Regulating Cdc42 and Its Signaling Pathways in Cancer: Small Molecules and MicroRNA as New Treatment Candidates

 ,
,
Abstract
:1. Introduction
2. The Expression of Cdc42 in Cancer
3. Regulators of Cdc42
3.1. Guanine Nucleotide Exchange Factors (GEFs)
3.2. GTPase-Activating Proteins (GAPs)
3.3. Guanosine Nucleotide Dissociation Inhibitors (GDIs)
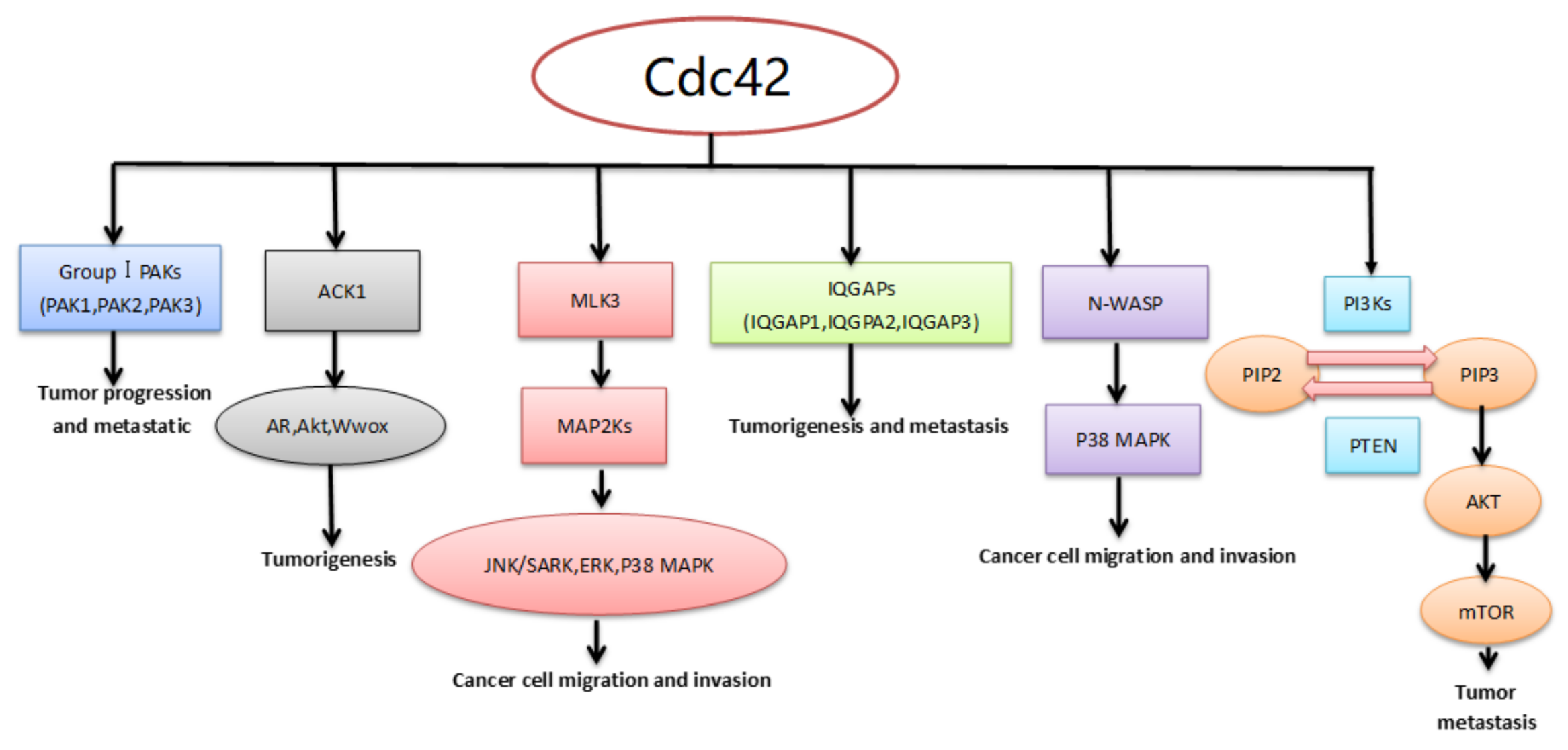
4. Downstream Effector/Adaptor Proteins of Cdc42 in Cancer
4.1. Activated Cdc42 Kinase 1 (ACK1)
4.2. P21-Activated Kinases (PAKs)
4.3. Mixed-Lineage Kinase 3 (MLK3)
4.4. Isoleucine-Glutamine-Motif Containing GTPase-Activating Proteins (IQGAPS)
4.5. Neural Wiskott–Aldrich Syndrome Protein (N-WASP)
4.6. Phosphoinositide 3-Kinases (PI3Ks)
5. Small Molecule Inhibitors of Cdc42
5.1. ZCL278
5.2. Secramine
5.3. CASIN
5.4. AZA197 and AZA1
5.5. ML141
5.6. PAK1-Specific Inhibitors
6. The Role of MicroRNAs in Cancer by Targeting Cdc42
6.1. The Role of MicroRNAs in Lung Cancer by Targeting Cdc42
6.2. The Role of MicroRNAs in Breast Cancer by Targeting Cdc42
6.3. The Role of MicroRNA-195 Targeting of Cdc42 in Esophageal Squamous Cell Carcinoma (ESCC)
6.4. The Role of MicroRNAs in Gastric Cancer by Targeting Cdc42
6.5. The Role of MicroRNAs in Colorectal Cancer by Targeting Cdc42
6.6. The Role of MicroRNAs in the Downstream Signaling Pathways of Cdc42
6.7. The Role of MicroRNA-224 in Hepatocellular Carcinoma by Targeting Cdc42
7. Concluding Remarks and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Melendez, J.; Grogg, M.; Zheng, Y. Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 2011, 286, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Cotteret, S.; Chernoff, J. The evolutionary history of effectors downstream of Cdc42 and Rac. Genome Biol. 2002, 3, Reviews0002. [Google Scholar] [CrossRef] [PubMed]
- Arias-Romero, L.E.; Chernoff, J. Targeting Cdc42 in cancer. Expert Opin. Ther. Targets 2013, 17, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.F.; Lim, W.A. The minimal autoinhibited unit of the guanine nucleotide exchange factor intersectin. PLoS ONE 2010, 5, e11291. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bi, F.; Zhou, X.; Zheng, Y. Rho gtpase regulation by mirnas and covalent modifications. Trends Cell Biol. 2012, 22, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. Cosmic: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Fidyk, N.; Wang, J.B.; Cerione, R.A. Influencing cellular transformation by modulating the rates of gtp hydrolysis by Cdc42. Biochemistry 2006, 45, 7750–7762. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, R.; Salem, O.; Krizova, H.; Mcfeeters, R.; Adams, P.D. A switch I mutant of Cdc42 exhibits decreased conformational freedom. Biochemistry 2011, 50, 6196–6207. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, B.J.; Zhou, H.; Lu, Q. Cdc42 signaling pathway inhibition as a therapeutic target in ras- related cancers. Curr. Med. Chem. 2017, 24, 3485–3507. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R.; Givan, S.A.; Cullen, P.; Sprague, G.F., Jr. Gtpase-activating proteins for Cdc42. Eukaryot. Cell 2002, 1, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Itoh, R.E.; Kurokawa, K.; Ohba, Y.; Yoshizaki, H.; Mochizuki, N.; Matsuda, M. Activation of Rac and Cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol. Cell. Biol. 2002, 22, 6582–6591. [Google Scholar] [CrossRef] [PubMed]
- Rameh, L.E.; Arvidsson, A.; Carraway, K.L., 3rd; Couvillon, A.D.; Rathbun, G.; Crompton, A.; VanRenterghem, B.; Czech, M.P.; Ravichandran, K.S.; Burakoff, S.J.; et al. A comparative analysis of the phosphoinositide binding specificity of pleckstrin homology domains. J. Biol. Chem. 1997, 272, 22059–22066. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.L.; Hall, A. Rho gtpases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. Web-based molecular graphics for large complexes. In Proceedings of the ACM 21st International Conference on Web3D Technology, Anaheim, CA, USA, 22–24 July 2016; pp. 185–186. [Google Scholar]
- Hoffman, G.R.; Nassar, N.; Cerione, R.A. Structure of the rho family gtp-binding protein Cdc42 in complex with the multifunctional regulator rhogdi. Cell 2000, 100, 345–356. [Google Scholar] [CrossRef]
- Mahajan, K.; Challa, S.; Coppola, D.; Lawrence, H.; Luo, Y.; Gevariya, H.; Zhu, W.; Chen, Y.A.; Lawrence, N.J.; Mahajan, N.P. Effect of ACK1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate 2010, 70, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.P.; Liu, Y.; Majumder, S.; Warren, M.R.; Parker, C.E.; Mohler, J.L.; Earp, H.S.; Whang, Y.E. Activated Cdc42-associated kinase ACK1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl. Acad. Sci. USA 2007, 104, 8438–8443. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated tyrosine kinase ACK1 promotes prostate tumorigenesis: Role of ACK1 in polyubiquitination of tumor suppressor WWOX. Cancer Res. 2005, 65, 10514–10523. [Google Scholar] [CrossRef] [PubMed]
