H.; Wang, Y. Sirt2 reverses 4-oxononanoyl lysine modification on histones. J. Am. Chem. Soc. 2016, 138, 12304–12307. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-oxadiazoles as potent and selective inhibitors of the human deacetylase sirtuin 2: Structure-activity relationship, X-ray crystal structure, and anticancer activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef] [PubMed]
- Sundriyal, S.; Moniot, S.; Mahmud, Z.; Yao, S.; Di Fruscia, P.; Reynolds, C.R.; Dexter, D.T.; Sternberg, M.J.; Lam, E.W.; Steegborn, C.; et al. Thienopyrimidinone based sirtuin-2 (sirt2)-selective inhibitors bind in the ligand induced selectivity pocket. J. Med. Chem. 2017, 60, 1928–1945. [Google Scholar] [CrossRef] [PubMed]
- Knyphausen, P.; de Boor, S.; Kuhlmann, N.; Scislowski, L.; Extra, A.; Baldus, L.; Schacherl, M.; Baumann, U.; Neundorf, I.; Lammers, M. Insights into lysine deacetylation of natively folded substrate proteins by sirtuins. J. Biol. Chem. 2016, 291, 14677–14694. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-based development of an affinity probe for sirtuin2. Angew. Chem. Int. Ed. Engl. 2016, 55, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as potent and selective sirt2 inhibitors: A structure-activity relationship study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Kudo, N.; Thelen, J.N.; Ito, A.; Yoshida, M.; Denu, J.M. Kinetic and structural basis for acyl-group selectivity and NAD(+) dependence in sirtuin-catalyzed deacylation. Biochemistry 2015, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Gerhardt, S.; Einsle, O.; Jung, M. Seeding for sirtuins: Microseed matrix seeding to obtain crystals of human Sirt3 and Sirt2 suitable for soaking. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.B.; Jing, H.; Aramsangtienchai, P.; He, B.; Khan, S.; Hu, J.; Lin, H.; Hao, Q. Efficient demyristoylase activity of Sirt2 revealed by kinetic and structural studies. Sci. Rep. 2015, 5, 8529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagata, K.; Goto, Y.; Nishimasu, H.; Morimoto, J.; Ishitani, R.; Dohmae, N.; Takeda, N.; Nagai, R.; Komuro, I.; Suga, H.; et al. Structural basis for potent inhibition of Sirt2 deacetylase by a macrocyclic peptide inducing dynamic structural change. Structure 2014, 22, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Pavletich, N.P. Structure of the histone deacetylase Sirt2. Nat. Struct. Biol. 2001, 8, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 Å crystal structure of the Sirt1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J. Med. Chem. 2013, 56, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule Sirt1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef] [PubMed]
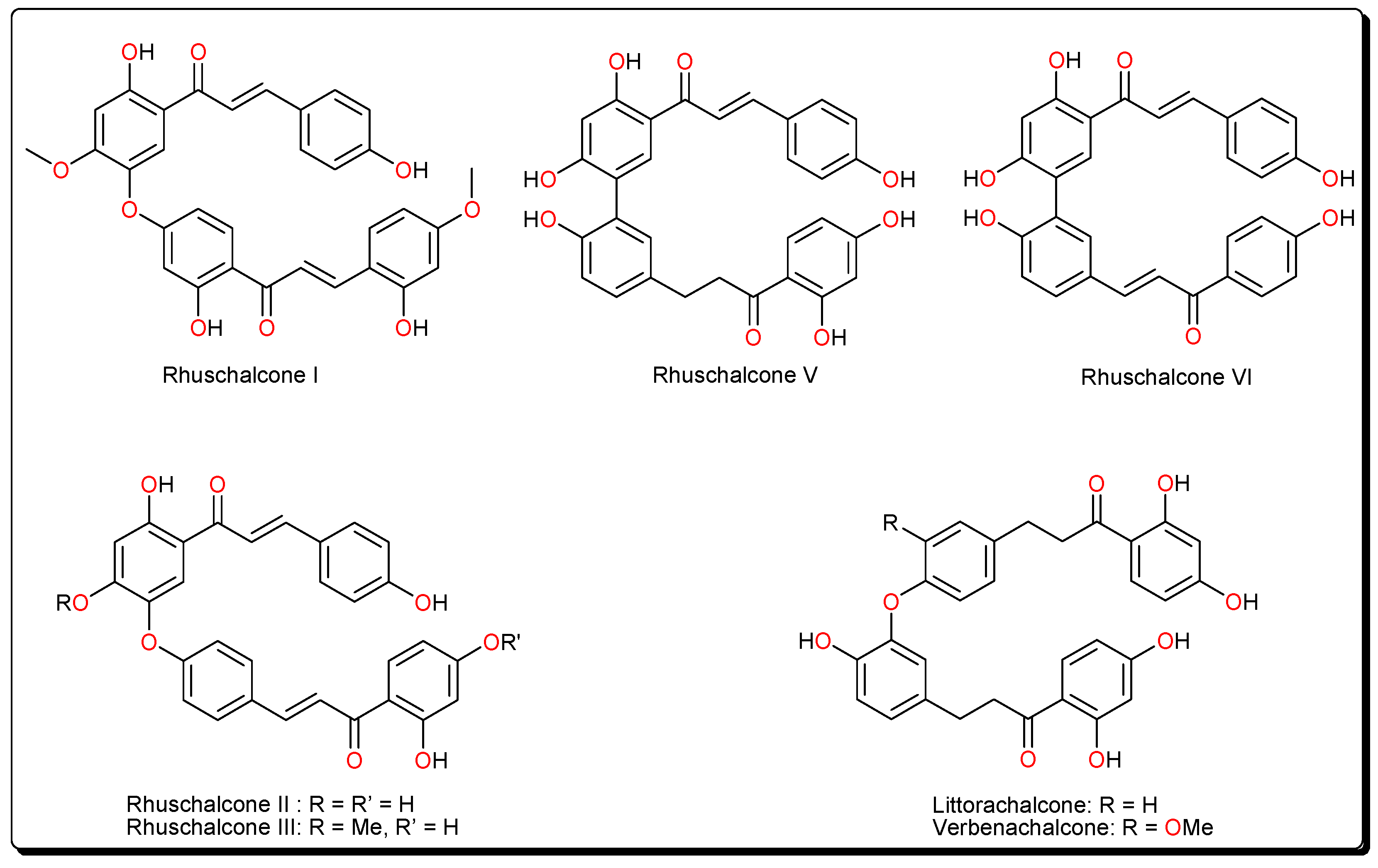
- Masesane, I.B.; Yeboah, S.O.; Liebscher, J.; Mügge, C.; Abegaz, B.M. A bichalcone from the twigs of Rhus pyroides. Phytochemistry 2000, 53, 1005–1008. [Google Scholar] [CrossRef]
- Mdee, L.K.; Yeboah, S.O.; Abegaz, B.M. Rhuschalcones II-VI, five new bichalcones from the root bark of Rhus pyroides. J. Nat. Prod. 2003, 66, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Shetonde, O.; Mdee, L.; Bezibih, M.; Mammo, W.; Abegaz, B. The characterization, total synthesis and antiprotozoal activities of novel bichalcones from Rhus pyroides. Planta Med. 2009, 75, SL11. [Google Scholar] [CrossRef]
- Mihigo, S.O.; Mammo, W.; Bezabih, M.; Andrae-Marobela, K.; Abegaz, B.M. Total synthesis, antiprotozoal and cytotoxicity activities of rhuschalcone VI and analogs. Bioorg. Med. Chem. 2010, 18, 2464–2473. [Google Scholar] [CrossRef] [PubMed]
