Enantioselective Organocatalysis-Based Synthesis of 3-Hydroxy Fatty Acids and Fatty γ-Lactones




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
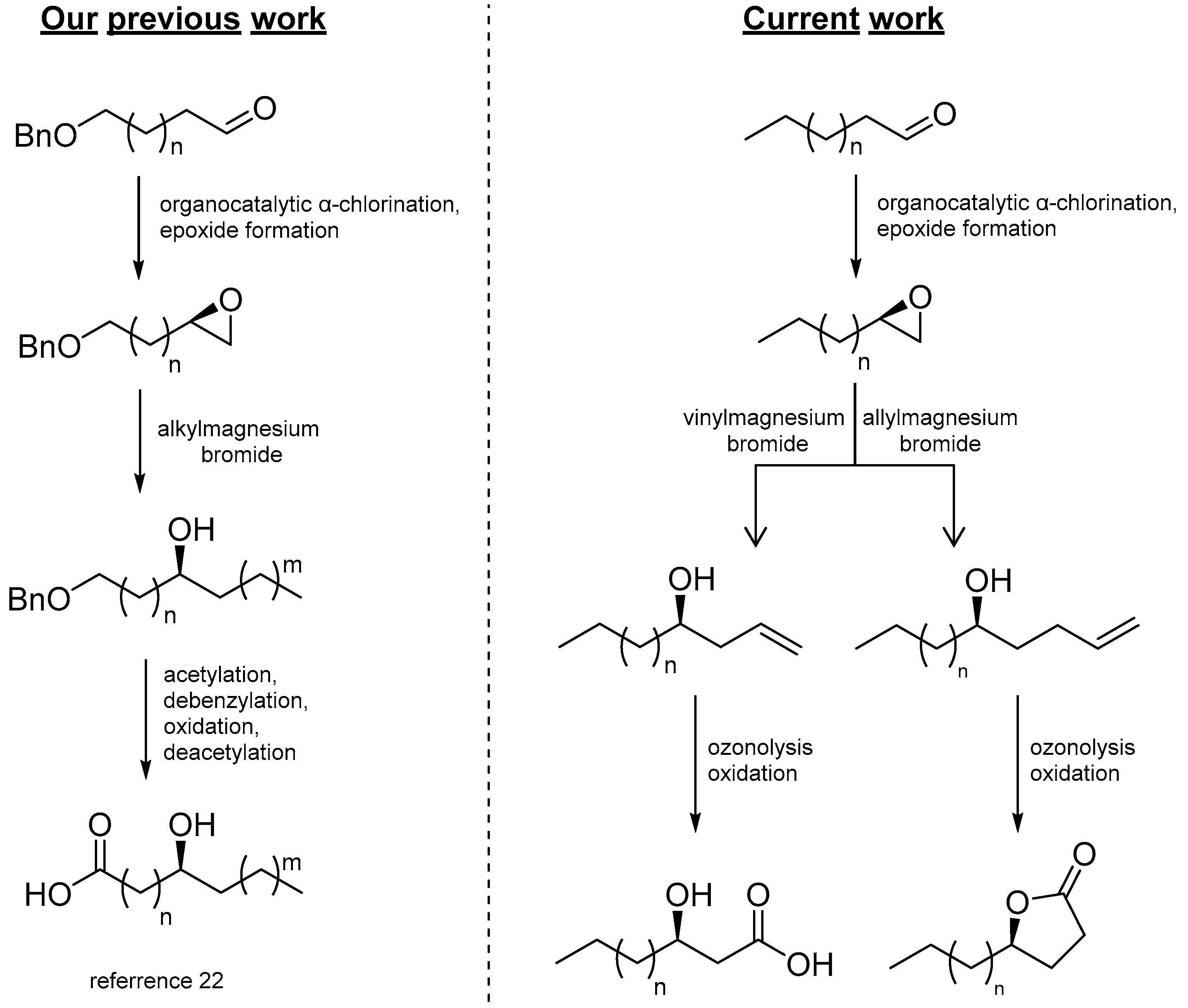
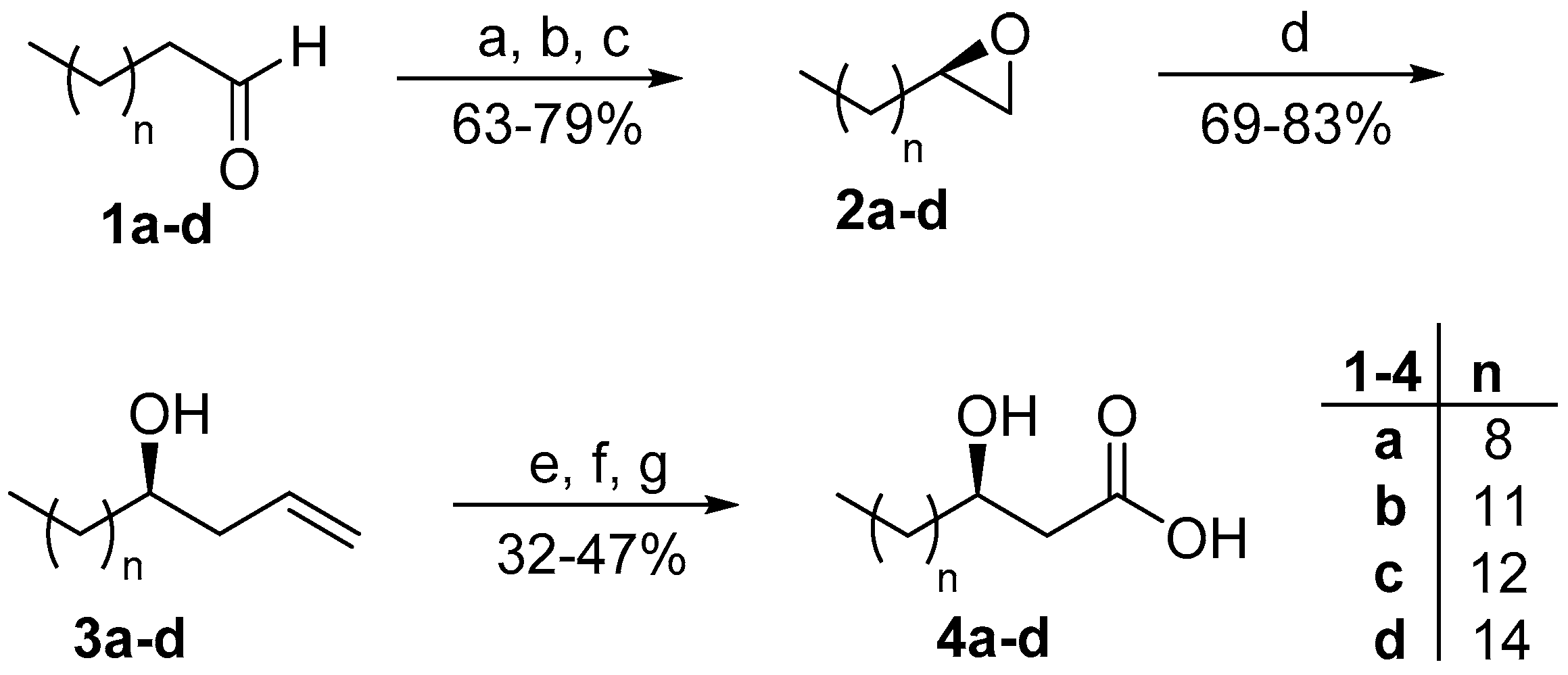
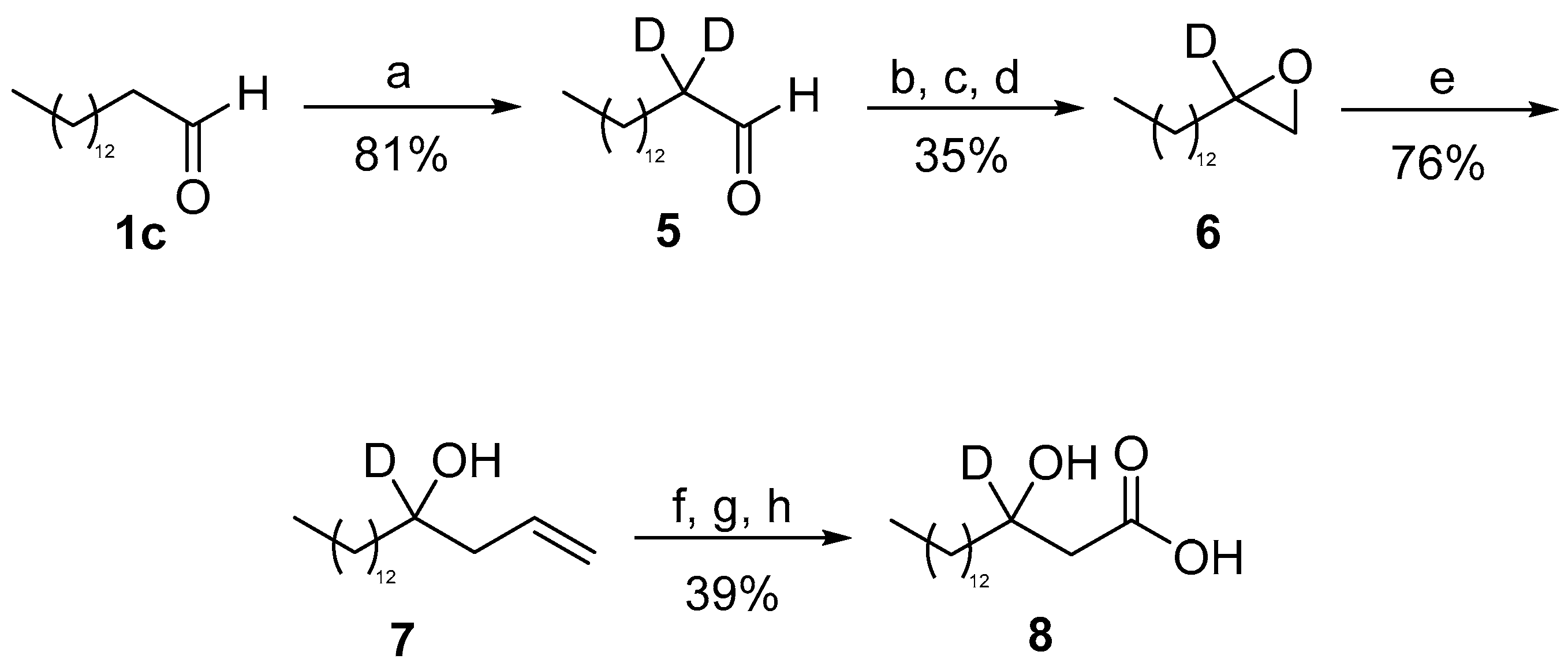
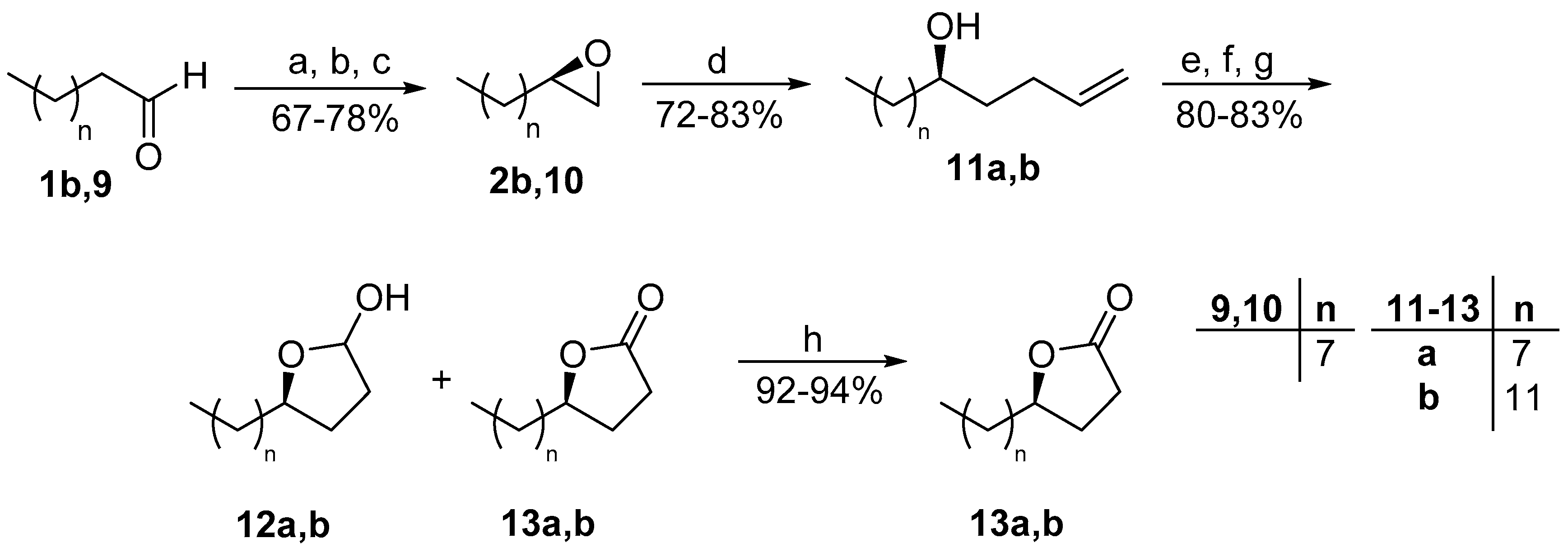
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis and Characterization
3.2.1. General Procedure for the Synthesis of Chiral Epoxides 2a–d, 10
3.2.2. General Procedure for the Synthesis of Racemic Epoxides 6, 15
3.2.3. General Procedure for the Synthesis of 4-Hydroxy Terminal Alkenes 3a–d, 7
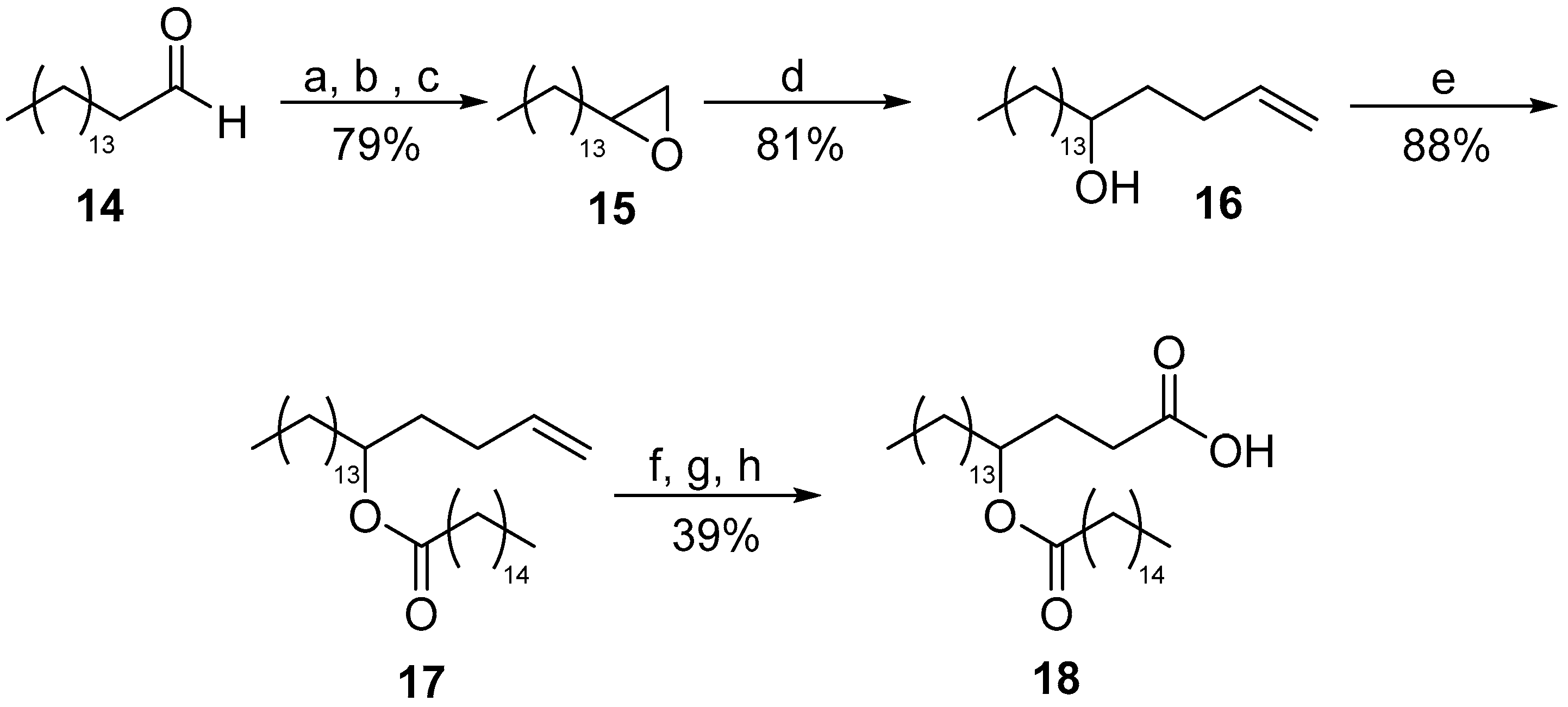
3.2.4. General Procedure for the Synthesis of 5-Hydroxy Terminal Alkenes 11a,b, 16
3.2.5. General Procedure for the Synthesis of 3-Hydroxy Fatty Acids 4a–d, 8
3.2.6. General Procedure for the Synthesis of Lactones 13a,b
3.2.7. Nonadec-1-en-5-yl Palmitate (17)
3.2.8. 4-(Palmitoyloxy)octadecanoic acid (18)
3.2.9. 2,2-d2-Pentadecanal (5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Offermanns, S. Free fatty acid (FFA) and hydroxy carboxylic acid (HCA) receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 407–434. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Takaishi, S.; Nagasaki, M.; Onozawa, Y.; Iino, I.; Maeda, H.; Komai, T.; Oda, T. Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J. Biol. Chem. 2013, 288, 10684–10691. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-J.; Hoppel, C.L.; Tserng, K.-Y. Incomplete fatty acid oxidation. The production and epimerization of 3-hydroxy fatty acids. J. Biol. Chem. 1992, 267, 119–125. [Google Scholar] [PubMed]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Jones, P.M.; Bennett, M.J. Clinical applications of 3-hydroxy fatty acid analysis by gas chromatography–mass spectrometry. Biochim. Biophys. Acta 2011, 1811, 657–662. [Google Scholar] [CrossRef]
