Abstract
In this study, we report the synthesis, antibacterial and anticancer evaluation of 38 novel phenanthridines that were designed as analogs of the benzo[c]phenanthridine alkaloids. The prepared phenanthridines differ from the benzo[c]phenanthridines in the absence of a benzene A-ring. All novel compounds were prepared from 6-bromo-2-hydroxy-3-methoxybenzaldehyde in several synthetic steps through reduction of Schiff bases and accomplished by radical cyclization. Twelve derivatives showed high antibacterial activity against Bacillus subtilis, Micrococcus luteus and/or Mycobacterium vaccae at single digit micromolar concentrations. Some compounds also displayed cytotoxicity against the K-562 and MCF-7 cancer cell lines at as low as single digit micromolar concentrations and were more potent than chelerythrine and sanguinarine. The active compounds caused cell-cycle arrest in cancer cells, increased levels of p53 protein and caused apoptosis-specific fragmentation of PARP-1. Biological activity was connected especially with the presence of the N-methyl quaternary nitrogen and 7-benzyloxy substitution (compounds 7i, 7j, 7k, and 7l) of phenanthridine.
1. Introduction
Benzo[c]phenanthridine alkaloids contain a chrysene-skeleton-based heterocyclic core and are classified as isoquinoline alkaloids (Figure 1). They are distributed especially in the plant family Papaveraceae and display a broad spectrum of biological activities, including anti-inflammatory, antimicrobial, antifungal and antitumor effects [1,2,3,4,5,6,7]. Sanguinarine and chelerythrine are the best-known benzo[c]phenanthridine alkaloids, most frequently studied for their antitumor effects [8]. As flat polyaromatic compounds they directly interact with DNA [9]. However, cell cycle arrest and induction of cell death caused by these alkaloids probably occurs not only due to DNA damage alone, but as a combined result of targeting other cellular structures, including topoisomerases, tubulin and antiapoptotic protein Bcl-XL [10,11]. Importantly, these alkaloids act at concentrations comparable to those of cytostatics used as anticancer drugs.
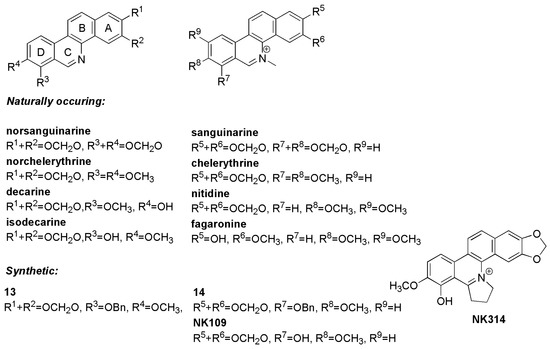
Figure 1.
Structures of some benzo[c]phenanthridines.
Benzo[c]phenanthridine alkaloids have been subjected to chemical modifications with the aim of understanding their structure-activity relationships and to improve their biological functions. Early studies indicated that the activity of benzo[c]phenanthridine alkaloids can be linked to the presence of their cationic quaternary nitrogen [12]. The quaternary iminium may undergo nucleophilic addition to biological amines or thiols, modify DNA and proteins and as a consequence induce cell death. In contrast, a recent report suggests that the iminium group is not essential for cytotoxicity in cancer cell lines [13]. Other systematic studies examined the correlation between the type of substituents at positions 2, 3, 7, 8 and 9 of their skeletons and antiproliferative activity [14] and revealed that cationic iminium alkaloids displayed stronger activity than their corresponding uncharged bases. The cytotoxic activity was further increased if these quaternary bases were 7,8-oxygenated [15].
These studies led to identification of NK109, the 7-O-demethylated synthetic analogue of chelerythrine, that was shown to have submicromolar dose growth-inhibitory activity against cancer cell lines, i.e., significantly lower than chelerythrine [16,17,18,19] (Figure 1). Further modification of NK109, in which ring C was fused to a pyrrolidine cycle, yielded NK314 with even stronger activities in several cancer models [20,21,22]. The molecular mechanism of action of these compounds has been often attributed to inhibition of topoisomerases, which are also targeted by the related compounds nitidine and fagaronine [23].
To our knowledge, there have not been many attempts to modify the heterocyclic skeleton of benzo[c]phenanthridines up until now in order to modify their biological activity. With the exception of benzo[h]quinolones, which are structurally related to the benzo[c]phenanthridines but lack the D-ring [24], and phenanthridines lacking the A-ring [25], most other reports describe the synthesis of various isomeric and aza-analogous structures such as benzo[i]phenanthridines and dibenzo[c,h]cinnolines [26], benzo[c]phenanthrolines [27], pyridophenanthrolines and azapyrimido-phenanthrolines [28] or compounds containing the 3,4-dihydroisoquinolin-2-ium scaffold [29,30].
We therefore decided to prepare novel phenanthridines related to benzo[c]phenanthridines lacking the benzene A-ring (Figure 2), because previous work has suggested that phenanthridines may retain biological activity [25]. The series contains derivatives differing in the presence and position of hydroxy and methoxy groups, 2,3- or 3,4-methylenedioxy bridges and 7-benzyloxy groups, but all compounds contain an 8-methoxy group. Some derivatives have been prepared either as free bases or protonated salts. In some compounds, the heterocyclic nitrogen is available in a quaternary form with methyl substitution. Thus, such variability allowed us to build a preliminary structure-activity relationship for antibacterial and antiproliferative activity, and to compare the phenanthridines with known benzo[c]phenanthridines (Figure 1).
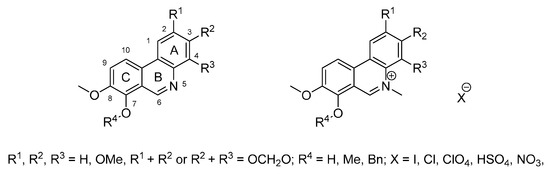
Figure 2.
Desired phenanthridines for biological evaluation (for details see Table 1).
For the preparation of novel phenanthridine derivatives we adapted methodologies from benzo[c]phenanthridine chemistry. Many synthetic approaches to the benzo[c]phenanthridine nucleus have been reported, with most routes involving the construction of either the B or C ring in the final or semifinal stage. These synthetic methods are summarized in reviews [31,32]. Our synthesis of desired phenanthridine compounds is based on our previous experience with the synthesis of isodecarine [33] and metabolites of benzo[c]phenanthridines [34], where a radical cyclization leading to ring C closure was used to accomplish the construction of the benzo[c]phenanthridine heterocyclic system. By this method we prepared phenanthridine derivatives where the B ring is formed.
2. Results and Discussion
2.1. Chemistry
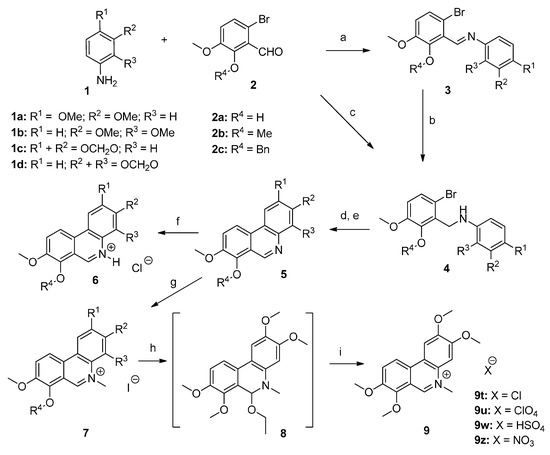
For the preparation of phenanthridines, selected anilines possessing substitution in positions 2,3 (1b [35], 1d [36]) and 3,4 (1a, 1c) with methylenedioxy or dimethoxy groups were used for the reactions. Commercially available 6-bromo-2-hydroxy-3-methoxybenzaldehyde (2a) needed for introduction of the C-ring aromatic core was used as is or after modification of the hydroxyl group to obtain the methoxyaldehyde 2b [25] or benzyloxyaldehyde 2c [16].
Reductive amination of aldehydes 2a–2c with selected anilines 1a–1d by NaBH(OAc)3 in toluene proceeded quantitatively and afforded the corresponding novel secondary amines 4. We have also verified an alternative two-step route leading to the same secondary amines through their isolated Schiff bases 3. The preparation of Schiff bases 3a, 3c, and 3d possessing unsubstituted hydroxyl groups proceeded smoothly providing products that readily precipitated from the reaction mixture with excellent nearly quantitative yields. Facile reduction of their iminium double bonds by NaBH4 in ethanol led to the corresponding secondary amines 4a, 4c, and 4d. Surprisingly, the condensation leading to the formation of Schiff bases with substituted hydroxyl group (R4 = benzyl or methyl) under the same reaction conditions was not quantitative (conversions ranged from 50–75%) and subsequent isolation attempts by conventional methods were complicated. Therefore, we tried to influence the reaction equilibrium by increasing the ratio of aniline to aldehyde, prolonging the reaction time, performing the reactions in alternative solvents or by use of a microwave reactor, but unfortunately, without any satisfactory improvement. In summary, reductive amination seems to be a more suitable method for the preparation of these sorts of amines 4.
To obtain compounds possessing a phenanthridine core, amines 4 needed to be cyclized. For this purpose, several synthetic approaches including palladium-catalyzed cross-coupling cyclization [37,38,39,40,41] or base-promoted homolytic aromatic substitution [42,43] were tested with amines 4a and 4e, unfortunately, the reactions failed or yields were very low. During these experiments debromination was the main reaction observed. Finally, radical cyclization by Bu3SnH using AIBN (azobisisobutyronitrile) as a radical initiator in toluene and subsequent aromatization by activated MnO2 [44] afforded the desired novel phenanthridine derivatives 5 in various yields. During all radical cyclization reactions we observed formation of debrominated starting amines 10 as side products in amounts of 10–20% (Scheme 2). Formation of these reduced compounds is in accordance with earlier results [33]. Further side products were detected when 3,4-disbstituted (R1 and R2) amines 4 were subjected to the radical cyclization conditions. Apart from reduced amines 10, cyclized regioisomers 11 were also observed as minor products. In the case of substituted amines 4, where R4 is benzyl or methyl the amount of 11 was around 5%, whereas the ratio of these regioisomers was much higher (up to 30%, based on HPLC analysis) for the unsubstituted hydroxylamines 4a–4d (Scheme 2).
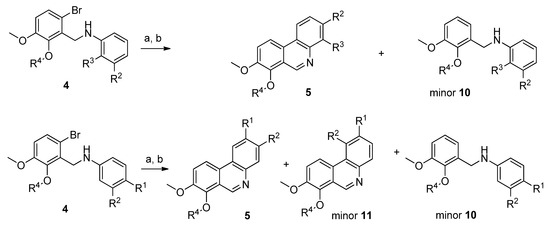
Scheme 2.
Side reaction products formed during radical cyclization of 4 to 5. Reagents and conditions: (a) Bu3SnH, AIBN, toluene, 104–106 °C, 3–6 h; (b) activated MnO2, r.t., 18 h.
Crystallization was utilized here as a useful method for the separation of most phenanthridines 5 from their reaction by-products. Unfortunately, a problem concerning the isolation of pure compounds was observed with hydroxyphenanthridines 5a–5d. The byproducts formed had similar properties to the targeted molecules, with only silica gel column chromatography providing pure compound 5d, but again with difficulties. For these reasons the remaining hydroxyphenanthridines 5a–5c were obtained by catalytic hydrogenation of the corresponding benzyl derivatives 5i–5k. This benzyl group deprotection proceeded under atmospheric pressure of hydrogen on 10% Pd/C. Under these conditions reduction of the iminium double bond can occur, so subsequent stirring of the reaction mixture in air or by addition of MnO2 was necessary for rearomatization. Alternatively, to avoid the need for this re-oxidation step benzyl group removal could also be achieved by acid hydrolysis with HCl followed by treatment with aqueous NH3 to obtain the free bases (Scheme 3).
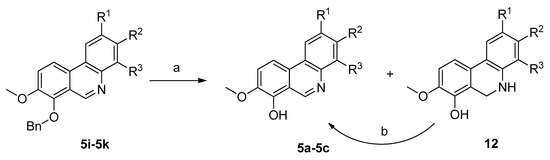
Scheme 3.
