Indolylboronic Acids: Preparation and Applications


Abstract
:1. Introduction
2. Synthetic Approaches to Indolylboronic Acids
2.1. Introduction
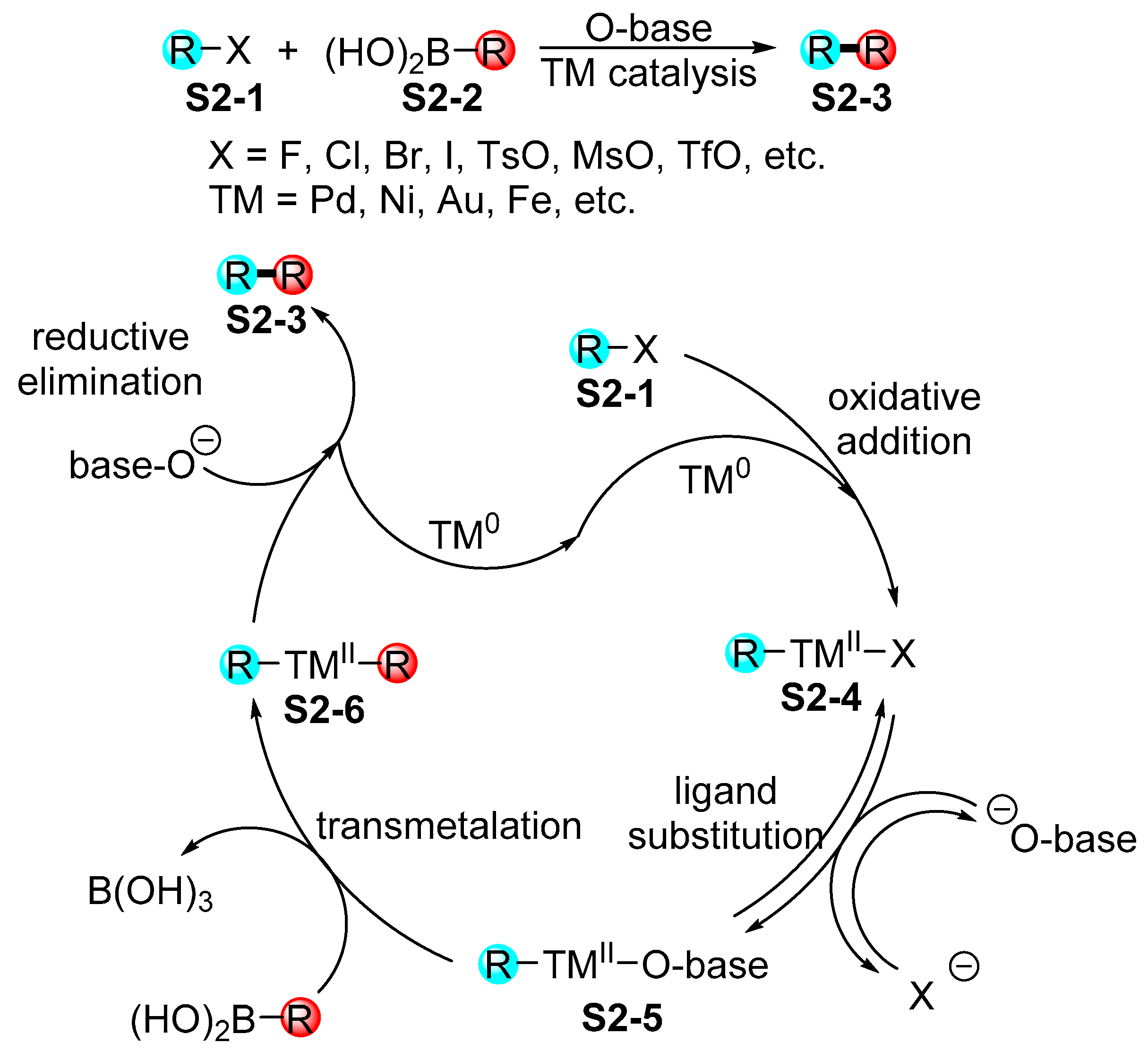
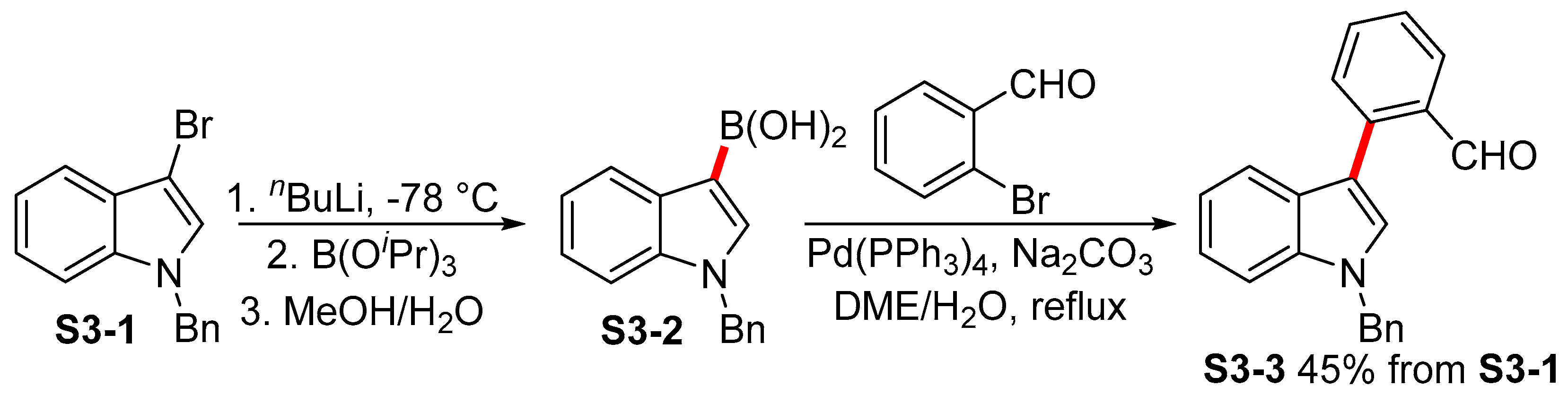
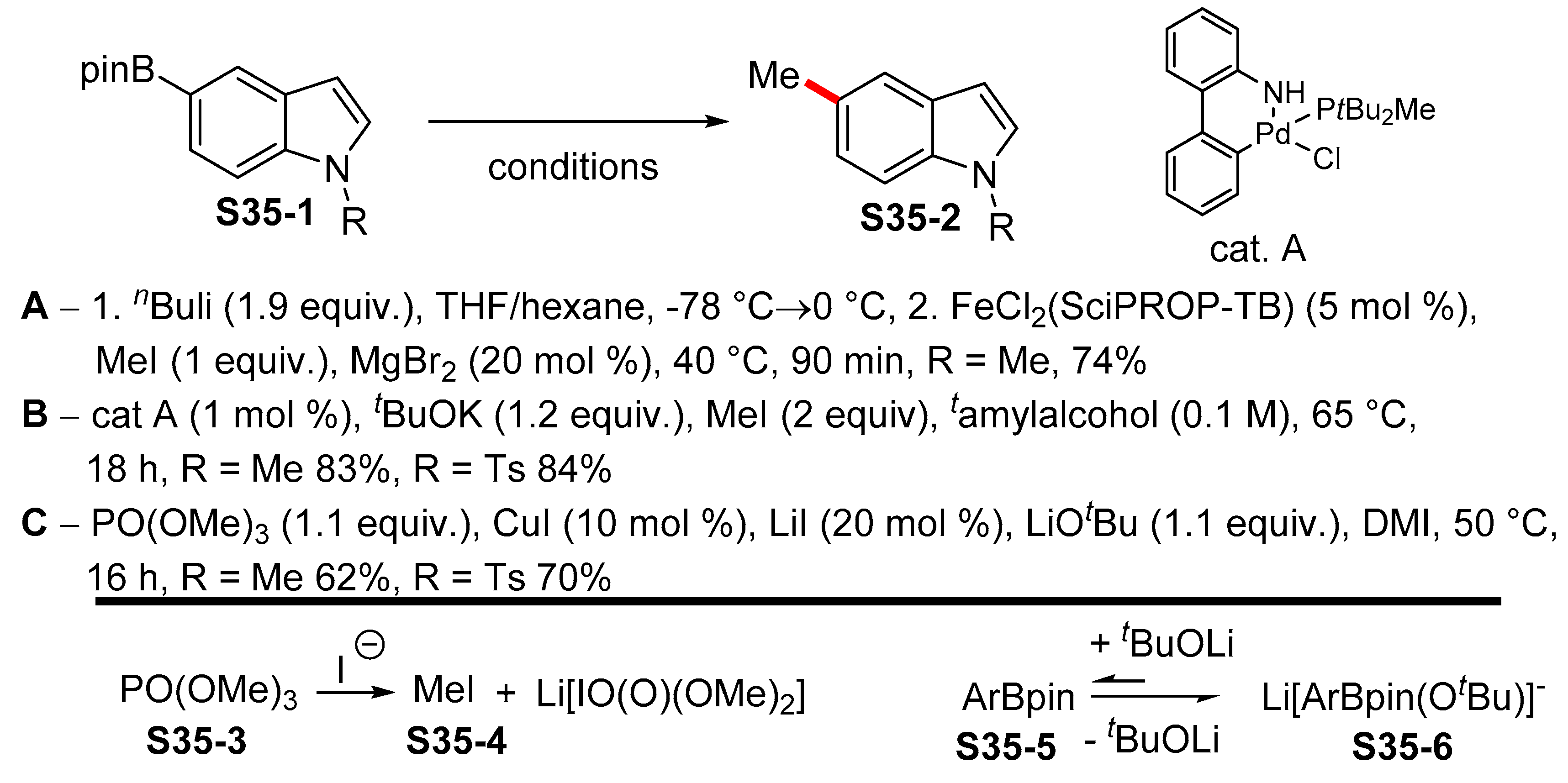