- Uhlig, S.; Negård, M.; Heldal, K.K.; Straumfors, A.; Madsø, L.; Bakke, B.; Eduard, W. Profiling of 3-hydroxy fatty acids as environmental markers of endotoxin using liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. A 2016, 1434, 119–126. [Google Scholar] [CrossRef]
- Oikawa, Y.; Sugano, K.; Yonemitsu, O. Meldrum’s acid in organic synthesis. 2. A general and versatile synthesis of α-keto esters. J. Org. Chem. 1978, 43, 2087–2088. [Google Scholar] [CrossRef]
- Ratovelomanana-Vidal, V.; Girard, C.; Touati, R.; Tranchier, J.P.; Ben Hassine, B.; Genêt, J.P. Enantioselective hydrogenation of α-keto esters using chiral diphosphine-ruthenium complexes: Optimization for academic and industrial purposes and synthetic applications. Adv. Synth. Catal. 2003, 345, 261–274. [Google Scholar] [CrossRef]
- Concellon, J.M.; Concellon, C. Aldol-type reactions of unmasked iodoacetic acid with carbonyl compounds promoted by samarium diiodide: Efficient synthesis of carboxylic 3-hydroxyacids and their derivatives. J. Org. Chem. 2006, 71, 4428–4432. [Google Scholar] [CrossRef]
- Sailer, M.; Dubicki, K.I.; Sorensen, J.L. The synthesis of medium-chain-length β-hydroxy esters via the Reformatsky reaction. Synthesis 2015, 47, 79–82. [Google Scholar]