Convenient route for the preparation of hydroxy phenanthridines 5a–5c. Reagents and conditions: (a) H2/10% Pd(C), atm. pressure, r.t. or HCl/EtOH, reflux 2.5 h, then aq. NH3; (b) air (O2) or MnO2; r.t.
Phenanthridine derivatives 5 were converted to their hydrochlorides 6 by addition of HCl into dioxane solutions of the free bases (Scheme 1). Surprisingly these compounds have very low solubility in water, and even in other polar solvents (DMSO, EtOH, DMF) which hindered biological testing.
Scheme 1.
The synthetic route for the preparation of phenanthridine derivatives. Reagents and conditions: (a) EtOH, reflux, 1 h; (b) NaBH4, EtOH, r.t., 2 h; (c) NaBH(OAc)3, toluene, r.t., 1 h; (d) Bu3SnH, AIBN, toluene, 104–106 °C, 3–6 h; (e) activated MnO2, r.t., 18 h; (f) HCl, dioxane, r.t.; (g) MeI, ACN, r.t., 7 days; (h) NaOH, EtOH, r.t., 1 h; (i) HX, r.t., 10 min.
N-methylation of phenanthridines 5 was carried out with methyl iodide in acetonitrile under mild conditions to give N-methylphenanthridinium iodides 7. These conditions enabled the selective methylation, even of the phenanthridines possessing a free hydroxyl group. Similarly to the hydrochlorides 6, the prepared quaternary iodides 7 were also surprisingly very poorly soluble in water. To verify the possibility if the anion exchange can influence the solubility of these compounds, selected quaternary iodides were transformed in a NaOH/EtOH solution into their unisolated colorless pseudobases 8. Different salts (chloride, hydrogensulfate, perchlorate, and nitrate) were obtained by addition of large excess of corresponding acids to provide the anion modified N-methylphenanthridine precipitates. It was found that anion exchange did not improve solubility of these mentioned compounds significantly. For illustration, we demonstrate experimentally the preparation of various salts of compound 9, which were confirmed by elemental analysis.
2.2. Biological Assays
2.2.1. Antibacterial Activity
Prepared derivatives were screened for antibacterial activity against representative Gram-positive bacteria (Bacillus. subtilis, ATCC 6633; Micrococcus luteus, ATCC 10240; Mycobacterium vaccae, DSM 43514; Staphylococcus aureus, CCM 2524) and Gram-negative bacteria (Pseudomonas aeruginosa, CCM 3955; Escherichia coli, CCM 3954) using agar diffusion assays [45]. Derivatives with zones of inhibition ≥20 mm were subjected to a further assay [46] that determines their minimum inhibitory concentrations (MIC). From the measured data (Table 2) it is possible to make a few general conclusions regarding their structure-activity relationships:

Table 2.
Antibacterial activity.
- (1)
- Antibacterial activity was observed only for derivatives containing a phenanthridine skeleton. No tested strain was susceptible to representative intermediates 3 and 4.
- (2)
- Derivatives with benzyl substituent as R4 (5k, 5l, 7i, 7j, 7k, and 7l) showed high antibacterial activity against B. subtilis, M. luteus and/or M. vaccae with MIC in single digit micromolar values.
- (3)
- Compounds with a charged nitrogen bearing methyl group (7i, 7j, 7k and 7l) demonstrated high activity as well. To summarize, the most active compounds have a similar structural motif—a phenanthridine skeleton with a charged N-methyl nitrogen and a benzyl group as a R4 substituent.
These points are noteworthy for potential structural exploitation in further antibacterial agent development. To compare the antibacterial activity results for well-known natural (chelerythrine, sanguinarine, isodecarine, norchelerythrine) or synthetic compounds (NK-109, 13, 14) with similar structure motifs are presented in Table 2 as well. From the measured results it is evident that some prepared derivatives provided similar or even better activity than these compounds (MIC in micromolar or submicromolar values). All newly reported compounds were less active than a previously developed analogue 14, with 7j performing better than that reference compound only against M. luteus. No compound displayed any relevant activity against Gram-negative bacteria. If the structures are compared the importance of the presence of a charged N-methyl nitrogen in the molecule is confirmed.
2.2.2. Anticancer Activity In Vitro
Benzo[c]phenanthridine alkaloids are known to display antiproliferative and anticancer activities [8,18]. Due to their structural analogy, the preliminary in vitro anticancer activity of the newly prepared compounds was evaluated on two established cancer cell lines, MCF-7 (breast carcinoma) and K-562 (chronic myelogeneous leukemia). The resulting data are presented in Table 3 and show that several of the new compounds have significant activity in both cell lines, with single digit micromolar EC50 values.
Table 3.
Anticancer activity in vitro.
Derivative 7j reached submicromolar values in K-562 cells and was even more potent than chelerythrine and sanguinarine. The most active compounds bear 7-benzyloxy substitution and either 2,3-dimethoxy or an isosteric 2,3-methylenedioxy bridge and are either N5-methylated (7j, 7k) or contain an unsubstituted N5 (5k). Compounds without a benzyl moiety or with 3,4-dimethoxy/3,4-methylenedioxy substitution were less potent. Interestingly, removal of the methyl from N5 of 7j, that also uncharged the nitrogen, decreased potency (5j). In contrast, presence of the quaternary nitrogen (methylated) significantly reduced the activity of several compounds (compare especially pairs 5f and 7f, 5g and 7g, 5h or 7h). According to previous studies, the activity of benzo[c]phenanthridine alkaloids can be explained by the presence of a cationic quaternary nitrogen [12,15]. These conclusions however probably cannot be applied to phenanthridines, because many of these compounds with a quaternary nitrogen have poor activity and one of the most potent compounds (5k) does not contain a quaternary nitrogen at all.
With the aim of directly comparing the activity of phenanthridines with the corresponding benzo[c]phenanthridines, we prepared also four variously substituted benzo[c]phenanthridines—13 [16,33], 14 [33], isodecarine [33] and norchelerythrine (Figure 1). In line with the abovementioned findings for phenanthridines, the presence of the benzyloxy functionality at position 7 correlated with in vitro anticancer activity for compounds 13 and 14. However, previous studies indicated that the activity of benzo[c]phenanthridine alkaloids can be explained by the presence of a cationic quaternary nitrogen atom [12]. We also observed lack of cellular activity with isodecarine and norchelerythrine, which were prepared as demethylated derivatives of the very potent NK-109 and chelerythrine, respectively. Compound 14, killing both cancer cell lines with EC50 values around 1 µM, contains not only a benzyloxy functionality, but also a quaternary nitrogen that could be essential for its activity. We cannot rule out the possibility that this cation is even more important for the activity.
2.2.3. Mechanism of Cellular Activity
Anticancer activity of benzo[c]phenanthridine alkaloids relates at least partly to their ability to inhibit topoisomerase I or II. For example, nitidine and fagaronine inhibit the topoisomerase I—mediated DNA relaxation [23].
Their synthetic derivatives NK109 and NK314 exerted their cytotoxic activity through inhibition of topoisomerase II, followed by DNA breaks [19,22]. DNA damage usually results in a cell cycle arrest and eventually leads to apoptosis. We therefore treated MCF-7 cells with several compounds and NK109 as a control. Interestingly, these compounds influenced the cell cycle profile in different manners (Figure 3). The most potent 7k reduced S and G2/M phases, 7g reduced S phase and caused slight accumulation in G2/M, whereas the only weakly cytotoxic 6g resulted in strong G2/M arrest. NK109 used as a control also arrested cells in G2/M. These differences suggest that the compounds may have different molecular targets.
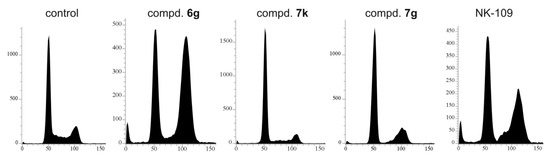
Figure 3.
Cell cycle effects of compounds 7k, 7g, 6g and NK-109 in MCF-7 cells treated for 24 h with doses corresponding to 3 × EC50 values (60, 5.4, 33 and 13.8 µM, respectively). Cells were harvested and then flow cytometric analysis of DNA stained by propidium iodide (10,000 counts) was performed as described in the Materials and Methods section.
DNA damage induces various responses, including stabilization and activation of tumor suppressor protein p53 [47]. In the following experiments, we analyzed levels and activities of tumor suppressor protein p53, which is typically activated in cells upon topoisomerase inhibition. We performed immunoblotting of proteins extracted from MCF-7 cells treated for 24 h with compounds 6g, 7k and 7g. Levels of p53 and its typical downstream target p21waf1 were both increased (Figure 4). In addition, fragmentation of PARP producing a 89 kDa band was observed in cells treated with 7k and NK109, indicating ongoing apoptosis. This fragmentation was dose-dependent for compound 7k. In contrast, neither 6g nor 7g triggered PARP fragmentation, which is in line with their weaker cytotoxicities.
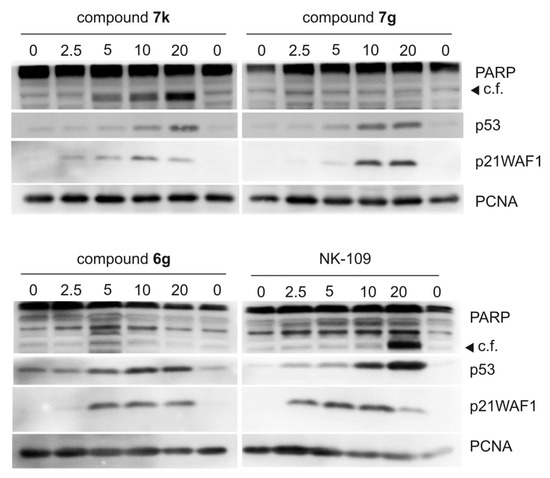
Figure 4.
Dose-dependent effect of compounds 7k, 7g, 6g and NK-109 on p53 and its activity in MCF-7 cells. The cells were treated for 24 h with indicated doses of compounds (given in µM) and then specific proteins were analyzed by immunoblotting as described in the Materials and Methods section. C.f., 89 kDa cleavage fragment of PARP.
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
All commercially available reagents were used without further purification and purchased from standard chemical suppliers. Reactions were monitored by LC/MS analyses on a UHPLC-MS system (Thermo Scientific, Waltham, MA, USA) consisting of a UHPLC chromatograph equipped with a photodiode array detector and a triple quadrupole mass spectrometer using a C18 column at 30 °C and flow rate of 800 μL/min. 1H and 13C-NMR spectra were measured on an ECA 400II (1H: 399.78 MHz, 13C: 100.53 MHz) NMR spectrometer (JEOL Resonance, Tokyo, Japan). Samples were dissolved and subsequently measured in DMSO-d6 or CDCl3. Chemical shifts (δ) are reported in ppm and referenced to the middle of the solvent signal (DMSO-d6: 2.50 ppm, 39.51 ppm; CDCl3: 7.27 ppm, 77.00 ppm. Data are reported as follows: chemical shift (multiplicity [singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), multiplet (m), broad resonance (br)], coupling constants [Hz], integration). All the NMR spectra were acquired at ambient temperature. All recorded 1H and 13C-NMR spectra are available as Supplementary material. High resolution mass spectra (HRMS) measurements were performed on an Orbitrap mass analyzer (Thermo Scientific, Waltham, MA, USA) equipped with Heated Electrospray Ionization (HESI). The spectrometer was tuned to obtain a maximum response for m/z 70–700. Elemental analyses were performed on an EA 1108 Elemental Analyser (Fisons Instruments, Thermo Scientific, Waltham, MA, USA). Thin layer chromatography (TLC) were performed on pre-coated silica gel 60 F254 plates (Merck, Prague, Czech Republic) and visualized by exposure to UV light (254 or 366 nm). Melting points were measured on a Boetius stage apparatus (WEB Analytik, Dresden, Germany) and are uncorrected.