- Howlin, J.; Rosenkvist, J.; Andersson, T. TNK2 preserves epidermal growth factor receptor expression on the cell surface and enhances migration and invasion of human breast cancer cells. Breast Cancer Res. BCR 2008, 10, R36. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, E.H.; Degenhardt, Y.Y.; Strelow, A.; Slavin, A.; Chinn, L.; Orf, J.; Rong, M.; Li, S.; See, L.H.; Nguyen, K.Q.C.; et al. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc. Natl. Acad. Sci. USA 2005, 102, 15901–15906. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Echague, V.; Gucwa, A.; Craddock, B.P.; Brown, D.A.; Miller, W.T. Cancer-associated mutations activate the nonreceptor tyrosine kinase ACK1. J. Biol. Chem. 2010, 285, 10605–10615. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xu, J.; Yin, M.X.; Zhang, L.; Lu, Y.; Wu, W.; Xue, Z.; Ho, M.S.; Gao, G.; Zhao, Y.; et al. Ack promotes tissue growth via phosphorylation and suppression of the hippo pathway component expanded. Cell Discov. 2016, 2, 15047. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, H.; Xia, L.; Oyang, L.; Zhou, Y.; Zhang, B.; Chen, X.; Luo, X.; Liao, Q.; Liang, J. Overexpression of PAK1 correlates with aberrant expression of emt markers and poor prognosis in non-small cell lung cancer. J. Cancer 2017, 8, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Al-Azayzih, A.; Gao, F.; Somanath, P.R. P21 activated kinase-1 mediates transforming growth factor Beta1-induced prostate cancer cell epithelial to mesenchymal transition. Biochim. Biophys. Acta 2015, 1853, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Q.; Song, Y.; Wang, X.; Guo, Q.; Zhang, J.; Li, J.; Han, Y.; Miao, Z.; Li, F. Pak1 regulates RUFY3-mediated gastric cancer cell migration and invasion. Cell Death Dis. 2015, 6, e1682. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Song, Y.; Liu, T.; Wang, C.; Zhang, Q.; Liu, F.; Cai, X.; Miao, Z.; Xu, H.; Xu, H.; et al. PAK1-mediated MORC2 phosphorylation promotes gastric tumorigenesis. Oncotarget 2015, 6, 9877–9886. [Google Scholar] [CrossRef] [PubMed]
- Al-Maghrabi, J.; Emam, E.; Gomaa, W.; Al-Qaydy, D.; Al-Maghrabi, B.; Buhmeida, A.; Abuzenadah, A.; Al-Qahtani, M.; Al-Ahwal, M. Overexpression of PAK-1 is an independent predictor of disease recurrence in colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 15895–15902. [Google Scholar] [PubMed]
- Parvathy, M.; Sreeja, S.; Kumar, R.; Pillai, M.R. Potential role of p21 activated kinase 1 (PAK1) in the invasion and motility of oral cancer cells. BMC Cancer 2016, 16 (Suppl. S1), 293. [Google Scholar] [CrossRef] [PubMed]
- Yanase, S.; Luo, Y.; Maruta, H. PAK1-deficiency/down-regulation reduces brood size, activates HSP16.2 gene and extends lifespan in caenorhabditis elegans. Drug Discov. Ther. 2013, 7, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Maruta, H. Herbal therapeutics that block the oncogenic kinase PAK1: A practical approach towards PAK1-dependent diseases and longevity. Phytother. Res. PTR 2014, 28, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ma, T.; Pang, L.; Xie, R. Activation of P21-activated protein kinase 2 is an independent prognostic predictor for patients with gastric cancer. Diagn. Pathol. 2014, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.W.; Wu, L.; Bu, L.L.; Liu, J.F.; Li, Y.C.; Ma, S.R.; Yu, G.T.; Mao, L.; Zhang, W.F.; Sun, Z.J. PAK2 promotes migration and proliferation of salivary gland adenoid cystic carcinoma. Am. J. Transl. Res. 2016, 8, 3387–3397. [Google Scholar] [PubMed]
- Flate, E.; Stalvey, J.R. Motility of select ovarian cancer cell lines: Effect of extra-cellular matrix proteins and the involvement of PAK2. Int. J. Oncol. 2014, 45, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Ding, B.; Agler, M.; Cockett, M.; McPhee, F. Lethality of PAK3 and SGK2 shrnas to human papillomavirus positive cervical cancer cells is independent of PAK3 and SGK2 knockdown. PLoS ONE 2015, 10, e0117357. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.Y.; Ching, Y.P. The role of P21-activated kinases in hepatocellular carcinoma metastasis. J. Mol. Signal. 2014, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, G.C.; Tangney, M.; Casey, G.; Ambrose, M.; Houston, A.; Barry, O.P. Modulation of P21-activated kinase 1 alters the behavior of renal cell carcinoma. Int. J. Cancer 2007, 121, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of P21-activated kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Nicholas, N.S.; Wells, C.M. Role of P-21-activated kinases in cancer progression. Int. Rev. Cell Mol. Biol. 2014, 309, 347–387. [Google Scholar] [PubMed]