- Kaspersen, M.H.; Jenkins, L.; Dunlop, J.; Milligan, G.; Ulven, T. Succinct synthesis of saturated hydroxy fatty acids and in vitro evaluation of all hydroxylauric acids at FFA1, FFA4 and GPR84. Med. Chem. Comm. 2017, 8, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Guaragna, A.; De Nisco, M.; Pedatella, S.; Palumbo, G. Studies towards lipid A: A synthetic strategy for the enantioselective preparation of 3-hydroxy fatty acids. Tetrahedron: Asymmetry 2006, 17, 2839–2841. [Google Scholar] [CrossRef]
- Jakob, B.; Voss, G.; Gerlach, H. Synthesis of (S)- and (R)-3-hydroxyhexadecanoic acid. Tetrahedron: Asymmetry 1996, 7, 3255–3262. [Google Scholar] [CrossRef]
- Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Dalko, P.I. (Ed.) Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Brochu, M.P.; Brown, S.P.; MacMillan, D.W.C. Direct and enantioselective organocatalytic α-chlorination of aldehydes. J. Am. Chem. Soc. 2004, 126, 4108–4109. [Google Scholar] [CrossRef]
- Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jørgensen, K.A. Direct organocatalytic asymmetric α-chlorination of aldehydes. J. Am. Chem. Soc. 2004, 126, 4790–4791. [Google Scholar] [CrossRef]
- Amatore, M.; Beeson, T.D.; Brown, S.P.; MacMillan, D.W.C. Enantioselective linchpin catalysis by SOMO catalysis: An approach to the asymmetric alpha-chlorination of aldehydes and terminal epoxide formation. Angew. Chem. Int. Ed. 2009, 48, 5121–5124. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Britton, R. A general method for the synthesis of nonracemic trans-epoxides: Concise syntheses of trans-epoxide-containing insect sex pheromones. Org. Lett. 2007, 24, 5083–5086. [Google Scholar] [CrossRef]
- Wang, L.; Cai, C.; Curran, D.P.; Zhang, W. Enantioselective α-chlorination of aldehydes with recyclable fluorous (S)-pyrrolidine–thiourea bifunctional organocatalyst. Synlett 2010, 433–436. [Google Scholar] [CrossRef]