3.1.2. Synthesis of Compounds 3–9
3-Bromo-2-{[(3,4-dimethoxyphenyl)imino]methyl}-6-methoxyphenol (3a). To a solution of aniline 1a (410 mg; 2.67 mmol) in anhydrous EtOH (15 mL) was added benzaldehyde 2a (600 mg; 2.60 mmol). The reaction mixture was stirred at reflux for 1 h and then slowly cooled to 0–5 °C. Resultant precipitate was filtered off, washed with ice-cold EtOH (2 mL) and dried. Compound 3a (921 mg, 97%) was obtained as an orange microcrystalline solid. Mp. = 157–159 °C; 1H-NMR (400 MHz, CDCl3) δ = 15.27 (s, 1H), 9.05 (s, 1H), 7.02 (d, J = 8.8 Hz, 1H), 6.98–6.89 (m, 3H), 6.78 (d, J = 8.8 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 3.90 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 160.4, 154.6, 149.6, 148.8, 148.7, 140.0, 121.8, 116.4, 115.7, 114.7, 113.1, 111.4, 105.1, 56.2, 56.1, 56.0; HRMS (HESI, m/z): [M + H]+ calcd. for C16H17BrNO4, 366.0341; found 366.0335.
2-[(1,3-Benzodioxole-5-ylimino)methyl]-3-bromo-6-methoxyphenol (3c). Schiff base 3c was obtained in a similar manner to compound 3a using benzaldehyde 2a (750 mg; 3.25 mmol), aniline 1c (445 mg; 3.25 mmol) and anhydrous EtOH (20 mL). Compound 3c (1.09 g; 92%) was obtained as a dark red microcrystalline solid. Mp. = 176–178 °C; 1H-NMR (400 MHz, CDCl3) δ = 15.07 (br. s, 1H), 9.02 (s, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.92–6.89 (m, 1H), 6.88–6.85 (m 2H), 6.78 (d, J = 8.7 Hz, 1H), 6.03 (s, 2H), 3.91 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 160.6, 154.4, 148.8, 148.7, 147.4, 141.4, 122.0, 116.5, 116.0, 115.9, 114.9, 108.7, 101.8, 101.5, 56.3; HRMS (HESI, m/z): [M + H]+ calcd. for C15H13BrNO4, 350.0028; found 350.0023.
2-[(1,3-Benzodioxole-4-ylimino)methyl]-3-bromo-6-methoxyphenol (3d). Schiff base 3d was obtained in a similar manner to compound 3a using benzaldehyde 2a (572 mg; 2.48 mmol), aniline 1d (350 mg; 2.55 mmol) and anhydrous EtOH (15 mL). Compound 3d (737 mg; 85%) was obtained as a pale orange microcrystalline solid. Mp. = 195–196 °C; 1H-NMR (400 MHz, CDCl3) δ = 15.18 (s, 1H), 9.40 (s, 1H), 7.05 (d, J = 8.6 Hz, 1H), 6.96–6.88 (m, 2H), 6.82–6.77 (m, 2H), 6.09 (s, 2H), 3.92 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 164.3, 154.7, 148.9, 148.7, 140.3, 129.3, 122.2, 121.9, 116.7, 116.3, 116.0, 115.0, 107.7, 101.7, 56.2; HRMS (HESI, m/z): [M + H]+ calcd. for C15H13BrNO4, 350.0028; found 350.0022.
3-Bromo-2-{[(3,4-dimethoxyphenyl)amino]methyl}-6-methoxyphenol (4a). To a suspension of the Schiff base 3a (921 mg, 2.51 mmol) in anhydrous EtOH (25 mL) was added NaBH4 (95 mg; 2.51 mmol). After 2 h of stirring at room temperature the reaction mixture was neutralized by adding an aqueous solution of 10% AcOH (1 mL). The suspension dissolved into solution, which was concentrated under reduced pressure. The residue was extracted with a mixture of EtOAc (20 mL) and water (20 mL). The aqueous phase was washed with EtOAc (20 mL). The combined EtOAc phases were dried over Na2SO4, filtered and concentrated under reduced pressure to provide a yellow crude oil (737 mg, 80%). TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, CDCl3) δ = 7.05 (d, J = 9.3 Hz, 1H), 6.74 (d, J = 8.3 Hz, 1H), 6.66 (d, J = 8.3 Hz, 1H), 6.47 (d, J = 2.1 Hz, 1H), 6.37 (dd, J = 2.1, 8.3, Hz, 1H), 5.89 (brs, 2H), 4.53 (s, 2H), 3.85 (s, 3H), 3.83 (s, 3H), 3.80 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 149.7, 146.6, 146.2, 142.7, 141.7, 123.4, 123.2, 115.1, 112.7, 111.2, 106.1, 100.5, 56.5, 56.1, 55.7, 45.8; HRMS (HESI, m/z): [M + H]+ calcd. for C16H19BrNO4, 368.0492; found 368.0498.
2-[(1,3-Benzodioxole-5-ylamino)methyl]-3-bromo-6-methoxyphenol (4c). The secondary amine 4c was obtained in a similar manner as compound 4a using Schiff base 3c (1.0 g; 2.9 mmol), NaBH4 (109 mg; 2.9 mmol) and anhydrous EtOH (25 mL). The resulting crude product was a pale yellow oil (1.0 g, 99%). A sample for analysis was prepared by crystallization from toluene (100 mg/3 mL) and standing this solution at −20 °C. Mp. = 95–98 °C as a colorless crystalline compound; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.34 (s, 1H), 7.02 (d, J = 8.7 Hz, 1H), 6.86 (d, J = 8.7 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 6.42 (d, J = 1.8 Hz, 1H), 6.12 (dd, J = 8.3, 2.3 Hz, 1H), 5.82 (s, 2H), 5.21 (t, J = 5.2 Hz, 1H), 4.19 (d, J = 5.2 Hz, 2H), 3.79 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 147.6, 147.1, 146.2, 144.6, 138.1, 124.7, 122.2, 115.6, 112.2, 108.3, 103.4, 99.9, 95.4, 56.0, 42.9; HRMS (HESI, m/z): [M + H]+ calcd. for C15H15BrNO4, 352.0179; found 352.0182.
2-[(1,3-Benzodioxole-4-ylamino)methyl]-3-bromo-6-methoxyphenol (4d). The secondary amine 4d was obtained in a similar manner as compound 4a using Schiff base 3d (500 mg; 1.43 mmol), NaBH4 (54 mg; 1.43 mmol) and anhydrous EtOH (13 mL). The resulting crude product was a pale yellow oil (500 mg, 99%). A sample for analysis was prepared by crystallization from toluene (100 mg/3 mL) and standing this solution at −20 °C. Mp. = 89–90 °C as a colorless crystalline compound; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, CDCl3) δ = 7.07 (d, J = 8.7 Hz, 1H), 6.75 (t, J = 8.0 Hz, 1H), 6.68 (d, J = 8.7 Hz, 1H), 6.60 (d, J = 8.2 Hz, 1H), 6.36 (d, J = 7.8 Hz, 1H), 5.90 (s, 2H), 4.59 (s, 2H), 3.87 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 147.2, 146.2, 145.6, 134.6, 132.2, 123.9, 123.3, 122.2, 115.7, 111.1, 107.8, 100.6, 99.8, 56.1, 43.7; HRMS (HESI, m/z): [M + H]+ calcd. for C15H15BrNO4, 352.0179; found 352.0183.
N-(6-Bromo-2,3-dimethoxybenzyl)-3,4-dimethoxyaniline (4e). To a solution of bromobenzaldehyde 2b (1.0 g; 4.08 mmol) and aniline 1a (644 mg; 4.20 mmol) in anhydrous toluene (50 mL) was added NaBH(OAc)3 (2.6 g; 12.2 mmol). The resulting suspension was stirred at room temperature for 1 h, and then washed with water (2 × 35 mL) and brine (1 × 40 mL). The aqueous phase was extracted with toluene (1 × 40 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. To induce crystallization of the product, MeOH (8 mL) was added to the residue. The resulting solid was filtered off, washed with cold MeOH (1 mL) and dried. Compound 4e was obtained as a slightly pink crystalline solid (1.19 g; 76%). Mp. = 79–80 °C; TLC Rf = 0.2 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, CDCl3) δ = 7.26 (d, J = 8.8 Hz, 1H), 6.79–6.72 (m, 2H), 6.43 (d, J = 2.6 Hz, 1H), 6.33 (dd, J = 8.5, 2.6 Hz, 1H), 4.42 (s, 2H), 4.02 (br. s, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 152.3, 149.8, 148.7, 142.7, 141.7, 132.6, 128.0, 115.2, 113.0, 112.9, 104.6, 99.5, 61.5, 56.6, 55.9, 55.7, 43.9; HRMS (HESI, m/z): [M + H]+ calcd. for C17H21BrNO4, 382.0654; found 382.0648.
N-(6-Bromo-2,3-dimethoxybenzyl)-2,3-dimethoxyaniline (4f). The secondary amine 4f was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2b (400 mg; 1.63 mmol), aniline 1b (258 mg; 1.68 mmol) and NaBH(OAc)3 (1.04 g; 4.9 mmol) in anhydrous toluene (20 mL). The residue was crystallized from EtOH/water to provide compound 4f (212 mg; 34%) as a colorless microcrystalline solid. Mp. = 52–53 °C; TLC Rf = 0.5 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.34 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 1H), 6.85 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 8.2, 1.2 Hz, 1H), 6.33 (dd, J = 8.2, 1.2 Hz, 1H), 4.85 (t, J = 6.2 Hz, 1H), 4.33 (d, J = 6.2 Hz, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 3.72 (s, 3H), 3.60 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 148.4, 141.6, 135.0, 131.9, 127.9, 124.2, 119.5, 114.4, 113.8, 104.7, 101.6, 61.2, 59.5, 55.9, 55.5, 42.2; HRMS (HESI m/z): [M + H]+ calcd. for C17H21BrNO4, 382.0654; found 382.0648.
N-(6-Bromo-2,3-dimethoxybenzyl)-1,3-benzodioxole-5-amine (4g). The secondary amine 4g was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2b (400 mg; 1.63 mmol), aniline 1c (231 mg; 1.68 mmol) and NaBH(OAc)3 (1.04 g; 4.9 mmol) in anhydrous toluene (20 mL). The residue was crystallized from EtOH/water to provide compound 4g (275 mg; 46%) as a pale yellow microcrystalline solid. Mp. = 74–75 °C; TLC Rf = 0.4 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.34 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 6.65 (d, J = 8.3 Hz, 1H), 6.41 (d, J = 2.3 Hz, 1H), 6.12 (dd, J = 8.3, 2.3 Hz, 1H), 5.82 (s, 2H), 5.26 (t, J = 5.3 Hz, 1H), 4.16 (d, J = 5.2 Hz, 2H), 3.81 (s, 3H), 3.75 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 148.5, 147.7, 144.6, 138.1, 132.0, 127.7, 115.0, 113.7, 108.4, 103.2, 99.9, 95.2, 61.1, 55.9, 43.0; HRMS (HESI m/z): [M + H]+ calcd. for C16H17BrNO4, 366.0341; found 366.0337.
