Deep Learning in Drug Discovery and Medicine; Scratching the Surface

Abstract
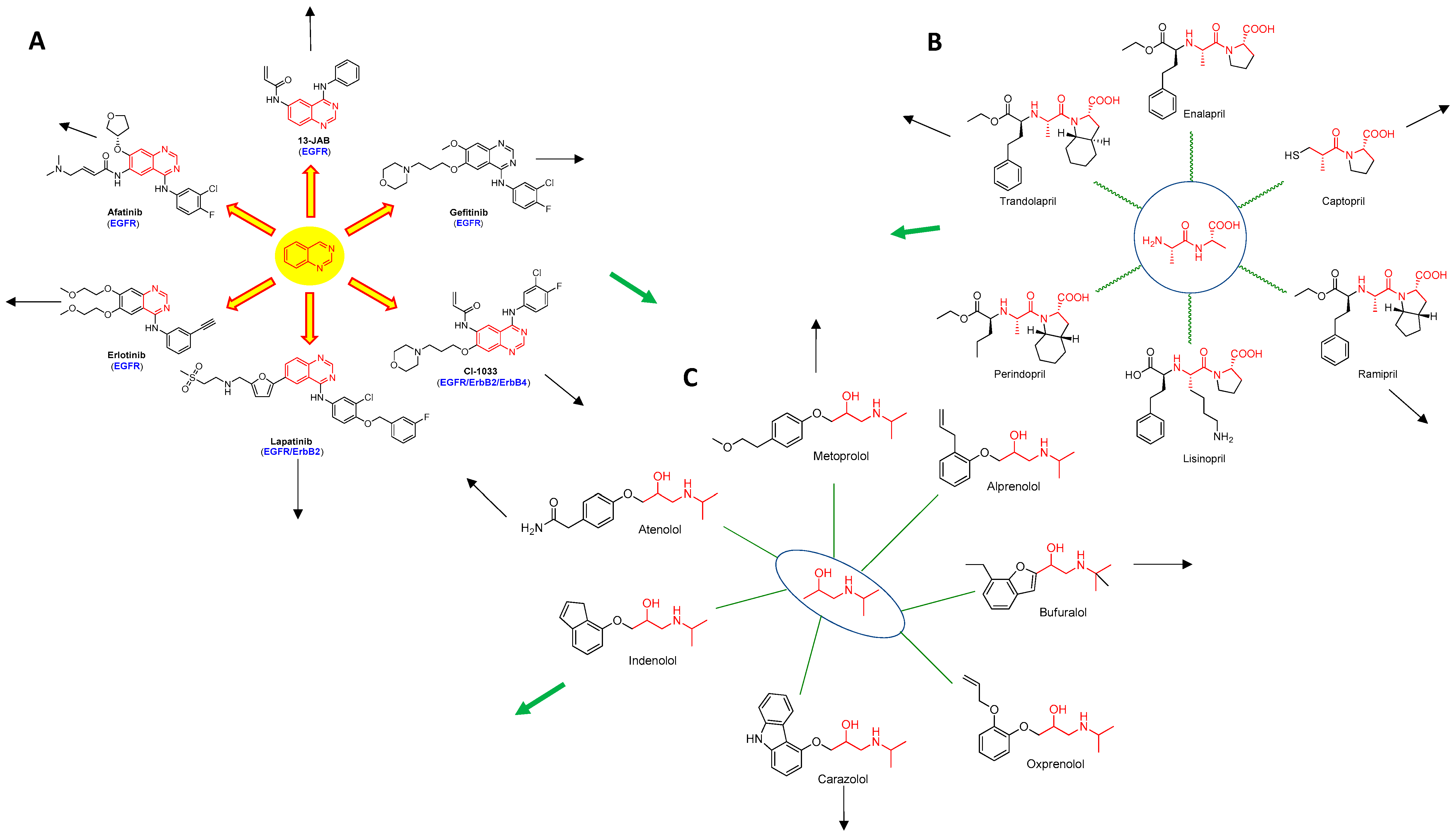
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Precision Medicine
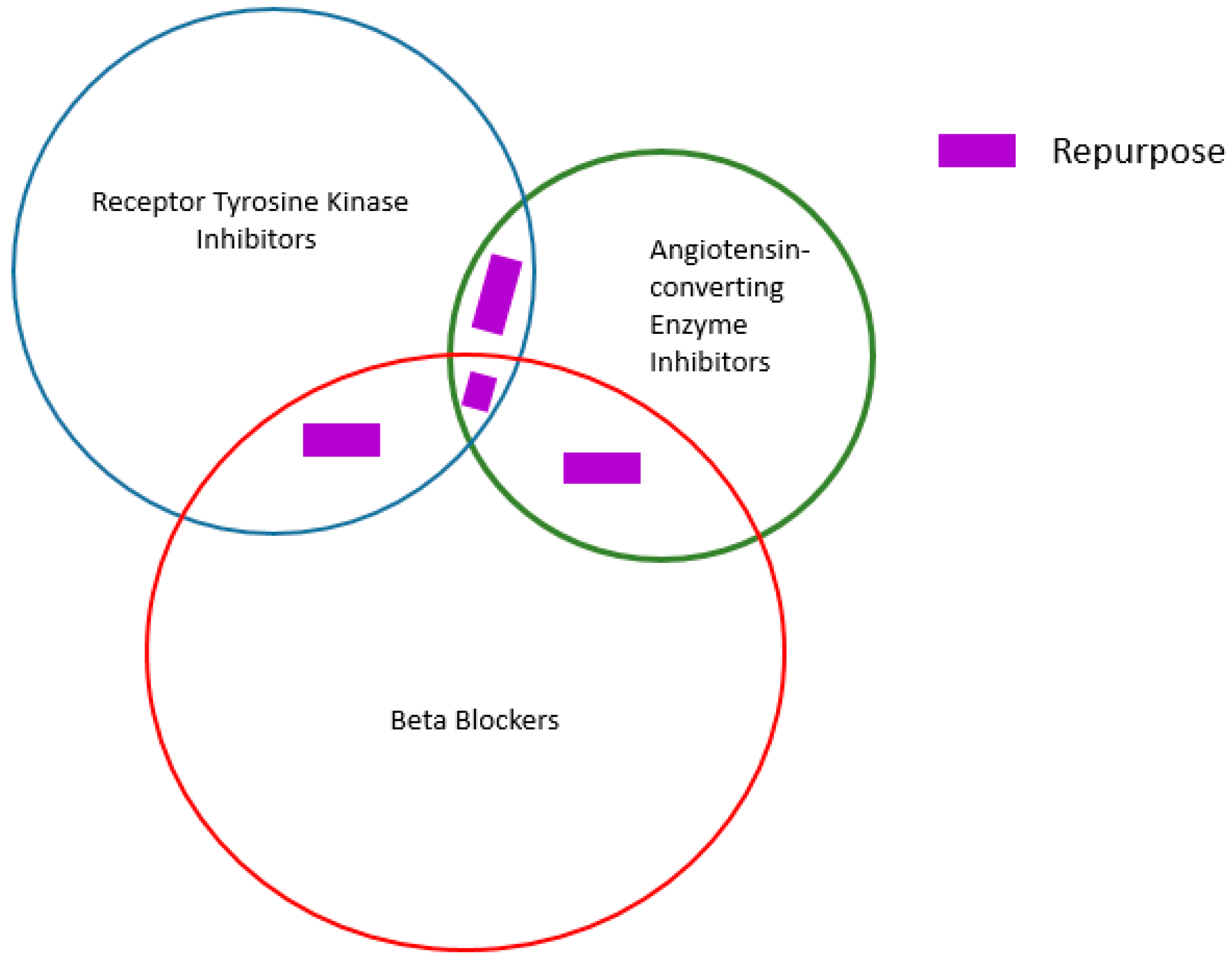
3. Drug Repositioning
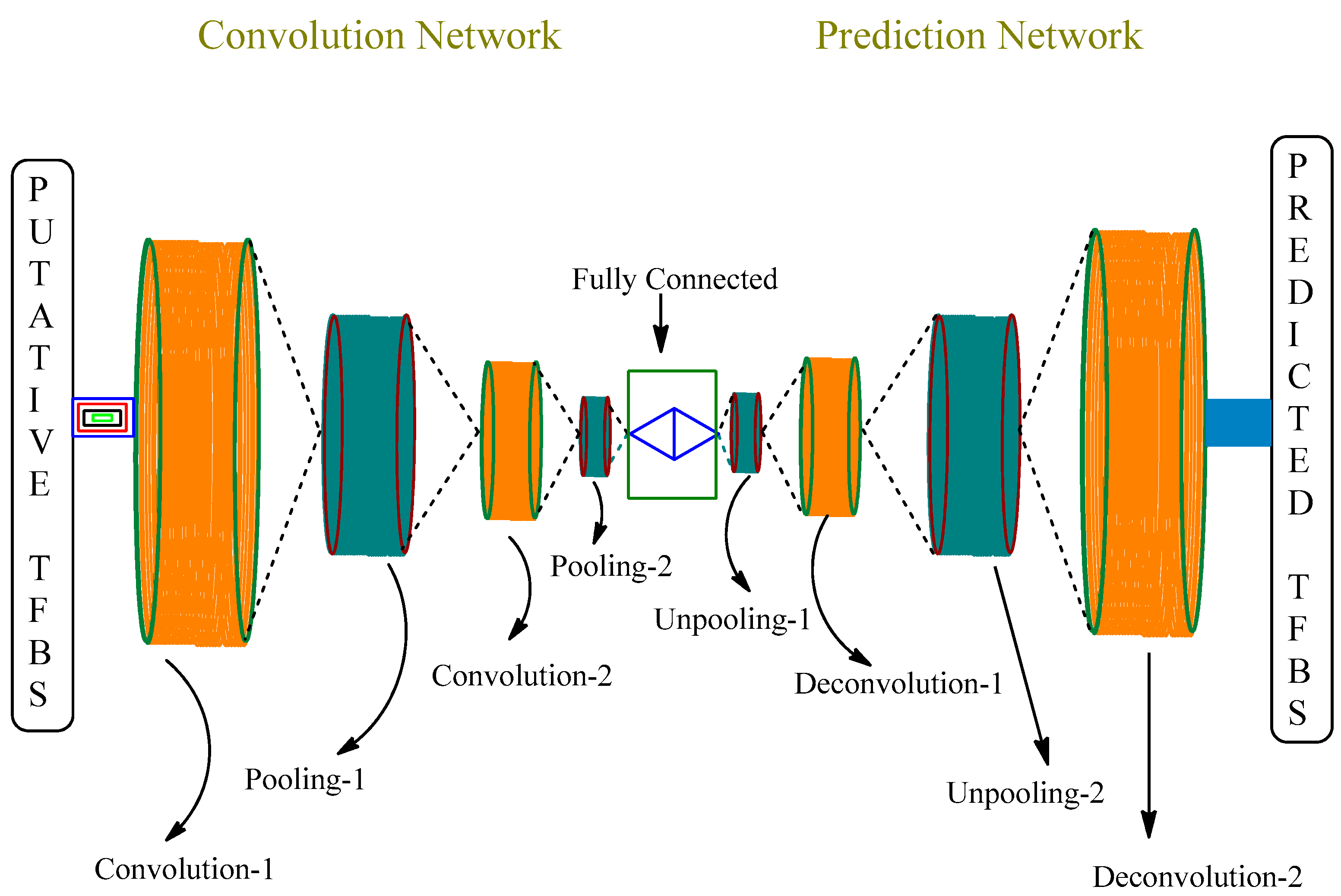
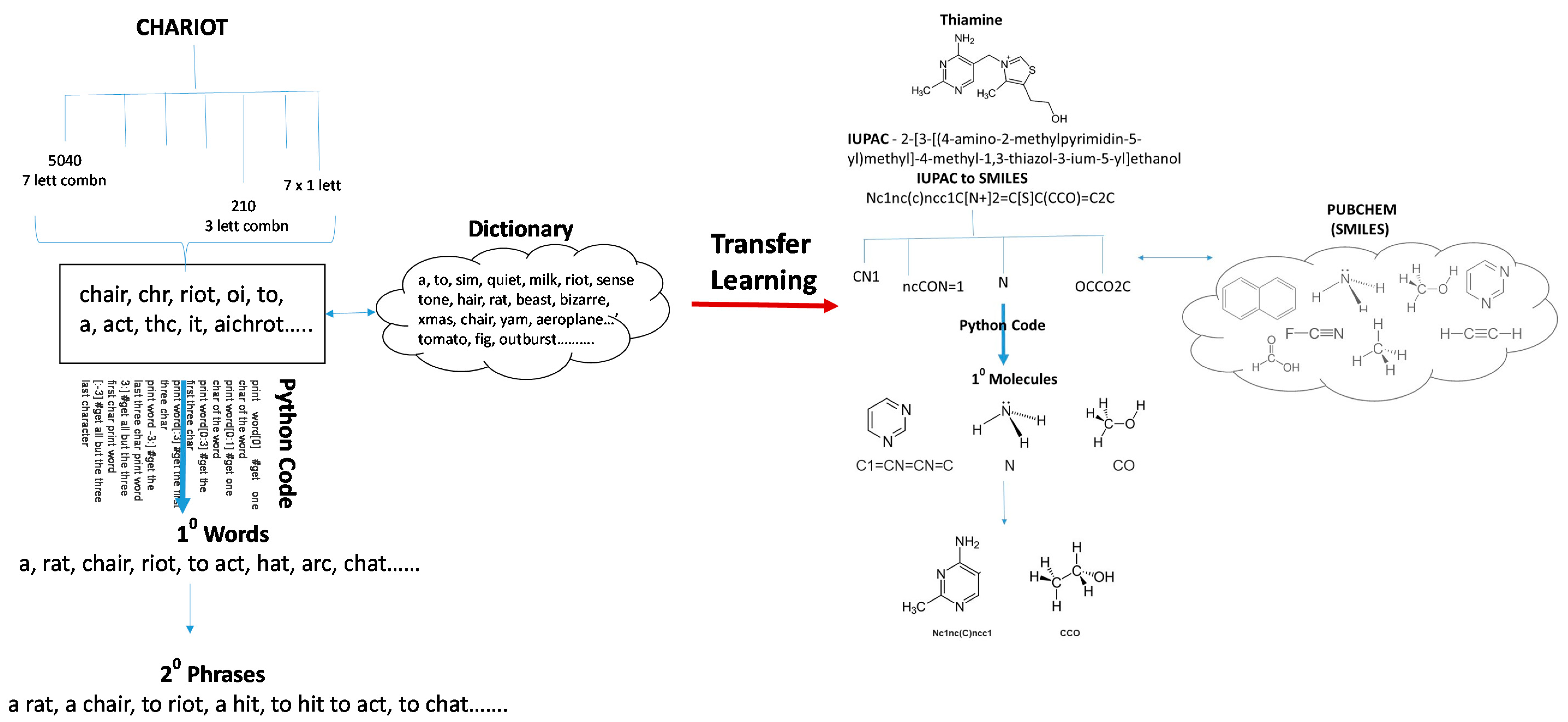
4. Artificial Intelligence (AI) in Drug Design and Molecular Medicine
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barkhem, T.; Carlsson, B.; Nilsson, Y.; Enmark, E.; Gustafsson, J.-Å.; Nilsson, S. Differential response of estrogen receptor α and estrogen receptor β to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Kirk, E.S. Differential response of large and small coronary arteries to nitroglycerin and angiotensin: Autoregulation and tachyphylaxis. Circ. Res. 1973, 33, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.A. Mindfulness training as a clinical intervention: A conceptual and empirical review. Clin. Psychol. Sci. Pract. 2003, 10, 125–143. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.L.; Longo, D.L. Precision medicine—Personalized, problematic, and promising. Obstet. Gynecol. Surv. 2015, 70, 612–614. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Snyder, M. Promise of personalized omics to precision medicine. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L. Obama Catapults Patient-Empowered Precision Medicine; Nature Publishing Group: London, UK, 2015. [Google Scholar]