2.2. Miyaura Borylation en route to Indolylboronates
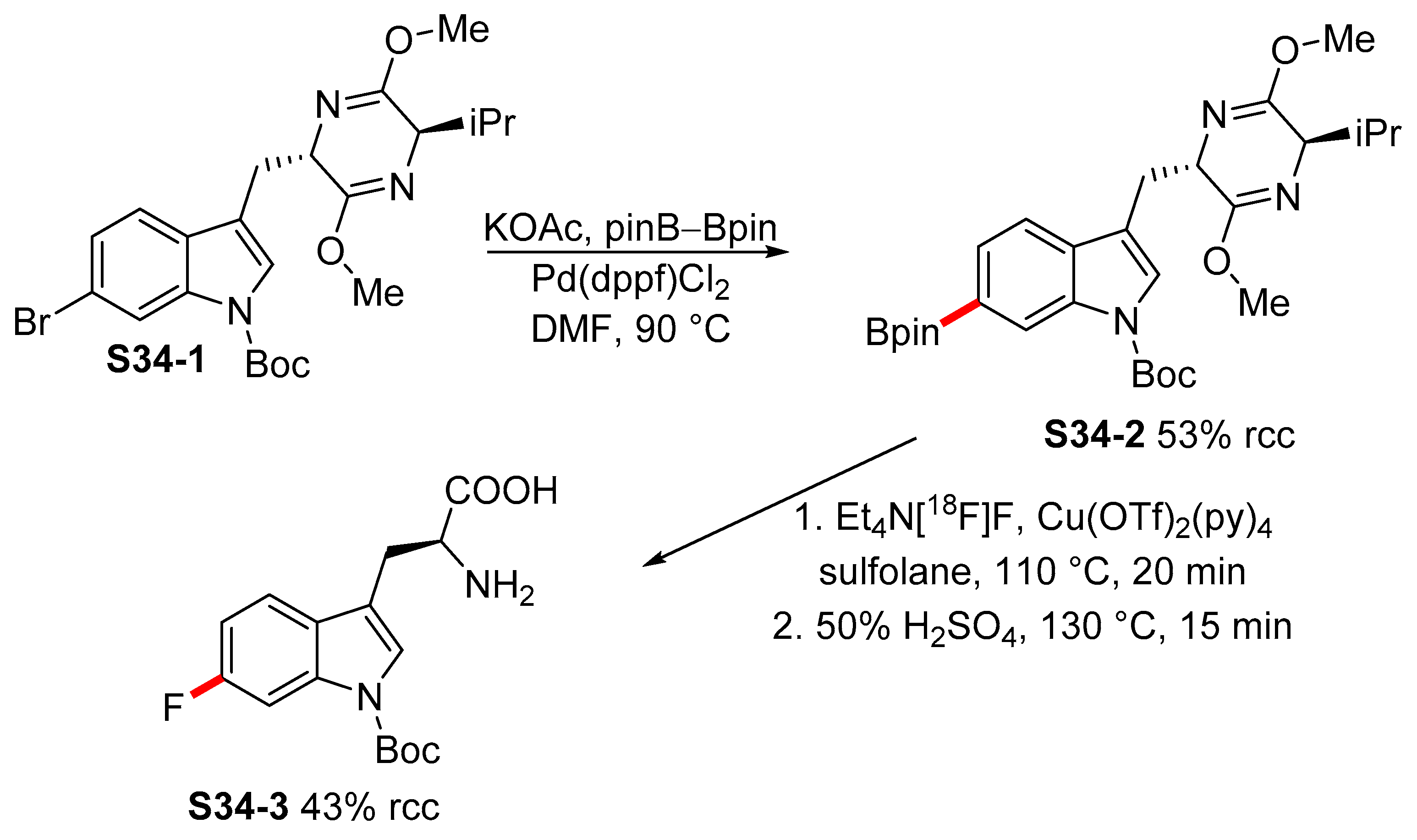
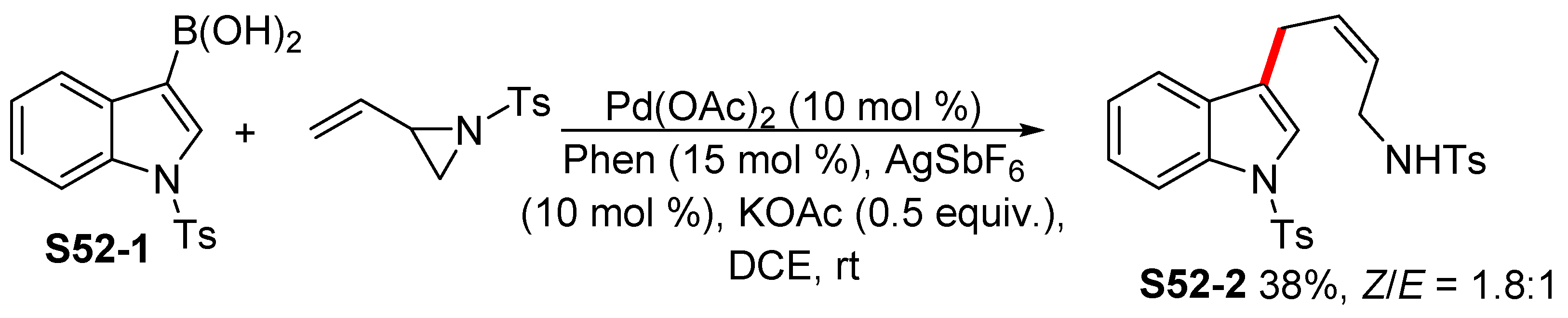