N-(6-Bromo-2,3-dimethoxybenzyl)-1,3-benzodioxole-4-amine (4h). The secondary amine 4h was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2b (253 mg; 1.03 mmol), aniline 1d (142 mg; 1.04 mmol) and NaBH(OAc)3 (660 mg; 3.1 mmol) in anhydrous toluene (12 mL). The residue was crystallized from MeOH to provide compound 4h (302 mg; 80%) as a colorless microcrystalline solid. Mp. = 90–92 °C; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.33 (d, J = 8.7 Hz, 1H), 6.98 (d, J = 9.2 Hz, 1H), 6.65 (t, J = 7.8 Hz, 1H), 6.46 (d, J = 8.2 Hz, 1H), 6.27 (dd, J = 7.8, 0.9 Hz, 1H), 5.90 (s, 2H), 4.82 (t, J = 6.0 Hz, 1H), 4.40 (d, J = 6.0 Hz, 2H), 3.80 (s, 3H), 3.77 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 148.4, 147.0, 133.2, 132.7, 131.8, 127.7, 122.1, 114.6, 113.7, 107.2. 100.0, 98.3, 61.0, 55.9, 42.6; HRMS (HESI m/z): [M + H]+ calcd. for C16H17BrNO4, 366.0341; found 366.0337.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-3,4-dimethoxyaniline (4i). The secondary amine 4i was prepared in a similar manner as amine 4e using bromobenzaldehyde 2c (1.0 g; 3.11 mmol), aniline 1a (491 mg; 3.21 mmol) and NaBH(OAc)3 (1.98 g; 9.4 mmol) in anhydrous toluene (50 mL). The residue was crystallized from MeOH to provide compound 4i (1.16 g; 81%) as a light pink microcrystalline solid. Mp.= 79–81 °C; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.43–7.29 (m, 6H), 7.03 (d, J = 8.8 Hz, 1H), 6.69 (d, J = 8.8 Hz, 1H), 6.39 (d, J = 8.6 Hz, 1H), 6.15 (dd, J = 8.6, 2.6 Hz, 1H), 5.10 (t, J = 5.6 Hz, 1H), 4.99 (s, 2H), 4.17 (d, J = 5.4 Hz, 2H), 3.86 (s, 3H), 3.61 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 149.8, 147.1, 143.7, 140.3, 137.1, 132.4, 128.4, 128.3, 128.1, 127.9, 115.0, 114.4, 113.6, 103.0, 98.7, 75.1, 56.6, 56.0, 55.1, 42.9; HRMS (HESI m/z): [M + H]+ calcd. for C23H25BrNO4, 458.0967; found 458.0961.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-2,3-dimethoxyaniline (4j). The secondary amine 4j was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2c (900 mg; 2.80 mmol), aniline 1b (442 mg; 2.89 mmol) and NaBH(OAc)3 (1.78 g; 8.4 mmol) in anhydrous toluene (45 mL). The residue was crystallized from EtOH to provide compound 4j (758 mg; 75%) as a pale yellow microcrystalline solid. Mp. = 86–87 °C; TLC Rf = 0.5 (hexane/EtOAc 10/3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.46–7.30 (m, 6H), 7.02 (d, J = 8.8 Hz, 1H), 6.81 (t, J = 8.2 Hz, 1H), 6.46 (d, J = 8.0 Hz, 1H), 6.32 (d, J = 8.3 Hz, 1H), 5.01 (s, 2H), 4.85 (t, J = 6.2 Hz, 1H), 4.27 (d, J = 4.9 Hz, 2H), 3.85 (s, 3H), 3.71 (s, 3H), 3.55 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 152.1, 147.0, 141.5, 137.1, 135.1, 132.2, 128.3, 128.2, 128.1, 128.0, 124.1, 114.4, 113.7, 104.6, 101.6, 74.9, 59.4, 56.0, 55.5, 42.4; HRMS (HESI m/z): [M + H]+ calcd. for C23H25BrNO4, 458.0967; found 458.0961.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-1,3-benzodioxole-5-amine (4k). The secondary amine 4k was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2c (600 mg; 1.87 mmol), aniline 1c (263 mg; 1.93 mmol) and NaBH(OAc)3 (1.19 g; 5.6 mmol) in anhydrous toluene (33 mL). The residue was crystallized from MeOH/acetone/water mixture to provide compound 4k (791 mg; 96%) as a pale brown microcrystalline solid. Mp. = 77–80 °C; TLC Rf = 0.5 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.41–7.27 (m, 6H), 7.04 (d, J = 9.1 Hz, 1H), 6.64 (d, J = 8.3 Hz, 1H), 6.38 (d, J = 2.3 Hz, 1H), 6.08 (dd, J = 8.3, 2.3 Hz, 1H), 5.82 (s, 2H), 5.23 (t, J = 5.2 Hz, 1H), 4.98 (s, 2H), 4.12 (d, J = 5.4 Hz, 2H), 3.86 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 147.7, 147.2, 144.6, 138.1, 137.1, 132.2, 128.3, 128.3, 128.1, 127.9, 115.1, 113.7, 108.4, 103.2, 99.9, 95.3, 75.0, 56.1, 43.2; HRMS (HESI m/z): [M + H]+ calcd. for C22H21BrNO4, 442.0654; found 442.0648.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-1,3-benzodioxole-4-amine (4l). The secondary amine 4l was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2c (1.0 g; 3.11 mmol), aniline 1d (439 mg; 3.20 mmol) and NaBH(OAc)3 (1.98 g; 9.3 mmol) in anhydrous toluene (45 mL). The residue was crystallized from EtOH to provide compound 4l (1.24 g; 90%) as a white microcrystalline solid. Mp. = 94–96 °C; TLC Rf = 0.4 (toluene); 1H-NMR (400 MHz, DMSO-d6) δ = 7.41–7.31 (m, 6H), 7.03 (d, J = 9.6 Hz, 1H), 6.62 (t, J = 8.3 Hz, 1H), 6.41 (d, J = 7.6 Hz, 1H), 6.26 (d, J = 7.6 Hz, 1H), 5.86 (s, 2H), 5.01 (s, 2H), 4.75 (t, J = 6.0 Hz, 1H), 4.33 (d, J = 6.0 Hz, 2H), 3.85 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 147.1, 147.0, 137.1, 133.2, 132.7, 132.1, 128.3 (2×), 128.1, 127.9, 122.1, 114.7, 113.8, 107.3, 100.0, 98.4, 74.9, 56.1, 42.7; HRMS (HESI m/z): [M + H]+ calcd. for C22H21BrNO4, 442.0654; found 442.0652.
2,3,8-Trimethoxyphenanthridin-7-ol (5a). To a suspension of benzylated phenanthridine 5i (52 mg; 0.14 mmol) in MeOH (8 mL) was added 10% Pd/C (5 mg) and the mixture was vigorously stirred under atmospheric pressure of hydrogen for 4 h. It was then filtered and concentrated under reduced pressure to afford compound 5a (28 mg; 71%) as an orange microcrystalline solid. Mp. = 212–215 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/2 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.88 (br. s., 1H), 9.40 (s, 1H), 8.18 (d, J = 9.1 Hz, 1H), 7.98 (s, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.47 (s, 1H), 4.00 (s, 3H), 3.95 (s, 3H), 3.93 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 149.9, 149.3, 145.5, 143.7, 142.7, 138.7, 126.4, 118.0, 117.7, 116.4, 112.8, 109.7, 102.7, 56.9, 55.9, 55.5; HRMS (HESI m/z): [M + H]+ calcd. for C16H16NO4 286.1079; found 286.1074.
3,4,8-Trimethoxyphenanthridin-7-ol (5b). Compound 5b was prepared in a similar manner as the base 5a using benzylated phenanthridine 5j (20 mg; 0.09 mmol), 10% Pd/C (2 mg) and MeOH (4 mL). After stirring for 4 h under atmospheric pressure of hydrogen, the reaction mixture was filtered. The filtrate contained both fully aromatized debenzylated product 5b and partially reduced debenzylated form 12b as a side product. Therefore, the mixture was vigorously stirred in air until only fully aromatized product was present (ca. 2 h). The solution was allowed to stand overnight at −20 °C and the resulting precipitate was filtered off to afford 5b (10 mg; 66%) as an orange crystalline solid. Mp.= 199–202 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/2 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.88 (s, 1H), 9.51 (d, J = 0.5 Hz, 1H), 8.35 (d, J = 9.3 Hz, 1H), 8.11 (d, J = 9.1 Hz, 1H), 7.63 (d, J = 9.1 Hz, 1H), 7.48 (d, J = 9.3 Hz, 1H), 3.96 (s, 3H), 3.95 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.0, 147.9, 144.2, 144.0, 143.1, 138.0, 126.6, 118.6, 118.2, 117.7, 115.8, 114.3, 112.5, 61.2, 56.7, 56.4; HRMS (HESI m/z): [M + H]+ calcd. for C16H16NO4 286.1079; found 286.1074.
3-Methoxy-[1,3]dioxolo[4,5-b]phenanthridin-4-ol (5c). Compound 5k (100 mg; 0.28 mmol) was added to ethanol (4 mL) containing aqueous 35% HCl (3 mL). This reaction mixture was heated in a sealed vial at 100 °C for 2.5 h. After removal of volatiles, the residue was diluted with water (5 mL) and alkalized with aqueous NH3 solution to pH = 8–9. The precipitated solid was filtered off, washed with water and dried to provide compound 5c (57 mg; 76%) as an orange crystalline solid. A sample for analysis was prepared by crystallization from larger amount of THF. Mp. = 260–270 °C (decomp.); Rf = 0.2 (CHCl3/MeOH 40/2 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.85 (s, 1H), 9.39 (s, 1H), 8.14 (s, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.63 (d, J = 9.0 Hz, 1H), 7.43 (s, 1H), 6.21 (s, 2H), 3.96 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 148.2, 147.84, 145.6, 143.8, 142.5, 140.04, 126.9, 119.5, 118.2, 116.56, 113.0, 106.9, 101.8, 100.0, 56.8; HRMS (HESI m/z): [M + H]+ calcd. for C15H12NO4, 270.0766; found 270.0760.
7-Methoxy-[1,3]dioxolo[4,5-c]phenanthridin-6-ol (5d). To a stirred solution of the secondary amine 4d (300 mg; 0.85 mmol) and Bu3SnH (460 μL; 1.70 mmol) in anhydrous toluene (15 mL) at 70 °C was added AIBN (210 mg, 1.28 mmol) and temperature was raised and maintained at 104–108 °C for a period of 3 h. Due to no further conversion of starting material (4d), another portion of Bu3SnH (160 μL; 0.60 mmol) and AIBN (50 mg, 0.30 mmol) were added to the reaction mixture and heating continued for a further 3 h. The reaction mixture was then cooled to room temperature, activated MnO2 [44] (222 mg, 2.56 mmol) was added and stirring continued overnight. The suspension was filtered and the filtrate concentrated under reduced pressure. Addition of cyclohexane (30 mL) to the residue resulted in the exclusion of a red precipitate, which was filtered off and further purified by silica gel gradient column chromatography (hexane/EtOAc 5/1–5/3 v/v) to afford compound 5d (30 mg; 13%) as an orange microcrystalline solid. Mp. = 246–248 °C; Rf = 0.2 (CHCl3/MeOH 40/2 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.99 (s, 1H), 9.44 (s, 1H), 8.16 (d, J = 8.8 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.35 (d, J = 8.8 Hz, 1H), 6.25 (s, 2H), 3.95 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 148.6, 146.1, 144.5, 143.3, 142.3, 129.3, 126.5, 119.5, 118.4, 115.8, 115.7, 112.7, 109.4, 102.0, 56.7; HRMS (HESI m/z): [M + H]+ calcd. for C15H12NO4, 270.0766; found 270.0761.
2,3,7,8-Tetramethoxyphenanthridin (5e). To a stirred solution of the secondary amine 4e (600 mg, 1.57 mmol) and Bu3SnH (845 μL; 3.14 mmol) in anhydrous toluene (30 mL) at 70 °C was added AIBN (387 mg, 2.36 mmol) and temperature of the reaction mixture was raised and maintained at 104–108 °C for a period of 3 h. The reaction was carried out under an inert atmosphere of argon. After 3 h the reaction mixture was slowly cooled to room temperature and activated MnO2 (409 mg; 4.71 mmol) was added. The resulting mixture was stirred overnight, then filtered and concentrated under reduced pressure. Addition of cyclohexane (8 mL) to the residue resulted in the exclusion of a brown precipitate. The suspension was left to stand overnight at 5 °C, and the next day it was filtered, washed with cyclohexane (2 mL) and recrystallized from CHCl3/hexane. Compound 5e (185 mg, 39%) was obtained as a beige crystalline solid. Mp. = 154–158 °C (CHCl3/hexane); Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.35 (s, 1H), 8.51 (d, J = 8.8 Hz, 1H), 8.03 (s, 1H), 7.74 (d, J = 9.1 Hz, 1H), 7.51 (s, 1H), 4.02 (s, 3H), 4.00 (s, 6H), 3.93 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 150.1, 149.6, 148.9, 145.0, 144.1, 139.0, 126.4, 120.2, 118.9, 118.5, 117.6, 109.8, 102.6, 61.4, 56.6, 56.0, 55.6; HRMS (HESI m/z): [M + H]+ calcd. for C17H18NO4, 300.1236; found 300.1230.