- Parekh, P.; Rao, K.V. Overexpression of cyclin d1 is associated with elevated levels of map kinases, Akt and PAK1 during diethylnitrosamine-induced progressive liver carcinogenesis. Cell Biol. Int. 2007, 31, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Rattanasinchai, C.; Gallo, K.A. MLK3 signaling in cancer invasion. Cancers 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, J.; Guo, Q.; Wang, Y.; Zhou, Y.; Peng, H.; Cheng, M.; Zhao, D.; Li, F. LCH-7749944, a novel and potent P21-activated kinase 4 inhibitor, suppresses proliferation and invasion in human gastric cancer cells. Cancer Lett. 2012, 317, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Miller, E.M.; Gallo, K.A. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene 2010, 29, 4399–4411. [Google Scholar] [CrossRef] [PubMed]
- Rattanasinchai, C.; Llewellyn, B.J.; Conrad, S.E.; Gallo, K.A. MLK3 regulates FRA-1 and mmps to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017, 6, e345. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Yu, M.C.; Tung, W.H.; Chen, T.T.; Yu, C.C.; Weng, C.M.; Tsai, Y.J.; Bai, K.J.; Hong, C.Y.; Chien, M.H.; et al. Connective tissue growth factor induces collagen I expression in human lung fibroblasts through the RAC1/MLK3/JNK/AP-1 pathway. Biochim. Biophys. Acta 2013, 1833, 2823–2833. [Google Scholar] [CrossRef] [PubMed]
- Blessing, N.A.; Brockman, A.L.; Chadee, D.N. The E3 ligase chip mediates ubiquitination and degradation of mixed-lineage kinase 3. Mol. Cell. Biol. 2014, 34, 3132–3143. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Abi Saab, W.F.; Modi, N.; Stewart, A.M.; Liu, J.; Chadee, D.N. Mixed lineage kinase 3 is required for matrix metalloproteinase expression and invasion in ovarian cancer cells. Exp. Cell Res. 2012, 318, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.F.; Allen, G.; Timson, D.J. Biochemical analysis of the interactions of IQGAP1 c-terminal domain with Cdc42. World J. Biol.Chem. 2012, 3, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.D.; Brown, M.D.; Sacks, D.B. Iqgaps in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Xie, C.; Lin, X.; Zhao, Y.; Han, Y.; Fan, C.; Zhang, X.; Du, J.; Han, Y.; Han, Q.; et al. Coexpression of iq-domain gtpase-activating protein 1 (IQGAP1) and dishevelled (DVL) is correlated with poor prognosis in non-small cell lung cancer. PLoS ONE 2014, 9, e113713. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, D.; Morales, M.; Maeso, M.D.C.; Garcia, M.D.P.; Gutierrez, R.; Valladares, F.; Avila, J.; Diaz-Flores, L.; Mobasheri, A.; Martin-Vasallo, P. Alterations in IQGAP1 expression and localization in colorectal carcinoma and liver metastases following oxaliplatin-based chemotherapy. Oncol. Lett. 2017, 14, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- White, C.D.; Khurana, H.; Gnatenko, D.V.; Li, Z.; Odze, R.D.; Sacks, D.B.; Schmidt, V.A. IQGAP1 and IQGAP2 are reciprocally altered in hepatocellular carcinoma. BMC Gastroenterol. 2010, 10, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Liu, D.; Bojdani, E.; El-Naggar, A.K.; Vasko, V.; Xing, M. IQGAP1 plays an important role in the invasiveness of thyroid cancer. Clin. Cancer Res. 2010, 16, 6009–6018. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Li, X.Z.; Zhai, L.Q.; Liu, Z.R.; Chen, X.J.; Pei, Y. Overexpression of IQGAP1 in human pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 540–545. [Google Scholar] [CrossRef]
- Schmidt, V.A.; Chiariello, C.S.; Capilla, E.; Miller, F.; Bahou, W.F. Development of hepatocellular carcinoma in IQGAP2-deficient mice is iqgap1 dependent. Mol. Cell. Biol. 2008, 28, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Jessen, W.J.; Al-Ahmadie, H.; Serio, A.M.; Lin, Y.; Shih, W.J.; Reuter, V.E.; Scardino, P.T.; Shen, M.M.; Aronow, B.J.; et al. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008, 68, 2132–2144. [Google Scholar] [CrossRef] [PubMed]
- Frugtniet, B.; Jiang, W.G.; Martin, T.A. Role of the wasp and wave family proteins in breast cancer invasion and metastasis. Breast Cancer (Dove Med. Press) 2015, 7, 99–109. [Google Scholar] [PubMed]
- Hou, J.; Yang, H.; Huang, X.; Leng, X.; Zhou, F.; Xie, C.; Zhou, Y.; Xu, Y. N-wasp promotes invasion and migration of cervical cancer cells through regulating P38 mapks signaling pathway. Am. J. Transl. Res. 2017, 9, 403–415. [Google Scholar] [PubMed]