- Kaplaneris, N.; Spyropoulos, C.; Kokotou, M.G.; Kokotos, C.G. Enantioselective organocatalytic synthesis of 2-oxopiperazines from aldehydes: Identification of the elusive epoxy lactone intermediate. Org. Lett. 2016, 18, 5800–5803. [Google Scholar] [CrossRef]
- Mountanea, O.G.; Limnios, D.; Kokotou, M.G.; Bourboula, A.; Kokotos, G. Asymmetric synthesis of saturated hydroxy fatty acids and fatty acid esters of hydroxy fatty acids. Eur. J. Org. Chem. 2019, 2019, 2010–2019. [Google Scholar] [CrossRef]
- Gant, T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef]
- Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 2017, 35, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Ariza, X.; Asins, G.; Garcia, J.; Hegardt, F.G.; Makowski, K.; Serra, D.; Velascoa, J. Preparation of α-labeled aldehydes by base-catalyzed exchange reactions. J. Label. Compd. Radiopharm. 2010, 53, 556–558. [Google Scholar] [CrossRef]
- Solladié, G.; Matloubi-Moghadam, F. Asymmetric synthesis of five- and six-membered lactones from chiral sulfoxides: Application to the asymmetric synthesis of insect pheromones, (R)-(+)-delta-n-hexadecanolactone and (R)-(+)-gamma-n-dodecanolactone. J. Org. Chem. 1982, 47, 91–94. [Google Scholar] [CrossRef]
- Wheeler, J.W.; Happ, G.M.; Araujo, J.; Pasteels, J.M. γ-Dodecalactone from rove beetles. Tetrahedron Lett. 1972, 46, 4635–4638. [Google Scholar] [CrossRef]