3,4,7,8-Tetramethoxyphenanthridine (5f). Phenanthridine 5f was prepared in a similar manner as the compound 5e using amine 4f (467 mg; 1.22 mmol), Bu3SnH (660 μL; 2.44 mmol), AIBN (301 mg, 1.83 mmol) and MnO2 (318 mg; 3.66 mmol) in anhydrous toluene (23 mL). The recrystallization from CHCl3/hexane afforded compound 5f (150 mg; 41%) as a beige crystalline solid. Mp. = 142–144 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.47 (s, 1H), 8.46 (d, J = 9.1 Hz, 1H), 8.42 (d, J = 9.1 Hz, 1H), 7.75 (d, J = 9.1 Hz, 1H), 7.52 (d, J = 9.1 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H), 3.96 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.2, 149.3, 147.5, 144.5, 144.0, 138.0, 126.7, 119.7, 119.3, 118.4, 118.2, 117.8, 114.6, 61.5, 61.3, 56.5, 56.4; HRMS (HESI m/z): [M + H]+ calcd. for C17H18NO4, 300.1236; found 300.1230.
3,4-Dimethoxy-[1,3]dioxolo[4,5-b]phenanthridine (5g). Phenanthridine 5g was prepared in a similar manner as compound 5e using secondary amine 4g (265 mg; 0.72 mmol), Bu3SnH (390 μL; 1.45 mmol), AIBN (178 mg, 1.09 mmol), MnO2 (189 mg; 2.17 mmol) in anhydrous toluene (13 mL). Compound 5g (55 mg, 27%) was obtained as a beige microcrystalline solid. Mp. = 172–174 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.33 (d, J = 0.5 Hz, 1H), 8.44 (d, J = 9.0 Hz, 1H), 8.20 (s, 1H), 7.73 (d, J = 9.3 Hz, 1H), 7.46 (s, 1H), 6.23 (s, 2H), 3.99 (s, 3H), 3.98 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 149.0, 148.4, 148.1, 145.2, 143.9, 140.2, 126.9, 119.5, 119.5, 119.1, 118.7, 107.0, 102.0, 100.0, 61.4, 56.6; HRMS (HESI m/z): [M + H]+ calcd. for C16H14NO4, 284.0923; found 284.0917.
6,7-Dimethoxy-[1,3]dioxolo[4,5-c]phenanthridine (5h). Phenanthridine 5h was prepared in a similar manner as the product 5e using amine 4h (478 mg; 1.31 mmol), Bu3SnH (700 μL; 2.61 mmol), AIBN (322 mg; 1.96 mmol) and MnO2 (340 mg; 3.92 mmol) in anhydrous toluene (23 mL). The recrystallization from CHCl3/hexane afforded compound 5h (135 mg; 37%) as a brown crystalline solid. Mp. = 163–166 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.40 (s, 1H), 8.42 (d, J = 9.1 Hz, 1H), 8.23 (d, J = 8.8 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.39 (d, J = 8.6 Hz, 1H), 6.27 (s, 2H), 4.00 (s, 3H), 3.99 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 149.5, 148.1, 146.2, 144.7, 142.3, 129.1, 126.4, 122.3, 119.8, 119.5, 118.3, 115.7, 109.7, 102.1, 61.5, 56.5; HRMS (HESI m/z): [M + H]+ calcd. for C16H14NO4, 284.0923; found 284.0917.
7-(Benzyloxy)-2,3,8-trimethoxyphenanthridine (5i). Phenanthridine 5i was prepared in a similar manner as phenanthridine 5e using secondary amine 4i (1.27 g, 2.78 mmol), Bu3SnH (1.5 mL; 5.55 mmol), AIBN (684 mg; 4.16 mmol) and MnO2 (724 mg, 8.33 mmol) in anhydrous toluene (50 mL). The recrystallization from CHCl3/hexane afforded compound 5i (240 mg; 23%) as a beige crystalline solid. Mp. = 155–156 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.25 (s, 1H), 8.51 (d, J = 9.1 Hz, 1H), 8.01 (s, 1H), 7.76 (d, J = 9.1 Hz, 1H), 7.53–7.49 (m, 2H), 7.46 (s, 1H), 7.41–7.30 (m, 3H), 5.25 (s, 2H), 4.04 (s, 3H), 4.01 (s, 3H), 3.92 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 150.0, 149.5, 149.0, 145.2, 142.6, 138.9, 137.1, 128.6, 128.4, 128.2, 126.3, 120.5, 118.7, 118.5, 117.5, 109.8, 102.6, 75.0, 56.7, 56.0, 55.5; HRMS (HESI m/z): [M + H]+ calcd. for C23H22NO4, 376.1549; found 376.1543.
7-(Benzyloxy)-3,4,8-trimethoxyphenanthridine (5j). Phenanthridine 5j was prepared in a similar manner as phenanthridine 5e using secondary amine 4j (950 mg, 2.07 mmol), Bu3SnH (1115 μL; 4.15 mmol), AIBN (511 mg, 3.11 mmol) and MnO2 (541 mg, 6.22 mmol) in anhydrous toluene (37 mL). The recrystallization from CHCl3/hexane afforded compound 5j (233 mg; 30%) as a slightly gray microcrystalline solid. Mp. = 153–155 °C; TLC Rf = 0.3 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.38 (s, 1H), 8.45 (d, J = 8.8 Hz, 1H), 8.40 (d, J = 9.1 Hz, 1H), 7.77 (d, J = 9.1 Hz, 1H), 7.55–7.48 (m, 3H), 7.42–7.30 (m, 3H), 5.26 (s, 2H), 4.04 (s, 3H), 3.96 (s, 3H), 3.93 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.2, 149.4, 147.6, 143.9, 143.0, 137.9, 137.0, 128.7, 128.4, 128.2, 126.6, 120.1, 119.1, 118.3, 118.2, 117.8, 114.5, 75.1, 61.3, 56.6, 56.4; HRMS (HESI m/z): [M + H]+ calcd. for C23H22NO4, 376.1549; found 376.1543.
4-(Benzyloxy)-3-methoxy-[1,3]dioxolo[4,5-b]phenanthridine (5k). Phenanthridine 5k was prepared in a similar manner as phenanthridine 5e using secondary amine 4k (730 mg; 1.65 mmol), Bu3SnH (890 μL; 3.30 mmol), AIBN (407 mg, 2.48 mmol) and MnO2 (430 mg; 4.95 mmol) in anhydrous toluene (29 mL). The recrystallization from CHCl3/hexane afforded compound 5k (280 mg; 47%) as a brown crystalline solid. Mp.= 129–131 °C; TLC Rf = 0.3 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.25 (s, 1H), 8.43 (d, J = 9.1 Hz, 1H), 8.18 (s, 1H), 7.76 (d, J = 9.1 Hz, 1H), 7.53–7.49 (m, 2H), 7.43–7.31 (m, 4H), 6.21 (s, 2H), 5.24 (s, 2H), 4.03 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 149.1, 148.4, 148.0, 145.4, 142.4, 140.0, 137.0, 128.6, 128.4, 128.2, 126.7, 120.6, 119.3, 118.9, 118.6, 106.9, 101.9, 99.9, 75.0, 56.6; HRMS (HESI m/z): [M + H]+ calcd. for C22H18NO4, 360.1236; found 360.1230.
6-(Benzyloxy)-7-methoxy-[1,3]dioxolo[4,5-c]phenanthridine (5l). Phenanthridine 5l was prepared in a similar manner as phenanthridine 5e using secondary amine 4l (1.1 g; 2.48 mmol), Bu3SnH (1.34 mL; 4.95 mmol), AIBN (610 mg, 3.72 mmol) and MnO2 (645 mg; 7.42 mmol) in anhydrous toluene (40 mL). The recrystallization from CHCl3/hexane afforded compound 5l (555 mg; 62%) as a beige crystalline solid. Mp.= 163–164 °C; TLC Rf = 0.3 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.27 (s, 1H), 8.42 (d, J = 9.1 Hz, 1H), 8.21 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 9.1 Hz, 1H), 7.50 (d, J = 6.9 Hz, 2H), 7.41 – 7.30 (m, 4H), 6.25 (s, 2H), 5.26 (s, 2H), 4.04 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 149.62, 148.32, 146.25, 143.13, 142.34, 136.87, 129.06, 128.77, 128.40, 128.28, 126.38, 120.24, 119.31, 119.10, 118.41, 115.68, 109.71, 102.07, 75.07, 56.56; HRMS (HESI m/z): [M + H]+ calcd. for C22H18NO4, 360.1236; found 360.1230.
3.1.3. General Procedure for the Preparation of Phenanthridine Hydrochlorides 6a–6l
The above prepared phenanthridine derivatives 5 (ca. 20 mg) were dissolved in dioxane (2–3 mL). To the resulting solution a 4 M HCl in dioxane solution (1.5 mL) was added resulting in formation of a yellow-orange precipitate. To the suspension was added ether (4 mL). The precipitate was filtered off, washed with ether (2 mL) and dried under vacuum over KOH. The yields of hydrochlorides 6 were within 80–95%. All compounds were proved by elemental analysis; the recorded values were within +/−0.4%.
7-Hydroxy-2,3,8-trimethoxyphenanthridin-5-ium chloride (6a). Elemental analysis, calcd. for C16H16ClNO4 (321.8), C, 59.73; H, 5.01; N, 4.35; found C, 59.83; H, 5.11; N, 4.08.
7-Hydroxy-3,4,8-trimethoxyphenanthridin-5-ium chloride (6b). Elemental analysis, calcd. for C16H16ClNO4 (321.8), C, 59.73; H, 5.01; N, 4.35; found C, 59.74; H, 4.90; N, 4.18.
4-Hydroxy-3-methoxy-[1,3]dioxolo[4,5-b]phenanthridin-6-ium chloride (6c). Elemental analysis, calcd. for C15H12ClNO4 (305.7), C, 58.93; H, 3.96; N, 4.58; found C, 58.73; H, 3.77; N, 4.88.
6-Hydroxy-7-methoxy-[1,3]dioxolo[4,5-c]phenanthridin-4-ium chloride (6d). Elemental analysis, calcd. for C15H12ClNO4 (305.7), C, 58.93; H, 3.96; N, 4.58; found C, 58.65; H, 3.98; N, 4.73.
2,3,7,8-Tetramethoxyphenanthridin-5-ium chloride (6e). Elemental analysis, calcd. for C17H18ClNO4 (335.8), C, 60.81; H, 5.40; N, 4.17; found C, 61.15; H, 5.63; N, 4.26.
3,4,7,8-Tetramethoxyphenanthridin-5-ium chloride (6f). Elemental analysis, calcd. for C17H18ClNO4 (335.8), C, 60.81; H, 5.40; N, 4.17; found C, 61.01; H, 5.31; N, 4.02.
3,4-Dimethoxy-[1,3]dioxolo[4,5-b]phenanthridin-6-ium chloride (6g). Elemental analysis, calcd. for C16H14ClNO4 (319.7), C, 60.10; H, 4.41; N, 4.38; found C, 60.01; H, 4.71; N, 4.22.
6,7-Dimethoxy-[1,3]dioxolo[4,5-c]phenanthridin-4-ium chloride (6h). Elemental analysis, calcd. for C16H14ClNO4 (319.7), C, 60.10; H, 4.41; N, 4.38; found C, 60.24; H, 4.69; N, 4.48.
7-(Benzyloxy)-2,3,8-trimethoxyphenanthridin-5-ium chloride (6i). Elemental analysis, calcd. for C23H22ClNO4 (411.9), C, 67.07; H, 5.38; N, 3.40; found C, 67.17; H, 5.31; N, 3.46.