- Leon, S.; Shapiro, B.; Sklaroff, D.; Yaros, M. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Cho, H.-S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Opdivo (nivolumab): Second PD-1 inhibitor receives FDA approval for unresectable or metastatic melanoma. Am. Health Drug Benefits 2015, 8, 180–183. [Google Scholar] [PubMed]
- Garraway, L.A.; Verweij, J.; Ballman, K.V. Precision oncology: An overview. J. Clin. Oncol. 2013, 31, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; van Venrooij, W.J. Anti-CCP antibodies, a highly specific marker for (early) rheumatoid arthritis. Clin. Appl. Immunol. Rev. 2004, 4, 239–262. [Google Scholar] [CrossRef]
- Wegner, N.; Lundberg, K.; Kinloch, A.; Fisher, B.; Malmström, V.; Feldmann, M.; Venables, P.J. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 2010, 233, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.K.S.; Silva, A.S.; Evangelista, I.W.Q.; Modesto Filho, J.; Gomes, C.N.A.P.; Nascimento, R.A.F.; Luna, R.C.P.; Costa, M.J.C.; Oliveira, N.F.P.; Persuhn, D.C. Hypermethylation in the promoter of the MTHFR gene is associated with diabetic complications and biochemical indicators. Diabetol. Metab. Syndr. 2017, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Santos, J.M.; Mishra, M. Epigenetic modifications and diabetic retinopathy. BioMed Res. Int. 2013, 2013, 635284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Cheng, X.; Connolly, B.; Dickman, M.; Hurd, P.; Hornby, D. Zebularine: A novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J. Mol. Biol. 2002, 321, 591–599. [Google Scholar] [CrossRef]