- Bittencourt, M.L.F.; Ribeiro, P.R.; Franco, R.L.P.; Hilhorst, H.W.M.; De Castro, R.D.; Fernandez, L.G. Metabolite profiling, antioxidant and antibacterial activities of Brazilian propolis: Use of correlation and multivariate analyses to identify potential bioactive compounds. Food Res. Int. 2015, 76, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef]
- Moraes-Vieira1, P.M.; Saghatelian, A.; Kahn, B.B. GLUT4 Expression in adipocytes regulates de novo lipogenesis and levels of a novel class of lipids with antidiabetic and anti-inflammatory effects. Diabetes 2016, 65, 1808–1815. [Google Scholar] [CrossRef]
- Kuda, M.; Brezinova, M.; Rombaldova, B.; Slavikova, M.; Posta, P.; Beier, P.; Janovska, J.; Veleba, J.; Kopecky, J., Jr.; Kudova, E.; et al. Docosahexaenoic acid-derived fatty acid esters of hydroxy fatty acids (FAHFAs) with anti-inflammatory properties. Diabetes 2016, 65, 2580–2590. [Google Scholar] [CrossRef]
- Nelson, A.T.; Kolar, M.J.; Chu, Q.; Syed, I.; Kahn, B.B.; Saghatelian, A.; Siegel, D. Stereochemistry of endogenous palmitic acid ester of 9-hydroxystearic acid and relevance of absolute configuration to regulation. J. Am. Chem. Soc. 2017, 139, 4943–4947. [Google Scholar] [CrossRef] [PubMed]
- Pflimlin, E.; Bielohuby, M.; Korn, M.; Breitschopf, K.; Löhn, M.; Wohlfart, P.; Konkar, A.; Podeschwa, M.; Bärenz, F.; Pfenninger, A.; et al. Acute and repeated treatment with 5-PAHSA or 9-PAHSA isomers does not improve glucose control in mice. Cell Metab. 2018, 28, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Savle, P.S.; Lamoreaux, M.J.; Berry, J.F.; Gandour, R.D. A convenient resolution of long-chain alkyl epoxides with Jacobsen’s salen(Co)III(OAc) catalysts. Tetrahedron: Asymmetry 1998, 11, 1843–1846. [Google Scholar] [CrossRef]