7-(Benzyloxy)-3,4,8-trimethoxyphenanthridin-5-ium chloride (6j). Elemental analysis, calcd. for C23H22ClNO4 (411.9), C, 67.07; H, 5.38; N, 3.40; found C, 67.22; H, 5.46; N, 3.20.
4-(Benzyloxy)-3-methoxy-[1,3]dioxolo[4,5-b]phenanthridin-6-ium chloride (6k). Elemental analysis, calcd. for C22H18ClNO4 (395.8), C, 66.75; H, 4.58; N, 3.54; found C, 66.66; H, 4.75; N, 3.44.
6-(Benzyloxy)-7-methoxy-[1,3]dioxolo[4,5-c]phenanthridin-4-ium chloride (6l). Elemental analysis, calcd. for C22H18ClNO4 (395.8), C, 66.75; H, 4.58; N, 3.54; found C, 66.58; H, 4.80; N, 3.32.
4-Hydroxy-3-methoxy-6-methyl-[1,3]dioxolo[4,5-b]phenanthridin-6-ium iodide (7c). The free base 5c (20 mg; 0.08 mmol) was dissolved in ACN (10 mL) and MeI (19 μL; 0.3 mmol) was added. The reaction mixture was allowed to stand at r.t. in the dark place without stirring. After 7 days, the precipitated crystalline solid was filtered and washed with cold ACN (1 mL) to obtain poorly soluble substance 7c (18 mg; 61%) as a dark orange crystalline solid. Mp. = 250–255 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 11.35 (br. s, 1H), 9.95 (s, 1H), 8.50 (s, 1H), 8.35 (d, J = 9.0 Hz, 1H), 8.05 (d, J = 9.1 Hz, 1H), 7.97 (s, 1H), 6.41 (s, 2H), 4.56 (s, 3H), 4.05 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 150.7, 150.1, 147.42, 145.7, 145.2, 130.22, 127.6, 124.0, 122.7, 114.6, 113.5, 103.71, 101.4, 98.6, 57.0, 46.1; HRMS (HESI m/z): [M − I]+ calcd. for C16H14NO4, 284.0923; found 284.0917; elemental analysis, calcd. for C16H14INO4 (411.2), C, 46.74; H, 3.43; N, 3.41; found C, 46.70; H, 3.66; N, 3.35.
6-Hydroxy-7-methoxy-4-methyl-[1,3]dioxolo[4,5-c]phenanthridin-4-ium iodide (7d). The quaternary salt 7d was prepared in a similar manner as compound 7c using base 5d (10 mg; 0.04 mmol), MeI (9 μL; 0.15 mmol) and ACN (3 mL). Compound 7d (9 mg; 61%) was obtained as a dark orange crystalline solid. Mp. = 236–239 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 11.52 (br. s, 1H), 9.90 (s, 1H), 8.49 (d, J = 8.6 Hz, 1H), 8.26 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.69 (dd, J = 1.0, 8.8 Hz, 1H), 6.38 (s, 2H), 4.65 (s, 3H), 4.04 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 148.8, 147.4, 145.7, 138.1, 126.9, 124.2, 120.9, 120.0, 118.4, 113.7, 113.0, 112.4, 103.1, 56.8, 48.2; HRMS (HESI m/z): [M − I]+ calcd. for C16H14NO4, 284.0923; found 284.0917; elemental analysis, calcd. for C16H14INO4 (411.2), C, 46.74; H, 3.43; N, 3.41; found C, 46.88; H, 3.54; N, 3.32.
2,3,7,8-Tetramethoxy-5-methylphenanthridinium iodide (7e). The quaternary salt 7e was prepared in a similar manner as compound 7d using base 5e (280 mg, 0.94 mmol), MeI (230 μL; 3.74 mmol) and ACN (14 mL). Compound 7e (372 mg; 90%) was obtained as a dark orange needle-like crystalline solid. Mp. = 263–266 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.96 (s, 1H), 8.84 (d, J = 9.3 Hz, 1H), 8.33 (s, 1H), 8.20 (d, J = 9.3 Hz, 1H), 7.73 (s, 1H), 4.70 (s, 3H), 4.13 (s, 6H), 4.10 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 149.9, 146.9, 145.6, 129.1, 127.7, 125.3, 120.8, 119.2, 118.7, 103.9, 100.7, 62.1, 57.0, 56.8, 56.6, 46.0; HRMS (HESI m/z): [M − I]+ calcd. for C18H20NO4, 314.1392; found 314.1387; elemental analysis, calcd. for C18H20INO4 (441.3), C, 48.99; H, 4.57 N, 3.17; found C, 48.92; H, 4.15; N, 3.12.
3,4,7,8-Tetramethoxy-5-methylphenanthridinium iodide (7f). The quaternary salt 7f was prepared in a similar manner as compound 7d using base 5f (60 mg, 0.20 mmol), MeI (50 μL, 0.80 mmol) and ACN (5 mL). Compound 7f (42 mg; 47%) was obtained as an orange crystalline solid. Mp. = 186–188 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.95 (s, 1H), 8.81 (d, J = 9.3 Hz, 1H), 8.70 (d, J = 9.3 Hz, 1H), 8.19 (d, J = 9.3 Hz, 1H), 7.89 (d, J = 9.3 Hz, 1H), 4.81 (s, 3H), 4.13 (s, 3H), 4.09 (s, 3H), 4.08 (s, 3H), 3.99 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 154.4, 153.6, 150.0, 146.4, 140.1, 128.3, 128.1, 126.2, 120.5, 120.2, 118.5, 118.0, 116.9, 62.2, 61.8, 56.9, 51.1, 39.5; HRMS (HESI m/z): [M − I]+ calcd. for C18H20NO4, 314.1392; found 314.1387.
3,4-Dimethoxy-6-methyl-[1,3]dioxolo[4,5-b]phenanthridin-6-ium iodide (7g). The quaternary salt 7g was prepared in a similar manner as substance 7d using base 5g (30 mg, 0.11 mmol), MeI (25 μL; 0.42 mmol) and ACN (6 mL). Compound 7g (40 mg; 89%) was obtained as a bright yellow crystalline solid. Mp. > 300 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.96 (s, 1H), 8.71 (d, J = 9.3 Hz, 1H), 8.58 (s, 1H), 8.19 (d, J = 9.3 Hz, 1H), 8.04 (s, 1H), 6.44 (s, 2H), 4.62 (s, 3H), 4.11 (s, 3H), 4.08 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.1, 150.4, 150.0, 147.2, 145.5, 130.6, 128.1, 125.5, 122.8, 119.1, 118.7, 103.9, 101.2, 98.5, 62.1, 57.0, 46.4; HRMS (HESI m/z): [M − I]+ calcd. for C17H16NO4, 298.1079; found 298.1074.
6,7-Dimethoxy-4-methyl-[1,3]dioxolo[4,5-c]phenanthridin-4-ium iodide (7h). The quaternary salt 7h was prepared in a similar manner as the substance 7d using free base 5h (90 mg; 0.32 mmol), MeI (80 μL; 1.27 mmol) and ACN (5 mL). Compound 7h (26 mg; 19%) was obtained as a blood-red crystalline solid. Mp. = 223–224 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.95 (s, 1H), 8.64 (d, J = 9.1 Hz, 1H), 8.59 (d, J = 8.8 Hz, 1H), 8.16 (d, J = 9.3 Hz, 1H), 7.76 (d, J = 8.8 Hz, 1H), 6.42 (s, 2H), 4.71 (s, 3 H), 4.11 (s, 3H), 4.07 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 150.2, 149.1, 146.7, 138.2, 127.6, 126.0, 120.9, 120.2, 118.7, 118.6, 118.0, 112.8, 103.3, 62.2, 56.9, 48.7; HRMS (HESI m/z): [M − I]+ calcd. for C17H16NO4, 298.1079; found 298.1074.
7-(Benzyloxy)-2,3,8-trimethoxy-5-methylphenanthridinium iodide (7i). The quaternary salt 7i was prepared in a similar manner as compound 7d using free base 5i (91 mg; 0.24 mmol), MeI (60 μL; 0.97 mmol) and ACN (9 mL). Compound 7i (77 mg; 61%) was obtained as a bright yellow crystalline solid. Mp. = 207–210 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.79 (s, 1H), 8.83 (d, J = 9.3 Hz, 1H), 8.30 (s, 1H), 8.20 (d, J = 9.1 Hz, 1H), 7.70 (s, 1H), 7.58 (d, J = 7.5 Hz, 2H), 7.43–7.30 (m, 3H), 5.40 (s, 2H), 4.68 (s, 3H), 4.12 (s, 6H), 4.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 150.2, 146.7, 143.9, 136.3, 128.9, 128.9, 128.4, 128.3, 127.5, 125.0, 120.7, 119.4, 119.1, 103.9, 100.7, 75.4, 57.0, 56.7, 56.6, 46.2; HRMS (HESI m/z): [M − I]+ calcd. for C24H24NO4, 390.1705; found 390.1700.
7-(Benzyloxy)-3,4,8-trimethoxy-5-methylphenanthridinium iodide (7j). The quaternary salt 7j was prepared in a similar manner as compound 7d using free base 5j (150 mg; 0.40 mmol), MeI (100 μL; 1.6 mmol) and ACN (7 mL). Compound 7j (123 mg; 60%) was obtained as a light orange microcrystalline solid. Mp. = 169–173 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.79 (s, 1H), 8.80 (d, J = 9.3 Hz, 1H), 8.70 (d, J = 9.1 Hz, 1H), 8.20 (d, J = 9.1 Hz, 1H), 7.89 (d, J = 9.3 Hz, 1H), 7.59 (d, J = 7.0 Hz, 2 H), 7.48–7.26 (m, 3H), 5.41 (s, 2H), 4.79 (s, 3H), 4.10 (s, 3H), 4.08 (s, 3H), 3.98 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 154.4, 153.5, 150.3, 144.6, 140.1, 136.3, 128.9, 128.4, 128.3, 128.2, 127.9, 126.0, 120.4, 120.2, 118.8, 118.5, 117.0, 75.6, 61.8, 57.0, 56.9, 51.3; HRMS (HESI m/z): [M − I]+ calcd. for C24H24NO4, 390.1705; found 390.1700.
4-(Benzyloxy)-3-methoxy-6-methyl-[1,3]dioxolo[4,5-b]phenanthridin-6-ium iodide (7k). The quaternary salt 7k was prepared in a similar manner as compound 7d using free base 5k (91 mg, 0.25 mmol), MeI (60 μL, 1.0 mmol) and ACN (8 mL). The reaction was conducted at 50 °C for 36 h. Compound 7k (77 mg; 61%) was obtained as a pale yellow crystalline solid. Mp. = 198–201 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.81 (s, 1H), 8.69 (d, J = 9.1 Hz, 1H), 8.55 (s, 1H), 8.19 (d, J = 9.3 Hz, 1H), 8.01 (s, 1H), 7.61–7.56 (m, 2H), 7.42–7.31 (m, 3H), 6.44 (s, 2H), 5.38 (s, 2H), 4.60 (s, 3H), 4.10 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.1, 150.4, 150.2, 147.0, 143.7, 136.3, 130.5, 128.9, 128.4, 128.3, 128.0, 125.3, 122.8, 119.2, 119.2, 103.9, 101.3, 98.5, 75.4, 56.9, 46.7; HRMS (HESI m/z): [M − I]+ calcd. for C23 H20NO4, 374.1392; found 374.1387.
6-(Benzyloxy)-7-methoxy-4-methyl-[1,3]dioxolo[4,5-c]phenanthridin-4-ium iodide (7l). The quaternary salt 7l was prepared in a similar manner as compound 7d with using free base 5l (36 mg, 0.1 mmol), MeI (24 μL, 0.4 mmol) and ACN (4 mL). The reaction was conducted at 50 °C for 36 h. Compound 7l (28 mg; 55%) was obtained as a light orange solid. Mp. = 169–172 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.80 (s, 1H), 8.64 (d, J = 9.2 Hz, 1H), 8.58 (d, J = 8.7 Hz, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.75 (d, J = 8.7 Hz, 1H), 7.64–7.52 (m, 2H), 7.44–7.28 (m, 3H), 6.40 (s, 2H), 5.39 (s, 2H), 4.69 (s, 3H), 4.08 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 150.2, 149.2, 145.0, 138.2, 136.2, 128.9, 128.5, 128.4, 127.6, 125.8, 120.8, 120.2, 119.0, 118.6, 118.5, 112.9, 103.3, 75.6, 56.9, 48.9; HRMS (HESI m/z): [M − I]+ calcd. for C23 H20NO4, 374.1392; found 374.1387.