- Choi, K.-C.; Jung, M.G.; Lee, Y.-H.; Yoon, J.C.; Kwon, S.H.; Kang, H.-B.; Kim, M.-J.; Cha, J.-H.; Kim, Y.J.; Jun, W.J. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Exp. Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone Deacetylase Inhibitors. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef] [PubMed]
- Salarinia, R.; Sahebkar, A.; Peyvandi, M.; Reza Mirzaei, H.; Reza Jaafari, M.; Matbou Riahi, M.; Ebrahimnejad, H.; Sadri Nahand, J.; Hadjati, J.; Ostadi Asrami, M. Epi-drugs and Epi-miRs: Moving beyond current cancer therapies. Curr. Cancer Drug Targets 2016, 16, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Gadegbeku, C.A.; Gipson, D.S.; Holzman, L.B.; Ojo, A.O.; Song, P.X.; Barisoni, L.; Sampson, M.G.; Kopp, J.B.; Lemley, K.V.; Nelson, P.J. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int. 2013, 83, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Meersseman, W.; Lagrou, K.; Maertens, J.; Wilmer, A.; Hermans, G.; Vanderschueren, S.; Spriet, I.; Verbeken, E.; Van Wijngaerden, E. Galactomannan in bronchoalveolar lavage fluid: A tool for diagnosing aspergillosis in intensive care unit patients. Am. J. Respir. Crit. Care Med. 2008, 177, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Krsek-Staples, J.A.; Kew, R.R.; Webster, R.O. Ceruloplasmin and transferrin levels are altered in serum