- Harbindu, A.; Kumar, P. Synthesis of aculeatins A and B via iterative hydrolytic kinetic resolution. Synthesis 2010, 1479–1484. [Google Scholar]
- Hubieki, M.P.; Gandour, R.D.; Ashendel, C.L. Synthesis of optically pure cyclic lipoidal ammonium salts and evaluation of inhibition of protein kinase C. J. Org. Chem. 1996, 61, 9379–9384. [Google Scholar] [CrossRef]
- Matsumoto, K.; Usuda, K.; Okabe, H.; Hashimoto, M.; Shimada, Y. Synthesis of optically active heterocyclic compounds via deracemization of 1,2-diol monotosylate derivatives bearing a long aliphatic chain by a combination of enzymatic hydrolysis with Mitsunobu inversion. Tetrahedron: Asymmetry 2013, 24, 108–115. [Google Scholar] [CrossRef]
- Jacoby, C.; Braekman, J.-C.; Daloze, D. Asymmetric synthesis of (2R)- and (2S)- 2-iodohexadecanal, natural inhibitors of the thyroid gland metabolism. Tetrahedron 1996, 52, 10473–10484. [Google Scholar] [CrossRef]
- Kumar, S.C.; Sreedhar, E.; Venkateswar, R.; Suresh, B.; Rao, J.M. A simple and efficient enantioselective route to 2, 6-disubstituted piperidines: Synthesis of (2R, 6S)-isosolenopsin A and (2S, 6R)-isosolenopsin. Tetrahedron: Asymmetry 2009, 20, 1160–1163. [Google Scholar] [CrossRef]
- Boga, C.; Drioli, S.; Forzato, C.; Micheletti, G.; Nitti, P.; Prati, F. An easy route to enantiomerically enriched 7- and 8-hydroxy-stearic acids by olefin-metathesis-based approach. Synlett 2016, 27, 1354–1358. [Google Scholar]
- Wetzel, I.; Krauss, J.; Bracher, F. Enantiodivergent chemoenzymatic synthesis of (S)- and (R)-(Z)-9-dodecyl-4,5,8,9-tetrahydro-3H-oxonin-2-one as analogues of topsentolides. Lett. Org. Chem. 2012, 9, 169–174. [Google Scholar] [CrossRef]