3.1.4. Preparation of 2,3,7,8-tetramethoxy-5-methylphenanthridinium Salts 9t–9z
To a suspension of quaternary iodide 7e (54 mg; 0.11 mmol) in EtOH (18 mL) was added 1.25 N aqueous NaOH (180 μL; 0.22 mmol). After 1 h of stirring the resulting colorless solution was diluted with water (2 mL) and concentrated under reduced pressure to remove EtOH resulting in precipitation of a white crystalline solid (pseudobase 8), which was not isolated. To the suspension of pseudobase was added 20% aqueous solution of corresponding acid (10 mL) and the mixture was stirred for 10 min, and then cooled to 0 °C. The resulting yellow precipitate was filtered, washed with ice water (2 mL) and dried at 110 °C.
2,3,7,8-Tetramethoxy-5-methylphenanthridinium chloride (9t): Yield 35 mg (82%) as a yellow microcrystalline compound. Mp. = 220–222 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.96 (s, 1H), 8.83 (d, J = 9.2 Hz, 1H), 8.29 (s, 1H), 8.17 (d, J = 9.3 Hz, 1H), 7.72 (s, 1H), 4.71 (s, 3H), 4.12 (s, 3H), 4.11 (s, 3H), 4.10 (s, 3H), 4.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 149.9, 146.8, 145.6, 129.1, 127.6, 125.2, 120.8, 119.18, 118.68, 103.9, 100.7, 62.1, 57.1, 56.7, 56.6, 46.0; elemental analysis, calcd. for C18H20ClNO4 (349.8), C, 61.80; H, 5.76; N, 4.00; found C, 61.83; H, 5.69; N, 4.03.
2,3,7,8-Tetramethoxy-5-methylphenanthridinium perchlorate (9u): Yield 40 mg (80%) as a light orange microcrystalline compound. Mp. = above 300 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.93 (s, 1H), 8.80 (d, J = 9.2 Hz, 1H), 8.28 (s, 1H), 8.17 (d, J = 9.3 Hz, 1H), 7.70 (s, 1H), 4.69 (s, 3H), 4.12 (s, 3H), 4.12 (s, 3H), 4.09 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 149.9, 146.8, 145.6, 129.1, 127.6, 125.2, 120.8, 119.18, 118.68, 103.9, 100.7, 62.1, 57.1, 56.7, 56.6, 46.0; elemental analysis, calcd. for C18H20ClNO8 (413.8), C, 52.24; H, 4.87; N, 3.38; found C, 52.12; H, 4.68; N, 3.35.
2,3,7,8-Tetramethoxy-5-methylphenanthridinium hydrogensulfate (9w): Yield 38 mg (75%) as a yellow microcrystalline compound. Mp. = 272–280 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.94 (s, 1H), 8.82 (d, J = 9.2 Hz, 1H), 8.30 (s, 1H), 8.18 (d, J = 9.3 Hz, 1H), 7.72 (s, 1H), 4.70 (s, 3H), 4.12 (s, 3H), 4.12 (s, 3H), 4.10 (s, 3H), 4.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 149.9, 146.8, 145.6, 129.1, 127.6, 125.2, 120.8, 119.18, 118.68, 103.9, 100.7, 62.1, 57.1, 56.7, 56.6, 46.0; elemental analysis, calcd. for C18H21NO8S (411.4), C, 52.55; H, 5.14; N, 3.40; found C, 52.40; H, 5.24; N, 3.43.
2,3,7,8-Tetramethoxy-5-methylphenanthridinium nitrate (9z): Yield 37 mg (80%) as a light orange microcrystalline compound. Mp. = 266–269 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.95 (s, 1H), 8.83 (d, J = 9.2 Hz, 1H), 8.32 (s, 1H), 8.19 (d, J = 9.3 Hz, 1H), 7.72 (s, 1H), 4.70 (s, 3H), 4.12 (s, 6H), 4.10 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 149.9, 146.8, 145.6, 129.1, 127.6, 125.2, 120.8, 119.18, 118.68, 103.9, 100.7, 62.1, 57.1, 56.7, 56.6, 46.0; elemental analysis, calcd. for C18H20N2O7 (376.1), C, 57.44; H, 5.36; N, 7.44; found C, 57.36; H, 5.45; N, 7.48.
3.2. Antibacterial Activity Testing
3.2.1. Agar Diffusion Test
Overnight cultures of test organisms were grown in LB broth for 18–24 h and standard suspensions of 1.5 × 108 CFU/mL were prepared in a sterile saline solution (0.9% NaCl) according to a BaSO4 0.5 McFarland Standard. The standardized suspension (0.1 mL) was added to 34 mL of sterile, melted and tempered (47–50 °C) Mueller-Hinton No. 2 agar. After gentle mixing, the inoculated melted agar was poured into a sterile Petri dish (145 mm × 20 mm, Greiner Bio-One, Kremsmünster, Austria) and allowed to solidify next to the flame with lids slightly ajar. Wells of 9 mm diameter were cut from the Petri dish agar and filled with 50 μL of the test sample solution. Solutions were made at 20 mM in DMSO and diluted in MeOH to 2 mM. The Petri dish was incubated at 37 °C for 18–24 h and the inhibition zone diameters were measured (mm) with an electronic caliper after 24–48 h.
3.2.2. MIC Determination
Antibacterial activity of the compounds was determined by measuring their minimum inhibitory concentrations (MIC) using the broth microdilution method. Each well of a 96-well microtiter plate was filled with 50 μL of sterile iron deficient MH2 broth. Each test compound was dissolved in DMSO making a 10 mM solution, then diluted with sterile MH2 broth to 800 µM. Exactly 50 μL of the compound solution was added to the first well of the microtiter plate and 2-fold serial dilutions were made down each row of the plate. A pre culture of bacteria was grown in Luria-Bertani broth overnight at 37 °C. This was diluted to McFarland standard 0.5 (1.5 × 108 CFU) with saline. 100 µL of the bacterial suspension was further diluted with 14.9 mL of MHII broth. Exactly 50 μL of bacterial (in broth) inoculum (1 × 106 CFU/mL) was then added to each well giving a total volume of 100 μL/well and 5 × 105 CFU/mL and a compound concentration gradient of 200–0.1 µM. The plate was incubated at 37 °C. After 18 h, each well was examined visually for bacterial growth. The MIC was recorded as the lowest compound concentration required to inhibit bacterial growth as judged by turbidity relative to a row of wells diluted with a the solvent DMSO as a growth control. Ciprofloxacin was included in a control row at a concentration gradient of 5 µg/mL–0.0025 µg/L.
3.3. Anticancer Activity In Vitro
The in vitro anticancer activity was determined using MCF-7 (breast adenocarcinoma) and K-562 (chronic myelogeneous leukemia) cell lines as described earlier [48]. Briefly, cells were treated in triplicate with three different doses of each compound for 72 h. After treatments, Calcein AM solution was added, and fluoresence from live cells was measured at 485 nm/538 nm (excitation/emission) using a Fluoroskan Ascent microplate reader (Thermo Scientific, Waltham, MA, USA). The EC50 value, that is, the drug concentration reducing number of live cells to 50%, was calculated from the dose response curves that resulted from the assays. The cells were maintained in DMEM medium supplemented with 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 µg/mL) and cultivated at 37 °C in 5% CO2.
3.3.1. Cell Cycle Analysis
Cell cycle analysis was performed as described earlier [48]. Briefly, subconfluent cells were treated with different concentrations of test compound for 24 h. The cultures were pulse-labeled with 10 μM 5-bromo-2′-deoxyuridine (BrdU) for 30 min at 37 °C prior to harvesting. The cells were then washed in PBS, fixed with 70% ethanol, and denatured in 2 M HCl. Following neutralization, the cells were stained with anti-BrdU fluorescein-labeled antibodies, washed, stained with propidium iodide, and analyzed by flow cytometry using a 488 nm laser (FACSVerse, Becton Dickinson, Franklin Lakes, NJ, USA).
3.3.2. Immunoblotting
Immunoblotting was performed as described earlier [48]. Briefly, cellular lysates were prepared by harvesting cells in Laemmli sample buffer. Proteins were separated on SDS-polyacrylamide gels and electroblotted onto nitrocellulose membranes. After blocking, the membranes were incubated with specific primary antibodies overnight, washed and then incubated with peroxidase-conjugated secondary antibodies. Finally, peroxidase activity was detected with ECL+ reagents (AP Biotech, Prague, Czech Republic) using a LAS-4000 CCD camera (AP Biotech, Prague, Czech Republic). Specific antibodies against PARP were purchased from Santa Cruz Biotechnology (Dallas, TX, USA) peroxidase-labeled secondary antibodies was from Sigma Aldrich (Prague, Czech Republic, peroxidase-labeled secondary antibodies) or were generously gifted by Dr. B. Vojtěšek (p53, p21WAF1, PCNA).
4. Conclusions
In this work we extended a previously developed method for the preparation of novel phenanthridine derivatives, which were derived from benzo[c]phenanthridines by deletion of the A-ring. Derivatives were prepared with substituents on the formed skeleton that mimic biologically active benzo[c]phenanthridines. The main principle for the preparation of the aforementioned compounds was based on the radical cyclization of reduced Schiff bases prepared by the condensation of appropriate aromatic aldehydes and amines. The prepared compounds were tested for antibacterial and anticancer cytotoxic effects and compared with several compounds containing a benzo[c]phenanthridine skeleton (e.g., chelerythrine, sanguinarine, isodecarine, norchelerythrine, NK-109). Several derivatives displayed antibacterial activity against Bacillus subtilis, Micrococcus luteus and/or Mycobacterium vaccae and cytotoxicity against K-562 and MCF-7 cancer cell lines at micromolar concentrations; these compounds typically contained a N-methylated quaternary nitrogen and 7-benzyloxy substitution. The mechanism of action of the novel phenanthridines however remains still unclear.
Supplementary Materials
The copies of 1H- and 13C-NMR spectra are available online.
Author Contributions
P.L. synthesized all compounds; K.M. evaluated the antimicrobial activity and analyzed the data; V.K. evaluated the anticancer activity and analyzed the data, J.S. conceived and designed the experiments and wrote the paper.
Funding
This research was funded by the Czech-National Program for Sustainability grant number LO1304, Palacky University grant numbers IGA_PrF_2018_029, IGA_LF_2018_032 and IGA_PrF_2018_006, and OP PIK grant number CZ.01.1.02/0.0/0.0/15_019/0004431.