- Wang, W.S.; Zhong, H.J.; Xiao, D.W.; Huang, X.; Liao, L.D.; Xie, Z.F.; Xu, X.E.; Shen, Z.Y.; Xu, L.Y.; Li, E.M. The expression of CFL1 and N-WASP in esophageal squamous cell carcinoma and its correlation with clinicopathological features. Dis. Esophagus 2010, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Ziv-Av, A.; Giladi, N.D.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Pauker, M.H.; Barda-Saad, M.; Poisson, L.; Brodie, C. RTVP-1 regulates glioma cell migration and invasion via interaction with N-WASP and HNRNPK. Oncotarget 2015, 6, 19826–19840. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Pereira, G.; Watkins, G.; Mansel, R.E.; Jiang, W.G. N-WASP is a putative tumour suppressor in breast cancer cells, in vitro and in vivo, and is associated with clinical outcome in patients with breast cancer. Clin. Exp. Metastasis 2008, 25, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Pichot, C.S.; Arvanitis, C.; Hartig, S.M.; Jensen, S.A.; Bechill, J.; Marzouk, S.; Yu, J.; Frost, J.A.; Corey, S.J. Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASP. Cancer Res. 2010, 70, 8347–8356. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Flamini, M.I.; Baldacci, C.; Goglia, L.; Genazzani, A.R.; Simoncini, T. Estrogen receptor-alpha promotes breast cancer cell motility and invasion via focal adhesion kinase and N-WASP. Mol. Endocrinol. 2010, 24, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, S.J.; Ferguson, D.T.; Mitchell, C.A.; Ooms, L.M. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of gtpases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.M.; Lengauer, C. Pten-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Kinase-dependent and -independent functions of the p110β phosphoinositide-3-kinase in cell growth, metabolic regulation and oncogenic transformation. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Dbouk, H.A.; Khalil, B.D.; Wu, H.; Shymanets, A.; Nurnberg, B.; Backer, J.M. Characterization of a tumor-associated activating mutation of the P110beta PI 3-kinase. PLoS ONE 2013, 8, e63833. [Google Scholar] [CrossRef] [PubMed]
- Pazarentzos, E.; Giannikopoulos, P.; Hrustanovic, G.; St John, J.; Olivas, V.R.; Gubens, M.A.; Balassanian, R.; Weissman, J.; Polkinghorn, W.; Bivona, T.G. Oncogenic activation of the PI3-kinase P110beta isoform via the tumor-derived Pik3cbeta(d1067v) kinase domain mutation. Oncogene 2016, 35, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.D.; Colman, L.; Lensun, L.; Rogers, H.L.; Shuttleworth, S.J. Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Signal Transduct. Target. Ther. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed]
- Pridham, K.J.; Le, L.; Guo, S.; Varghese, R.T.; Algino, S.; Liang, Y.; Fajardin, R.; Rodgers, C.M.; Simonds, G.R.; Kelly, D.F.; et al. Pik3CB/p110beta is a selective survival factor for glioblastoma. Neuro Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Friesland, A.; Zhao, Y.; Chen, Y.H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42-intersectin interaction disrupts golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Westwood, N.J.; Feng, Y.; Kirchhausen, T.; Shair, M.D. Use of biomimetic diversity-oriented synthesis to discover galanthamine-like molecules with biological properties beyond those of the natural product. J. A. Chem. Soc. 2001, 123, 6740–6741. [Google Scholar] [CrossRef]
- Pelish, H.E.; Peterson, J.R.; Salvarezza, S.B.; Rodriguez-Boulan, E.; Chen, J.L.; Stamnes, M.; Macia, E.; Feng, Y.; Shair, M.D.; Kirchhausen, T. Secramine inhibits cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat. Chem. Biol. 2006, 2, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Lebensohn, A.M.; Pelish, H.E.; Kirschner, M.W. Biochemical suppression of small-molecule inhibitors: A strategy to identify inhibitor targets and signaling pathway components. Chem. Biol. 2006, 13, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Bickford, L.C.; Morgan, D.; Kim, A.S.; Ouerfelli, O.; Kirschner, M.W.; Rosen, M.K. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat. Struct. Mol. Biol. 2004, 11, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Lokey, R.S.; Mitchison, T.J.; Kirschner, M.W. A chemical inhibitor of N-WASP reveals a new mechanism for targeting protein interactions. Proc. Natl. Acad. Sci. USA 2001, 98, 10624–10629. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Gunawardhana, S.; Lucas, T.; Abraham, D.; Aharinejad, S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of pak1 activity. J. Transl. Med. 2013, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Lucas, T.; Reichl, P.; Abraham, D.; Aharinejad, S. A RAC1/Cdc42 gtpase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS ONE 2013, 8, e74924. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Waller, A.; Strouse, J.J.; Bologa, C.; Ursu, O.; Salas, V.; Parkinson, J.F.; Phillips, G.K.; Romero, E.; Wandinger-Ness, A.; et al. A potent and selective inhibitor of Cdc42 GTPase. In Probe Reports from the Nih Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Noth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Liang, X.; Li, L.; Wang, B.; Ding, F.; Li, Y.; Wang, X.; Zhan, Q.; Liu, Z. MicroRNA-548j functions as a metastasis promoter in human breast cancer by targeting Tensin1. Mol. Oncol. 2016, 10, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Dolan, B.M.; Duron, S.G.; Campbell, D.A.; Vollrath, B.; Shankaranarayana Rao, B.S.; Ko, H.Y.; Lin, G.G.; Govindarajan, A.; Choi, S.Y.; Tonegawa, S. Rescue of fragile X syndrome phenotypes in Fmr1 ko mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 2013, 110, 5671–5676. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; He, H.; Patel, O.; Lowy, A.M.; Baldwin, G.S.; Nikfarjam, M. Frax597, a PAK1 inhibitor, synergistically reduces pancreatic cancer growth when combined with gemcitabine. BMC Cancer 2016, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Licciulli, S.; Maksimoska, J.; Zhou, C.; Troutman, S.; Kota, S.; Liu, Q.; Duron, S.; Campbell, D.; Chernoff, J.; Field, J.; et al. Frax597, a small molecule inhibitor of the P21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated schwannomas. J. Biol. Chem. 2013, 288, 29105–29114. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.C.; Gierke, S.; Pitt, C.; Sagolla, M.; Cheng, C.K.; Zhou, W.; Jubb, A.M.; Strickland, L.; Schmidt, M.; Duron, S.G.; et al. Small molecule inhibition of group I P21-activated kinases in breast cancer induces apoptosis and potentiates the activity of microtubule stabilizing agents. Breast Cancer Res. BCR 2015, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Ndubaku, C.O.; Crawford, J.J.; Drobnick, J.; Aliagas, I.; Campbell, D.; Dong, P.; Dornan, L.M.; Duron, S.; Epler, J.; Gazzard, L.; et al. Design of selective PAK1 inhibitor G-5555: Improving properties by employing an unorthodox Low-PK a polar moiety. ACS Med. Chem. Lett. 2015, 6, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Maruta, H.; Ahn, M.R. From bench (laboratory) to bed (hospital/home): How to explore effective natural and synthetic PAK1-blockers/longevity-promoters for cancer therapy. Eur. J. Med. Chem. 2017, 142, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Karpov, A.S.; Amiri, P.; Bellamacina, C.; Bellance, M.H.; Breitenstein, W.; Daniel, D.; Denay, R.; Fabbro, D.; Fernandez, C.; Galuba, I.; et al. Optimization of a dibenzodiazepine hit to a potent and selective allosteric PAK1 inhibitor. ACS Med. Chem. Lett. 2015, 6, 776–781. [Google Scholar] [CrossRef] [PubMed]
- McCoull, W.; Hennessy, E.J.; Blades, K.; Chuaqui, C.; Dowling, J.E.; Ferguson, A.D.; Goldberg, F.W.; Howe, N.; Jones, C.R.; Kemmitt, P.D.; et al. Optimization of highly kinase selective bis-anilino pyrimidine PAK1 inhibitors. ACS Med. Chem. Lett. 2016, 7, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Goreshnik, I.; Maly, D.J. A small molecule-regulated guanine nucleotide exchange factor. J. Am. Chem. Soc. 2010, 132, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.T.; Worthylake, D.K.; Rossman, K.L.; Betts, L.; Pruitt, W.M.; Siderovski, D.P.; Der, C.J.; Sondek, J. Structural basis for the selective activation of RHO gtpases by DBL exchange factors. Nat. Struct. Biol. 2002, 9, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Sorber, R.; Teper, Y.; Abisoye-Ogunniyan, A.; Waterfall, J.J.; Davis, S.; Killian, J.K.; Pineda, M.; Ray, S.; McCord, M.R.; Pflicke, H.; et al. Whole genome sequencing of newly established pancreatic cancer lines identifies novel somatic mutation (c.2587G>A) in axon guidance receptor plexin A1 as enhancer of proliferation and invasion. PLoS ONE 2016, 11, e0149833. [Google Scholar] [CrossRef] [PubMed]
- Sakamori, R.; Das, S.; Yu, S.; Feng, S.; Stypulkowski, E.; Guan, Y.; Douard, V.; Tang, W.; Ferraris, R.P.; Harada, A.; et al. Cdc42 and RAB8A are critical for intestinal stem cell division, survival, and differentiation in mice. J. Clin. Investig. 2012, 122, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Dorr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Akbar, H.; Cancelas, J.; Williams, D.A.; Zheng, J.; Zheng, Y. Rational design and applications of a RAC gtpase-specific small molecule inhibitor. Methods Enzymol. 2006, 406, 554–565. [Google Scholar] [PubMed]