- Padhi, B.; Reddy, G.S.; Mohapatra, D.K. Total synthesis of tetraketide and cryptorigidifoliol I via a sequential allylation strategy. J. Nat. Prod. 2016, 79, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Waldeck, A.R.; Krische, M.J. Total synthesis of cryptocaryol A via enantioselective iridium catalyzed alcohol C-H allylation. Angew. Chem. Int. Ed. 2016, 55, 5049–5052. [Google Scholar] [CrossRef]
- Tremblay, A.E.; Whittle, E.; Buist, P.H.; Shanklin, J. Stereochemistry of Δ4 dehydrogenation catalyzed by an ivy (Hedera helix) Δ9 desaturase homolog. Org. Biom. Chem. 2007, 5, 1270–1275. [Google Scholar] [CrossRef]
- Kumar, P.; Pandey, M.; Gupta, P.; Dhavale, D.D. Organocatalytic stereoselective synthesis of passifloricin A. Org. Biom. Chem. 2012, 10, 1820–1825. [Google Scholar] [CrossRef]
- Hardy, P.M.; Aubrey Prout, R.; Rydon, H.N. Polypeptides. Part XXIV. Synthesis of isariic acid. J. Chem. Soc. Perkin Trans. 1 1974, 796. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Zhang, F.; Ambadi, S.; Rao, Y.G. Total synthesis of fellutamides, lipopeptide proteasome inhibitors. More sustainable peptide bond formation. Org. Biomol. Chem. 2016, 14, 8367–8375. [Google Scholar] [CrossRef] [Green Version]
- Lammek, B. Resolution of racemic 3-hydroxypentadecanoic acid into its enantiomers and determination of their absolute configuration. Roczn. Chem. 1976, 50, 997. [Google Scholar] [CrossRef]
- Skogh, M. The higher normal chain DL-beta-hydroxy acids-synthesis and investigation of the crystal behavior of 17 homologous acids with 8 to 24 carbon atoms. Acta Chem. Scand. 1952, 6, 809–817. [Google Scholar] [CrossRef]