Acknowledgments
Authors thank H. Rezková, E. Řezníčková and P. Pospíšilová for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dvořák, Z.; Kubán, V.; Klejdus, B.; Vičar, J.; Ulrichová, J.; Hlaváč, J.; Šimánek, V. Quaternary benzo[c]phenanthridines sanguinarine and chelerythrine: A review of investigations from chemical and biological studies. Heterocycles 2006, 68, 2403. [Google Scholar] [CrossRef]
- Ahsan, H.; Reagan-Shaw, S.; Breur, J.; Ahmad, N. Sanguinarine induces apoptosis of human pancreatic carcinoma AsPC-1 and BxPC-3 cells via modulations in Bcl-2 family proteins. Cancer Lett. 2007, 249, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.-C.; Park, J.-G.; Song, D.-K.; Baek, W.-K.; Yoo, S.K.; Jung, K.-H.; Park, G.-Y.; Lee, T.-Y.; Suh, S.-I. Sanguinarine induces apoptosis in A549 human lung cancer cells primarily via cellular glutathione depletion. Toxicol. Vitro 2009, 23, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Yang, X.-J.; Zhou, L.; Hu, H.-J.; Zheng, F.; Ding, X.-D.; Sun, D.-M.; Zhou, C.-D.; Sun, W. Structural modification of sanguinarine and chelerythrine and their antibacterial activity. Nat. Prod. Res. 2011, 25, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. Benzo[c]phenanthridine bases and their antituberculosis activity. Med. Res. Rev. 2001, 21, 61–72. [Google Scholar] [CrossRef]
- Šimánek, V. Benzophenanthridine alkaloids. In The Alkaloids; Brossi, A., Ed.; Academic Press: New York, NY, USA, 1985; Volume 26, pp. 185–240. [Google Scholar]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Molecular targets and anticancer potential of sanguinarine—A benzophenanthridine alkaloid. Phytomedicine 2017, 34, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Slaninová, I.; Pěnčíková, K.; Urbanová, J.; Slanina, J.; Táborská, E. Antitumour activities of sanguinarine and related alkaloids. Phytochem. Rev. 2014, 13, 51–68. [Google Scholar] [CrossRef]
- Maiti, M.; Nandi, R.; Chaudhuri, K. Sanguinarine: A monofunctional intercalating alkaloid. FEBS Lett. 1982, 142, 280–284. [Google Scholar] [CrossRef]
- Wang, X.; Tanaka, M.; Krstin, S.; Peixoto, H.; Wink, M. The interference of selected cytotoxic alkaloids with the cytoskeleton: An insight into their modes of action. Molecules 2016, 21, 906. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.-L.; Lee, M.C.; Tan, K.O.; Yang, L.-K.; Lee, A.S.Y.; Flotow, H.; Fu, N.Y.; Butler, M.S.; Soejarto, D.D.; Buss, A.D.; et al. Identification of chelerythrine as an inhibitor of BclXL function. J. Biol. Chem. 2003, 278, 20453–20456. [Google Scholar] [CrossRef] [PubMed]
- Caolo, M.A.; Stermitz, F.R. Benzophenanthridinium salt equilibria. Heterocycles 1979, 12, 11. [Google Scholar] [CrossRef]
- Romo-Pérez, A.; Miranda, L.D.; Chávez-Blanco, A.D.; Dueñas-González, A.; del Rayo Camacho-Corona, M.; Acosta-Huerta, A.; García, A. Mild C(sp3)–H functionalization of dihydrosanguinarine and dihydrochelerythrine for development of highly cytotoxic derivatives. Eur. J. Med. Chem. 2017, 138, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zee-Cheng, R.K.Y.; Yan, S.-J.; Cheng, C.C. Antileukemic activity of ungeremine and related compounds. Preparation of analogs of ungeremine by a practical photochemical reaction. J. Med. Chem. 1978, 21, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Hatae, N.; Fujita, E.; Shigenobu, S.; Shimoyama, S.; Ishihara, Y.; Kurata, Y.; Choshi, T.; Nishiyama, T.; Okada, C.; Hibino, S. Antiproliferative activity of O4-benzo[c]phenanthridine alkaloids against HCT-116 and HL-60 tumor cells. Bioorg. Med. Chem. Lett. 2015, 25, 2749–2752. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Suzuki, M.; Mashiba, A.; Ishikawa, K.; Yokotsuka, T. Synthesis of NK109, an anticancer benzo[c]phenanthridine alkaloid. J. Org. Chem. 1998, 63, 4235–4239. [Google Scholar] [CrossRef]
- Kanzawa, F.; Nishio, K.; Ishida, T.; Fukuda, M.; Kurokawa, H.; Fukumoto, H.; Nomoto, Y.; Fukuoka, K.; Bojanowski, K.; Saijo, N. Anti-tumour activities of a new benzo[c]phenanthridine agent, 2,3-(methylenedioxy)-5-methyl-7-hydroxy-8-methoxybenzo[c]phenanthridinium hydrogensulphate dihydrate (NK109), against several drug-resistant human tumour cell lines. Br. J. Cancer 1997, 76, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Masuda, A.; Suwa, M.; Akiyama, Y.; Hoshino-Abe, N.; Suzuki, M. Synthesis of derivatives of NK109, 7-OH Benzo[c]phenanthridine alkaloid, and evaluation of their cytotoxicities and reduction-resistant properties. Bioorg. Med. Chem. Lett. 2000, 10, 2321–2323. [Google Scholar] [CrossRef]
- Fukuda, M.; Inomata, M.; Nishio, K.; Fukuoka, K.; Kanzawa, F.; Arioka, H.; Ishida, T.; Fukumoto, H.; Kurokawa, H.; Oka, M.; et al. A Topoisomerase II inhibitor, NK109, induces DNA single- and double-strand breaks and apoptosis. Jpn. J. Cancer Res. 1996, 87, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Sueoka-Aragane, N.; Sato, A.; Tomimasu, R.; Ide, M.; Kurimasa, A.; Okamoto, K.; Kimura, S.; Sueoka, E. NK314 potentiates antitumor activity with adult T-cell leukemia-lymphoma cells by inhibition of dual targets on topoisomerase II and DNA-dependent protein kinase. Blood 2011, 117, 3575–3584. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, E.; Kagaya, S.; Cowell, I.G.; Kurosawa, A.; Kamoshita, K.; Nishikawa, K.; Iiizumi, S.; Koyama, H.; Austin, C.A.; Adachi, N. NK314, a topoisomerase II inhibitor that specifically targets the α isoform. J. Biol. Chem. 2008, 283, 23711–23720. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, X.; Nishikawa, K.; Plunkett, W. Inhibition of topoisomerase II and G2 cell cycle arrest by NK314, a novel benzo[c]phenanthridine currently in clinical trials. Mol. Cancer Ther. 2007, 6, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Johnson, R.K.; Hecht, S.M. Inhibition of topoisomerase I function by nitidine and fagaronine. Chem. Res. Toxicol. 1993, 6, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Janin, Y.L.; Bisagni, E.; Carrez, D. Synthesis of some benzo[h]quinoline derivatives. J. Heterocycl. Chem. 1993, 30, 1129–1131. [Google Scholar] [CrossRef]
- Bernardo, P.H.; Wan, K.-F.; Sivaraman, T.; Xu, J.; Moore, F.K.; Hung, A.W.; Mok, H.Y.K.; Yu, V.C.; Chai, C.L.L. Structure−activity relationship studies of phenanthridine-based Bcl-X L inhibitors. J. Med. Chem. 2008, 51, 6699–6710. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Singh, S.K.; Liu, A.; Li, T.-K.; Liu, L.F.; LaVoie, E.J. Substituted dibenzo[c,h]cinnolines: Topoisomerase I-targeting anticancer agents. Bioorg. Med. Chem. 2003, 11, 1475–1491. [Google Scholar] [CrossRef]
- Yapi, A.-D.; Desbois, N.; Chezal, J.-M.; Chavignon, O.; Teulade, J.-C.; Valentin, A.; Blache, Y. Design and preparation of aza-analogues of benzo[c]phenanthridine framework with cytotoxic and antiplasmodial activities. Eur. J. Med. Chem. 2010, 45, 2854–2859. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, T.N.; Girreser, U.; Meier, C.; Cushman, M.; Clement, B. One-step synthetic access to isosteric and potent anticancer nitrogen heterocycles with the benzo[c]phenanthridine scaffold. Chem. A Eur. J. 2016, 22, 8301–8308. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Yang, R.; Zhang, C.; Zhu, L.-F.; Miao, F.; Yang, X.-J.; Zhou, L. 2-(Substituted phenyl)-3,4-dihydroisoquinolin-2-iums as novel antifungal lead compounds: Biological evaluation and structure-activity relationships. Molecules 2013, 18, 10413–10424. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yao, Y.; Qin, Y.; Hou, Z.; Yang, R.; Miao, F.; Zhou, L. Synthesis and in vitro antifungal activities of new 2-aryl-6,7-methylenedioxy-3,4-dihydroisoquinolin-2-ium bromides. Chem. Pharm. Bull. 2013, 61, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ishii, H. Recent advances on antitumor-active benzo[c]phenanthridine alkaloids. Heterocycles 1999, 50, 627. [Google Scholar] [CrossRef]
- Mackay, S.P.; Meth-Cohn, O.; Waigh, R.D. Synthesis of quaternary benzo[c]phenanthridine alkaloids and their analogues. Adv. Heterocycl. Chem. 1996, 67, 345–389. [Google Scholar] [CrossRef]
- Stýskala, J.; Cankař, P.; Soural, M.; Hradil, P.; Vičar, J.; Šimánek, V.; Hlaváč, J. Synthesis of isodecarine. Heterocycles 2007, 73, 769. [Google Scholar] [CrossRef]
- Stýskala, J.; Hlaváč, J.; Cankař, P. Synthesis of oxidative dihydroxy metabolites of benzo[c]phenanthridines. Tetrahedron 2013, 69, 4670–4678. [Google Scholar] [CrossRef]
- Larghi, E.L.; Obrist, B.V.; Kaufman, T.S. A formal total synthesis of the marine alkaloid aaptamine. Tetrahedron 2008, 64, 5236–5245. [Google Scholar] [CrossRef]
- Meisels, A.; Sondheimer, F. The constituents of casimiroa edulis llave et lex. III. 1 The structure of casimiroin 2. J. Am. Chem. Soc. 1957, 79, 6328–6333. [Google Scholar] [CrossRef]
- Majumdar, K.; Taher, A.; Debnath, P. palladium-catalyzed intramolecular biaryl coupling: A highly efficient avenue for benzannulated pyranoquinolines and julolidine derivatives. Synthesis 2009, 2009, 793–800. [Google Scholar] [CrossRef]
- Harayama, T.; Aktyama, T.; Kawano, K. A convenient synthesis of benzo[c]phenanthridine alkaloid, chelerythrine, by the palladium-assisted internal biaryl coupling reaction. Chem. Pharm. Bull. 1996, 44, 1634–1636. [Google Scholar] [CrossRef]
- Campeau, L.-C.; Parisien, M.; Leblanc, M.; Fagnou, K. Biaryl synthesis via direct arylation: Establishment of an efficient catalyst for intramolecular processes. J. Am. Chem. Soc. 2004, 126, 9186–9187. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Akiyama, T.; Akamatsu, H.; Kawano, K.; Abe, H.; Takeuchi, Y. Total synthesis of benzo[c]phenanthridine alkaloids, chelerythrine and 12-methoxydihydrochelerythrine, by a palladium-assisted internal biaryl coupling reaction. Synthesis 2001, 2001, 0444–0450. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C−H bond functionalizations: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Mishra, S.; Kakde, B.N.; Dey, D.; Bisai, A. Expeditious approach to pyrrolophenanthridones, phenanthridines, and benzo[c]phenanthridines via organocatalytic direct biaryl-coupling promoted by potassium tert -butoxide. J. Org. Chem. 2013, 78, 7823–7844. [Google Scholar] [CrossRef] [PubMed]
- Dewanji, A.; Murarka, S.; Curran, D.P.; Studer, A. Phenyl hydrazine as initiator for direct arene C–H arylation via base promoted homolytic aromatic substitution. Org. Lett. 2013, 15, 6102–6105. [Google Scholar] [CrossRef] [PubMed]
- Carpino, L.A. Simple preparation of active manganese dioxide from activated carbon. J. Org. Chem. 1970, 35, 3971–3972. [Google Scholar] [CrossRef]
- Murray, P.R.; Baron, E.J.; Pfaller, M.A.; Tenover, F.C.; Yolken, R.H. Manual of Clinical Microbiology, 7th ed.; American Society for Microbiology: Washington, DC, USA, 1999. [Google Scholar]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 8th ed.; Approved Standard Document M07-A7; Clinical and Laboratory Standards Institute (CLSI): Villanova, PA, USA, 2009.
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, M.; Jorda, R.; Gucký, T.; Řezníčková, E.; Voller, J.; Pospíšil, T.; Malínková, V.; Adamcová, H.; Kryštof, V.; Strnad, M. Synthesis and in vitro biological evaluation of 2,6,9-trisubstituted purines targeting multiple cyclin-dependent kinases. Eur. J. Med. Chem. 2013, 61, 61–72. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).