- Oprea, T.I.; Sklar, L.A.; Agola, J.O.; Guo, Y.; Silberberg, M.; Roxby, J.; Vestling, A.; Romero, E.; Surviladze, Z.; Murray-Krezan, C.; et al. Novel activities of select nsaid R-enantiomers against RAC1 and Cdc42 gtpases. PLoS ONE 2015, 10, e0142182. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, D.; Blelloch, R. From micrornas to targets: Pathway discovery in cell fate transitions. Curr. Opin. Genet. Dev. 2011, 21, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Detassis, S.; Grasso, M.; Del Vescovo, V.; Denti, M.A. MicroRNAs make the call in cancer personalized medicine. Front. Cell Dev.Biol. 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Li, Y.; Jing, R. MicroRNA-29a functions as a potential tumor suppressor through directly targeting Cdc42 in non-small cell lung cancer. Oncol. Lett. 2017, 13, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Jin, X.; Sun, Z.; Zhao, Y.; Song, X. Mir-186 inhibited migration of NSCLC via targeting Cdc42 and effecting emt process. Mol. Cells 2017, 40, 195–201. [Google Scholar] [PubMed]
- Zhu, X.; Li, Y.; Shen, H.; Li, H.; Long, L.; Hui, L.; Xu, W. Mir-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and CDK6. FEBS Lett. 2013, 587, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chen, T.; Li, Y.; Gao, L.; Zhang, S.; Wang, T.; Chen, M. Downregulation of miR-25 modulates non-small cell lung cancer cells by targeting Cdc42. Tumour Biol. 2015, 36, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Guo, W.; Qian, J.; Wang, B. Negative regulation of Cdc42 expression and cell cycle progression by miR-29A in breast cancer. Open Med. 2016, 11, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Wang, B.; Zheng, T.; Li, X.; Zheng, L.; Hu, J.; Zhang, Y.; Xing, Y.; Xi, T. Malat1 induced migration and invasion of human breast cancer cells by competitively binding miR-1 with Cdc42. Biochem. Biophys. Res. Commun. 2016, 472, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.G.; Li, S.; Yu, T.T.; Qian, L.J.; Cao, R.S.; Zhu, H.; Xiao, B.; Jiao, C.H.; Tang, N.N.; Ma, J.J.; et al. Differential expression of miR-195 in esophageal squamous cell carcinoma and miR-195 expression inhibits tumor cell proliferation and invasion by targeting of Cdc42. FEBS Lett. 2013, 587, 3471–3479. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Ye, L.; Chang, T.; Li, X.; Li, X. MicroRNA-195-Cdc42 axis acts as a prognostic factor of esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6871–6879. [Google Scholar] [PubMed]
- Zhang, Y.; Takahashi, S.; Tasaka, A.; Yoshima, T.; Ochi, H.; Chayama, K. Involvement of microRNA-224 in cell proliferation, migration, invasion, and anti-apoptosis in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2013, 28, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, X.; Zhang, M.; Fan, Q.; Luo, S.; Cao, X. MiR-137 is frequently down-regulated in gastric cancer and is a negative regulator of Cdc42. Dig. Dis. Sci. 2011, 56, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Liu, F.; Wang, G.; Li, Y.; Zhang, H.; Li, F. MiR-133 is a key negative regulator of Cdc42-PAK pathway in gastric cancer. Cell. Signal. 2014, 26, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, K.J.; McKinnon, R.A.; Michael, M.Z. MiR-18a inhibits Cdc42 and plays a tumour suppressor role in colorectal cancer cells. PLoS ONE 2014, 9, e112288. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lang, N.; Qiu, M.; Xu, F.; Li, Q.; Tang, Q.; Chen, J.; Chen, X.; Zhang, S.; Liu, Z.; et al. MiR-137 targets Cdc42 expression, induces cell cycle G1 arrest and inhibits invasion in colorectal cancer cells. Int. J. Cancer 2011, 128, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lang, N.; Chen, X.; Tang, Q.; Liu, S.; Huang, J.; Zheng, Y.; Bi, F. MiR-185 targets RHOA and Cdc42 expression and inhibits the proliferation potential of human colorectal cells. Cancer Lett. 2011, 301, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Chen, Y.R.; Liu, S.S.; Ye, Y.P.; Jiao, H.L.; Wang, S.Y.; Xiao, Z.Y.; Wei, W.T.; Qiu, J.F.; Liang, L.; et al. MiR-384 inhibits human colorectal cancer metastasis by targeting KRAS and Cdc42. Oncotarget 2016, 7, 84826–84838. [Google Scholar] [CrossRef] [PubMed]
- Sakamori, R.; Yu, S.; Zhang, X.; Hoffman, A.; Sun, J.; Das, S.; Vedula, P.; Li, G.; Fu, J.; Walker, F.; et al. Cdc42 inhibition suppresses progression of incipient intestinal tumors. Cancer Res. 2014, 74, 5480–5492. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.N.; Yu, X.T.; Tang, J.; Zhou, C.X.; Wang, C.L.; Yin, Q.Q.; Gong, X.F.; He, M.; He, J.R.; Chen, G.Q.; et al. MicroRNA-494 inhibits breast cancer progression by directly targeting PAK1. Cell Death Dis. 2017, 8, e2529. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.D.; Ohshiro, K.; Rayala, S.K.; Kumar, R. MicroRNA-7, a homeobox D10 target, inhibits P21-activated Kinase 1 and regulates its functions. Cancer Res. 2008, 68, 8195–8200. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zhang, X.; Yang, G.; Song, Y.; Cai, W. Decrement of miR-199A-5P contributes to the tumorigenesis of bladder urothelial carcinoma by regulating MLK3/NF-kappab pathway. Am. J. Transl. Res. 2015, 7, 2786–2794. [Google Scholar] [PubMed]