and bronchoalveolar lavage fluid of patients with the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1992, 145, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Gadhiya, S.; Ponnala, S.; Harding, W.W. A divergent route to 9, 10-oxygenated tetrahydroprotoberberine and 8-oxoprotoberberine alkaloids: Synthesis of (±)-isocorypalmine andáoxypalmatine. Tetrahedron 2015, 71, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Joy, M.S.; Gipson, D.S.; Powell, L.; MacHardy, J.; Jennette, J.C.; Vento, S.; Pan, C.; Savin, V.; Eddy, A.; Fogo, A.B. Phase 1 trial of adalimumab in Focal Segmental Glomerulosclerosis (FSGS): II. Report of the FONT (Novel Therapies for Resistant FSGS) study group. Am. J. Kidney Dis. 2010, 55, 50–60. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef] [PubMed]
- National Center for Advancing Translational Science. NIH–Industry Partnerships Projects. Available online: https://ncats.nih.gov/ntu/projects/2013 (accessed on 21 August 2018).
- Carroll, S.B. Chance and necessity: The evolution of morphological complexity and diversity. Nature 2001, 409, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Guo, J.T.; Li, T.; Xu, Y. Structure-based prediction of transcription factor binding sites using a protein-DNA docking approach. Proteins Struct. Funct. Bioinf. 