- Ansari, A.A.; Osman, S.M. Reaction of hydrogen bromide with diols of long chain α,β-unsaturated acids. J. Am. Oil Chem. Soc. 1976, 53, 118–121. [Google Scholar] [CrossRef]
- Negelmann, L.; Pisch, S.; Bornscheuer, U.; Schmid, R.D. Properties of unusual phospholipids. III: Synthesis, monolayer investigations and DSC studies of hydroxy octadeca(e)noic acids and diacylglycerophosphocholines derived therefrom. Chem. Phys. Lipids 1997, 90, 117–134. [Google Scholar] [CrossRef]
- Goossen, L.J.; Ohlmann, D.M.; Dierker, M. Silver triflate-catalysed synthesis of γ-lactones from fatty acids. Green Chem. 2010, 12, 197–200. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourboula, A.; Limnios, D.; Kokotou, M.G.; Mountanea, O.G.; Kokotos, G. Enantioselective Organocatalysis-Based Synthesis of 3-Hydroxy Fatty Acids and Fatty γ-Lactones. Molecules 2019, 24, 2081. https://doi.org/10.3390/molecules24112081
Bourboula A, Limnios D, Kokotou MG, Mountanea OG, Kokotos G. Enantioselective Organocatalysis-Based Synthesis of 3-Hydroxy Fatty Acids and Fatty γ-Lactones. Molecules. 2019; 24(11):2081. https://doi.org/10.3390/molecules24112081
Chicago/Turabian StyleBourboula, Asimina, Dimitris Limnios, Maroula G. Kokotou, Olga G. Mountanea, and George Kokotos. 2019. "Enantioselective Organocatalysis-Based Synthesis of 3-Hydroxy Fatty Acids and Fatty γ-Lactones" Molecules 24, no. 11: 2081. https://doi.org/10.3390/molecules24112081
APA StyleBourboula, A., Limnios, D., Kokotou, M. G., Mountanea, O. G., & Kokotos, G. (2019). Enantioselective Organocatalysis-Based Synthesis of 3-Hydroxy Fatty Acids and Fatty γ-Lactones. Molecules, 24(11), 2081. https://doi.org/10.3390/molecules24112081