- Liu, Z.; Liu, Z.; Zhang, Y.; Li, Y.; Liu, B.; Zhang, K. MiR-24 represses metastasis of human osteosarcoma cells by targeting ACK1 via AKT/MMPS pathway. Biochem. Biophys. Res. Commun. 2017, 486, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Jiao, D.M.; Yao, Q.H.; Yan, J.; Song, J.; Chen, F.Y.; Lu, G.H.; Zhou, J.Y. Expression analysis of Cdc42 in lung cancer and modulation of its expression by curcumin in lung cancer cell lines. Int. J. Oncol. 2012, 40, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho gtpase signalling in cell migration. Curr. Opin. Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Dacic, S.; Kelly, L.; Shuai, Y.; Nikiforova, M.N. Mirna expression profiling of lung adenocarcinomas: Correlation with mutational status. Mod. Pathol. 2010, 23, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Rosenberg, P.S.; Chen, W.Q.; Hartman, M.; Lim, W.Y.; Chia, K.S.; Wai-Kong Mang, O.; Chiang, C.J.; Kang, D.; Ngan, R.K.; et al. Female breast cancer incidence among asian and western populations: More similar than expected. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Craig, B.T.; Romain, C.V.; Qiao, J.; Chung, D.H. Silencing of Cdc42 inhibits neuroblastoma cell proliferation and transformation. Cancer Lett. 2014, 355, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.G.; Liu, Q.; Qin, X.; Geng, Y.H.; Zheng, S.T.; Liu, T.; Sheyhidin, I.; Lu, X.M. Clinicopathological pattern and annexin A2 and Cdc42 status in patients presenting with differentiation and lymphnode metastasis of esophageal squamous cell carcinomas. Mol. Biol. Rep. 2012, 39, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Gomez Del Pulgar, T.; Valdes-Mora, F.; Bandres, E.; Perez-Palacios, R.; Espina, C.; Cejas, P.; Garcia-Cabezas, M.A.; Nistal, M.; Casado, E.; Gonzalez-Baron, M.; et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int. J. Oncol. 2008, 33, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, T.; Schwartz, B. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int. J. Cancer 2008, 123, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Alwan, A. World health organization. Disaster Med. Public Health Preparedness 2007, 1, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, V.; Mishra, R.; Mehrotra, S.; Sondarva, G.; Ray, R.S.; Rao, A.; Chatterjee, M.; Rana, B.; Rana, A. Estrogen suppresses MLK3-mediated apoptosis sensitivity in ER + breast cancer cells. Cancer Res. 2010, 70, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Senthivinayagam, S.; Rangasamy, V.; Sondarva, G.; Rana, B. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol. Endocrinol. 2010, 24, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, Y.F.; Chen, G.D.; Ou, D.P.; Qiu, X.X.; Zuo, C.H.; Yang, L.Y. ACK1 overexpression promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Oncotarget 2015, 6, 40622–40641. [Google Scholar] [CrossRef] [PubMed]
- Van Hengel, J.; D’Hooge, P.; Hooghe, B.; Wu, X.; Libbrecht, L.; De Vos, R.; Quondamatteo, F.; Klempt, M.; Brakebusch, C.; van Roy, F. Continuous cell injury promotes hepatic tumorigenesis in Cdc42-deficient mouse liver. Gastroenterology 2008, 134, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Mitsushima, M.; Toyoshima, F.; Nishida, E. Dual role of Cdc42 in spindle orientation control of adherent cells. Mol. Cell. Biol. 2009, 29, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, M.A.; Bracci, M.; Lazzarini, R.; Tomasetti, M.; Amati, M.; Lucarini, G.; Di Primio, R.; Santarelli, L. Use of potential dietary phytochemicals to target mirna: Promising option for breast cancer prevention and treatment? J. Funct. Foods 2017, 28, 177–193. [Google Scholar] [CrossRef]
- Li, Q.; Yao, Y.; Eades, G.; Liu, Z.; Zhang, Y.; Zhou, Q. Downregulation of miR-140 promotes cancer stem cell formation in basal-like early stage breast cancer. Oncogene 2014, 33, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eades, G.; Yao, Y.; Zhang, Y.; Zhou, Q. Characterization of a stem-like subpopulation in basal-like ductal carcinoma in situ (DCIS) lesions. J. Biol. Chem. 2014, 289, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bian, S.; Yang, C.S. Green tea polyphenol egcg suppresses lung cancer cell growth through upregulating miR-210 expression caused by stabilizing HIF-1alpha. Carcinogenesis 2011, 32, 1881–1889. [Google Scholar] [CrossRef] [PubMed]

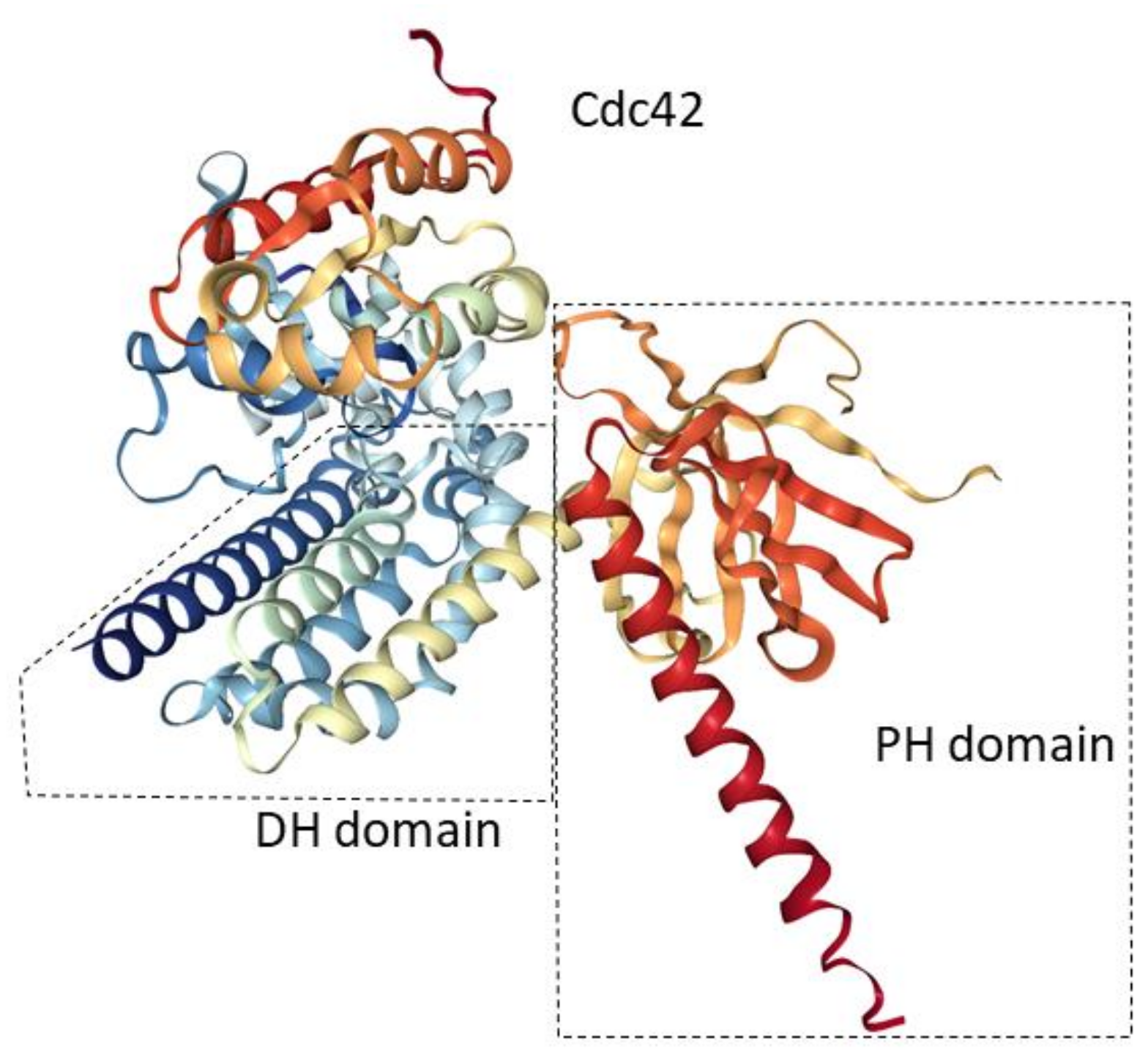
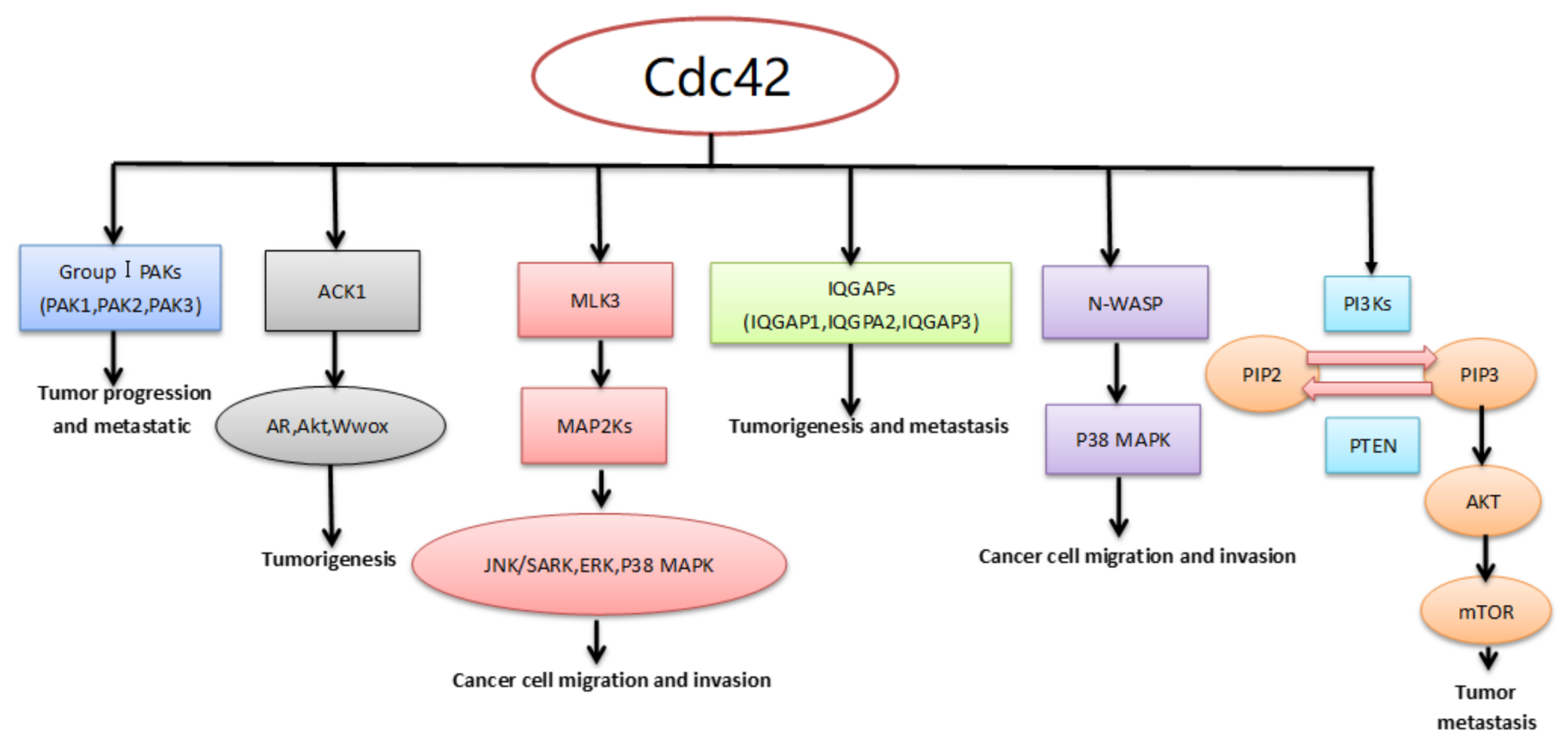