2008, 72, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP–seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Allhoff, M.; Seré, K.; Chauvistré, H.; Lin, Q.; Zenke, M.; Costa, I.G. Detecting differential peaks in ChIP-seq signals with ODIN. Bioinformatics 2014, 30, 3467–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.S.; Yu, J.; Shen, J.; Maher, C.A.; Hu, M.; Kalyana-Sundaram, S.; Yu, J.; Chinnaiyan, A.M. HPeak: An HMM-based algorithm for defining read-enriched regions in ChIP-Seq data. BMC Bioinform. 2010, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Valouev, A.; Johnson, D.S.; Sundquist, A.; Medina, C.; Anton, E.; Batzoglou, S.; Myers, R.M.; Sidow, A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods 2008, 5, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, P.F.; Chung, D.; Pan, G.; Thomson, J.A.; Stewart, R.; Keleş, S. A statistical framework for the analysis of ChIP-Seq data. J. Am. Stat. Assoc. 2011, 106, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gossling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCHTM: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Salekin, S.; Zhang, J.M.; Huang, Y. A deep learning model for predicting transcription factor binding location at single nucleotide resolution. In Proceedings of the 2017 IEEE EMBS International Conference on Biomedical & Health Informatics (BHI), Orlando, FL, USA, 16–19 February 2017; pp. 57–60. [Google Scholar]
- The New York Times. Available online: https://www.nytimes.com/2012/11/24/science/scientists-see-advances-in-deep-learning-a-part-of-artificial-intelligence.html (accessed on 21 August 2018).