- Arslan, T.; Çelik, G.; Çelik, H.; Sentürk, M.; Yayli, N.; Ekinci, D. Synthesis and biological evaluation of novel bischalcone derivatives as carbonic anhydrase inhibitors. Arch. Pharm. Chem. Life Sci. 2016, 349, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Svenningsen, A.B.; Madsen, K.D.; Liljefors, T.; Stafford, G.I.; van Staden, J.; Jäger, A.K. Biflavones from Rhus species with affinity for the GABAA/benzodiazepine receptor. J. Ethnopharmacol. 2006, 103, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Yang, Y.; Guan, X. The synthesis of bichalcone rhuschalcone I. Chin. J. Appl. Chem. 2011, 28, 652–656. [Google Scholar]
- Kahyo, T.; Ichikawa, S.; Hatanaka, T.; Yamada, M.K.; Setou, M. A novel chalcone polyphenol inhibits the deacetylase activity of Sirt1 and cell growth in HEK293T cells. J. Pharmacol. Sci. 2008, 108, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment, version 2014; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014.
- LigPrep Software; Version 2; LLC: New York, NY, USA, 2011.
- Halgren, T.A. Merck molecular forcefield. J. Comput. Chem. 1996, 17, 490–641. [Google Scholar] [CrossRef]
- Canvas, Schrödinger, version 3.1.011; LLC: New York, NY, USA, 2017.
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of Pan Assay Interference Compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Maestro; Version 9.2; LLC: New York, NY, USA, 2011.
- Glideprogram (Schrödinger Suite 2012-5.8), version 5.8; Schrodinger: New York, NY, USA, 2012.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Heltweg, B.; Trapp, J.; Jung, M. In vitro assays for the determination of histone deacetylase activity. Methods 2005, 36, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A resource for natural products from Northern African sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 8 and 9 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | Sirt 1 (µM) | Sirt 2 (µM) | Sirt 3 (µM or % Inhibition) |
|---|---|---|---|
| 1 b | n.d. c | n.d. c | n.d. c |
| 2 | n.i. a | n.i. a | n.i. a |
| 8 | 46.7 ± 6.0 | 48.5 ± 39.5 | 38% |
| 9 | 40.8 ± 8.5 | 44.8 ± 5.1 | 23% |
| 10 | n.i. a | n.i. a | n.i. a |
| 12 b | n.d. | n.d. | n.d. |
| 13 | n.i. a | n.i. a | n.i. a |
| NA | 142.4 ± 9.1 | 49.8 ± 4.6 | 67.9 ± 3.3 |
| EX-527 | 1.4 ± 0.1 | 10.6 ± 1.1 | 19% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karaman, B.; Alhalabi, Z.; Swyter, S.; Mihigo, S.O.; Andrae-Marobela, K.; Jung, M.; Sippl, W.; Ntie-Kang, F. Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing. Molecules 2018, 23, 416. https://doi.org/10.3390/molecules23020416
Karaman B, Alhalabi Z, Swyter S, Mihigo SO, Andrae-Marobela K, Jung M, Sippl W, Ntie-Kang F. Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing. Molecules. 2018; 23(2):416. https://doi.org/10.3390/molecules23020416
Chicago/Turabian StyleKaraman, Berin, Zayan Alhalabi, Sören Swyter, Shetonde O. Mihigo, Kerstin Andrae-Marobela, Manfred Jung, Wolfgang Sippl, and Fidele Ntie-Kang. 2018. "Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing" Molecules 23, no. 2: 416. https://doi.org/10.3390/molecules23020416
APA StyleKaraman, B., Alhalabi, Z., Swyter, S., Mihigo, S. O., Andrae-Marobela, K., Jung, M., Sippl, W., & Ntie-Kang, F. (2018). Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing. Molecules, 23(2), 416. https://doi.org/10.3390/molecules23020416