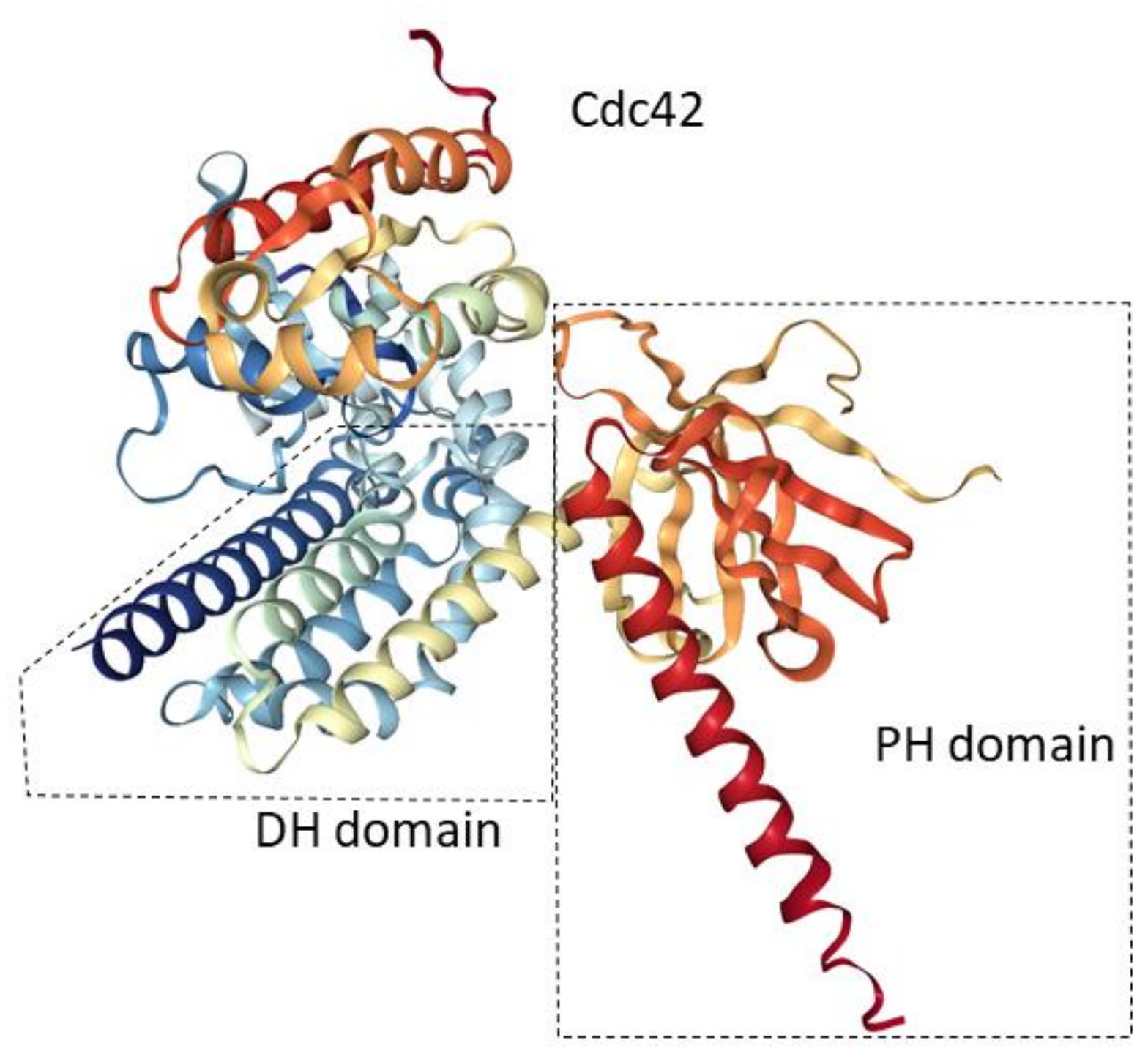
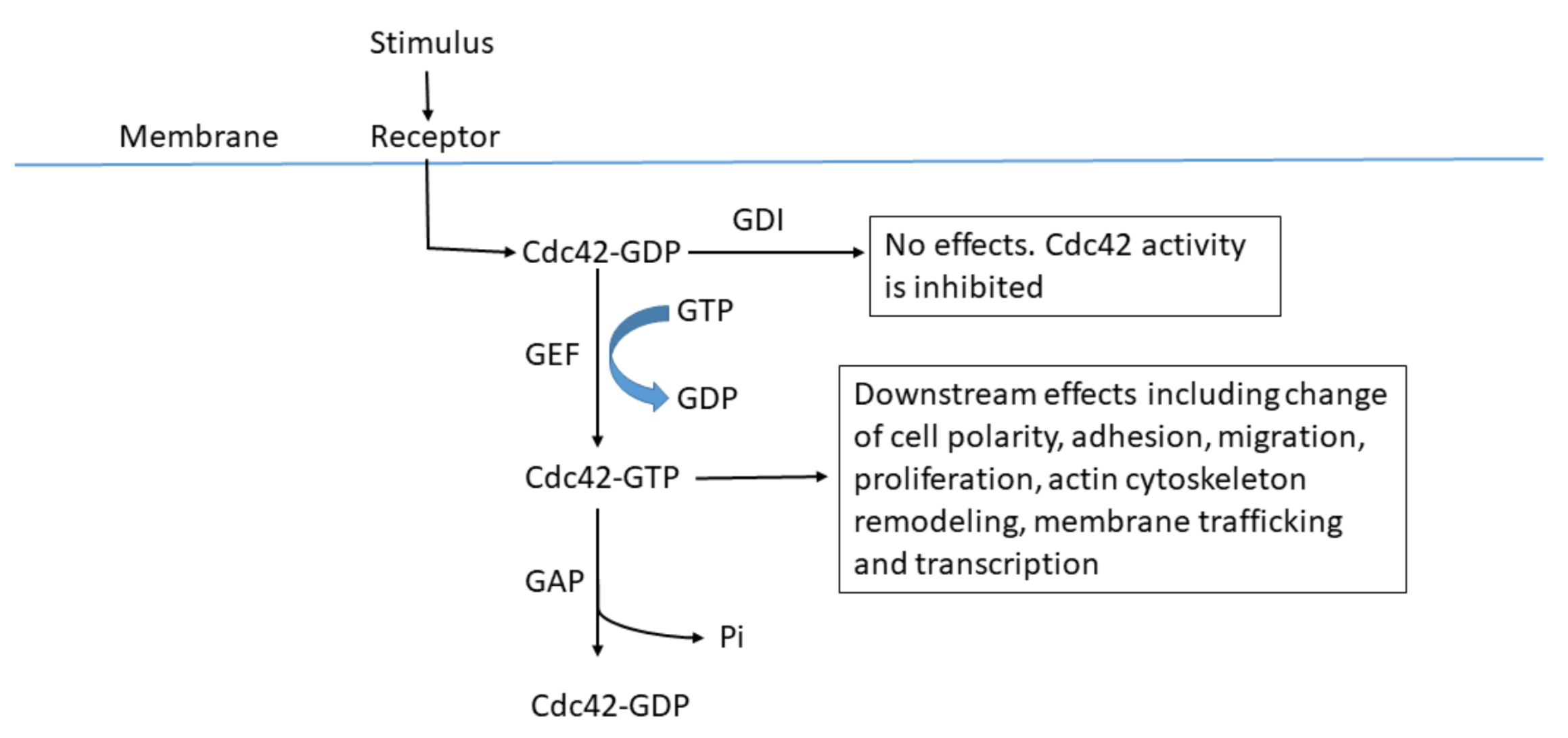{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Mode of Action | Reference |
|---|---|---|
| ZCL278 | act as intersectin (ITSN) by directly binding to Cdc42 | [74] |
| Secramine | reduce membrane association of prenylated Cdc42(GDP) in a Cdc42(GDP)–RhoGDI1 complex fashion | [75,76] |
| CASIN | inhibit nucleotide exchange on Cdc42 | [77,78,79] |
| AZA197 | disrupt the interaction between Cdc42 and GEFs | [80] |
| AZA1 | disrupt the interaction between Cdc42 and GEFs | [81] |
| ML141 | specifically block GTP binding to Cdc42 | [82,83,84] |
| FRAXs (486, 597, and 1036) | inhibit group I PAKs (PAK1, PAK2, PAK3) by competing with ATP | [85,86,87,88] |
| G-5555 | specific suppress PAK1 by competing with ATP | [89] |
| NOV-3 | specific suppress PAK1 by competing with ATP | [90,91] |
| AZ137-05339 | specific suppress PAK1 by competing with ATP | [92] |
| A | ||||
| miRNA | Target | Cancer Types | Functional Contribution | Reference |
| miR-29a | CDC42 3’UTR 5’…UGGUGCU… | NSCLC | inhibit proliferation, migration, invasion | [102] |
| miR-186 | CDC42 3’UTR 5’…UUAAGAA… | NSCLC | inhibit migration and effect EMT | [103] |
| miR-137 | CDC42 3’UTR 5’…GCAAUAA… | NSCLC | ||
| CDK6 3’UTR 5’…GCAAUA… | NSCLC | inhibit proliferation and induce cell cycle arrest | [104] | |
| miR-25 | CDC42 3’UTR 5’…UGCAAU… | NSCLC | reduce proliferation and induce G1 cell cycle arrest | [105] |
| miR-29a | CDC42 3’UTR | Breast cancer | inhibit growth through cell cycle regulation | [106] |
| miR-1 | CDC42 3’UTR 5’…ACAUUCC… | Breast cancer | inhibit migration and invasion | [107] |
| miR-195 | CDC42 3’UTR 5’…UGCUGCU… | ESCC | inhibit proliferation and invasion; act as a prognostic biomarker | [108,109] |
| miR-224 | CDC42, CDH1,PAK2,BLC-2,MAPK1 | Hepatocellular carcinoma | promote cell proliferation, migration, invasion; anti-apoptosis | [110] |
| miR-137 | CDC42 3’UTR | Gastric cancer | inhibit cell cycle progression and induce apoptosis | [111] |
| miR-133 | CDC42 3’UTR 5’…GGGGACCAG… | Gastric cancer | suppress cell growth, migration and invasion | [112] |
| miR-18a | CDC42 3’UTR 5’…CACCUU… | Colorectal cancer | reduce proliferation, migration; increase apoptosis; induce cell cycle arrest | [113] |
| miR-137 | CDC42 3’UTR 5’…AAGCAAT… | Colorectal cancer | inhibit invasion, proliferation; induce cell cycle G1 arrest | [114] |
| miR-185 | CDC42 3’UTR 5’…UGCCUUU… | |||
| RhoA 3’UTR 5’…UUCUCUCCA… | Colorectal cancer | inhibit proliferation, invasion; induce G0/G1 arrest | [115] | |
| miR-384 | CDC42 3’UTR 5’…UAGGAA… | |||
| KRAS 3’UTR 5’…UAGGAA… | Colorectal cancer | inhibit metastasis and invasive | [116] | |
| miR-224 | CDC42 3’UTR 5’…GUGACUU… | |||
| SMAD4 3’UTR 5’…GUGACUU… | Colorectal cancer | suppress migration and the formation of Actin Filaments | [117] | |
| B | ||||
| miRNA | Target | Cancer Types | Functional Contribution | Reference |
| miR-497 | PAK1 | Breast cancer | suppress colonogetic ability, metastasis, tumorigenesis, invasion | [118] |
| miR-7 | PAK1 | Breast cancer | inhibit motility, invasiveness, anchorage-independent growth | [119] |
| miR-137 | PAK1 | Colorectal cancer | suppress Cdc42/pak signaling pathway | [114] |
| MLC, ERK1/2 | ||||
| CyclinD1 | ||||
| miR-195 | ERK1/2 | ESCC | suppressed Cdc42/ERK/Cyclin D1 signaling pathway | [108] |
| CyclinD1 | ||||
| miR-199-5p | MLK3 | Bladder urothelial carcinoma | inhibit MLK3/IkB/NF-KB signaling pathway | [120] |
| miR-24 | ACK1 | Osteosarcoma | inhibit migration and invasion | [121] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, X.-H.; Lv, L.-C.; Duan, J.; Wu, Y.-M.; He, S.-J.; Hu, Z.-Z.; Xiong, L.-X. Regulating Cdc42 and Its Signaling Pathways in Cancer: Small Molecules and MicroRNA as New Treatment Candidates. Molecules 2018, 23, 787. https://doi.org/10.3390/molecules23040787
Xiao X-H, Lv L-C, Duan J, Wu Y-M, He S-J, Hu Z-Z, Xiong L-X. Regulating Cdc42 and Its Signaling Pathways in Cancer: Small Molecules and MicroRNA as New Treatment Candidates. Molecules. 2018; 23(4):787. https://doi.org/10.3390/molecules23040787
Chicago/Turabian StyleXiao, Xing-Hua, Lin-Chen Lv, Jing Duan, Ye-Meng Wu, Shu-Jin He, Zhen-Zhen Hu, and Li-Xia Xiong. 2018. "Regulating Cdc42 and Its Signaling Pathways in Cancer: Small Molecules and MicroRNA as New Treatment Candidates" Molecules 23, no. 4: 787. https://doi.org/10.3390/molecules23040787
APA StyleXiao, X.-H., Lv, L.-C., Duan, J., Wu, Y.-M., He, S.-J., Hu, Z.-Z., & Xiong, L.-X. (2018). Regulating Cdc42 and Its Signaling Pathways in Cancer: Small Molecules and MicroRNA as New Treatment Candidates. Molecules, 23(4), 787. https://doi.org/10.3390/molecules23040787