- Livingstone, D.J. Pattern recognition methods in rational drug design. In Methods in Enzymology; Elsevier: New York, NY, USA, 1991; Volume 203, pp. 613–638. [Google Scholar]
- Mitchell, J.B. Informatics, machine learning and computational medicinal chemistry. Future Med. Chem. 2011, 3, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Riebe, K.; Partl, A.M.; Enke, H.; Forero-Romero, J.; Gottlöber, S.; Klypin, A.; Lemson, G.; Prada, F.; Primack, J.R.; Steinmetz, M. The MultiDark database: Release of the Bolshoi and MultiDark cosmological simulations. Astron. Nachr. 2013, 334, 691–708. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing Distributions over Molecular Space. An Objective-Reinforced Generative Adversarial Network for Inverse-Design Chemistry (ORGANIC); Working Paper; Harvard University: Cambridge, MA, USA, 2017. [Google Scholar]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of generative autoencoder in de novo molecular design. Mol. Inform. 2018, 37, 1700123. [Google Scholar] [CrossRef] [PubMed]
- Madapa, S.; Gadhiya, S.; Kurtzman, T.; Alberts, I.L.; Ramsey, S.; Reith, M.; Harding, W.W. Synthesis and evaluation of C9 alkoxy analogues of (−)-stepholidine as dopamine receptor ligands. Eur. J. Med. Chem. 2017, 125, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Manuszak, M.; Harding, W.; Gadhiya, S.; Ranaldi, R. (−)-Stepholidine reduces cue-induced reinstatement of cocaine seeking and cocaine self-administration in rats. Drug Alcohol. Depend. 2018, 189, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Gadhiya, S.; Madapa, S.; Kurtzman, T.; Alberts, I.L.; Ramsey, S.; Pillarsetty, N.-K.; Kalidindi, T.; Harding, W.W. Tetrahydroprotoberberine alkaloids with dopamine and σ receptor affinity. Bioorg. Med. Chem. 2016, 24, 2060–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillisch, A.; Heinrich, N.; Wild, H. Computational chemistry in the pharmaceutical industry: From childhood to adolescence. ChemMedChem 2015, 10, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Guba, W.; Kansy, M. Integrating molecular design resources within modern drug discovery research: The Roche experience. Drug Discov. Today 2006, 11, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schneider, G. De novo design at the edge of chaos: Miniperspective. J. Med. Chem. 2016, 59, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep learning in drug discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De novo design of bioactive small molecules by artificial intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.J.; Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2010, 22, 1345–1359. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2017, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Van Herwerden, E.F.; Süssmuth, R.D. Sources for leads: Natural products and libraries. In New Approaches to Drug Discovery; Springer: Berlin, Germany, 2015; pp. 91–123. [Google Scholar]
- Gadhiya, S.V.; Hu, C.; Harding, W.W. An alternative synthesis and X-ray crystallographic confirmation of (−)-stepholidine. Tetrahedron Lett. 2016, 57, 2090–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dana, D.; Gadhiya, S.V.; St. Surin, L.G.; Li, D.; Naaz, F.; Ali, Q.; Paka, L.; Yamin, M.A.; Narayan, M.; Goldberg, I.D.; et al. Deep Learning in Drug Discovery and Medicine; Scratching the Surface. Molecules 2018, 23, 2384. https://doi.org/10.3390/molecules23092384
Dana D, Gadhiya SV, St. Surin LG, Li D, Naaz F, Ali Q, Paka L, Yamin MA, Narayan M, Goldberg ID, et al. Deep Learning in Drug Discovery and Medicine; Scratching the Surface. Molecules. 2018; 23(9):2384. https://doi.org/10.3390/molecules23092384
Chicago/Turabian StyleDana, Dibyendu, Satishkumar V. Gadhiya, Luce G. St. Surin, David Li, Farha Naaz, Quaisar Ali, Latha Paka, Michael A. Yamin, Mahesh Narayan, Itzhak D. Goldberg, and et al. 2018. "Deep Learning in Drug Discovery and Medicine; Scratching the Surface" Molecules 23, no. 9: 2384. https://doi.org/10.3390/molecules23092384
APA StyleDana, D., Gadhiya, S. V., St. Surin, L. G., Li, D., Naaz, F., Ali, Q., Paka, L., Yamin, M. A., Narayan, M., Goldberg, I. D., & Narayan, P. (2018). Deep Learning in Drug Discovery and Medicine; Scratching the Surface. Molecules, 23(9), 2384. https://doi.org/10.3390/molecules23092384