2.3. Transition-Metal-Catalyzed Borylation of Indoles by Means of C–H Activation
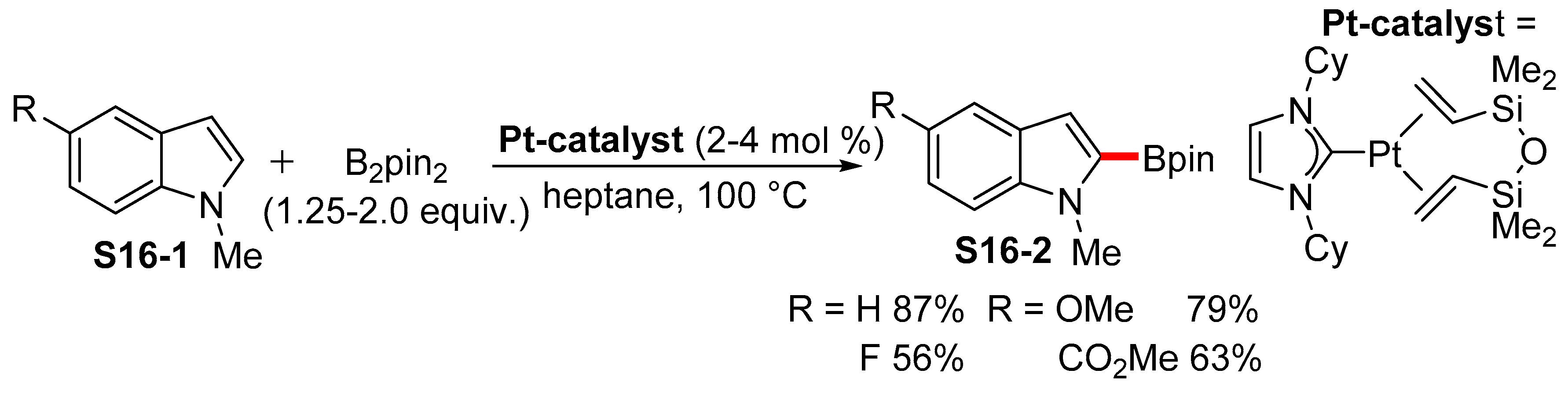
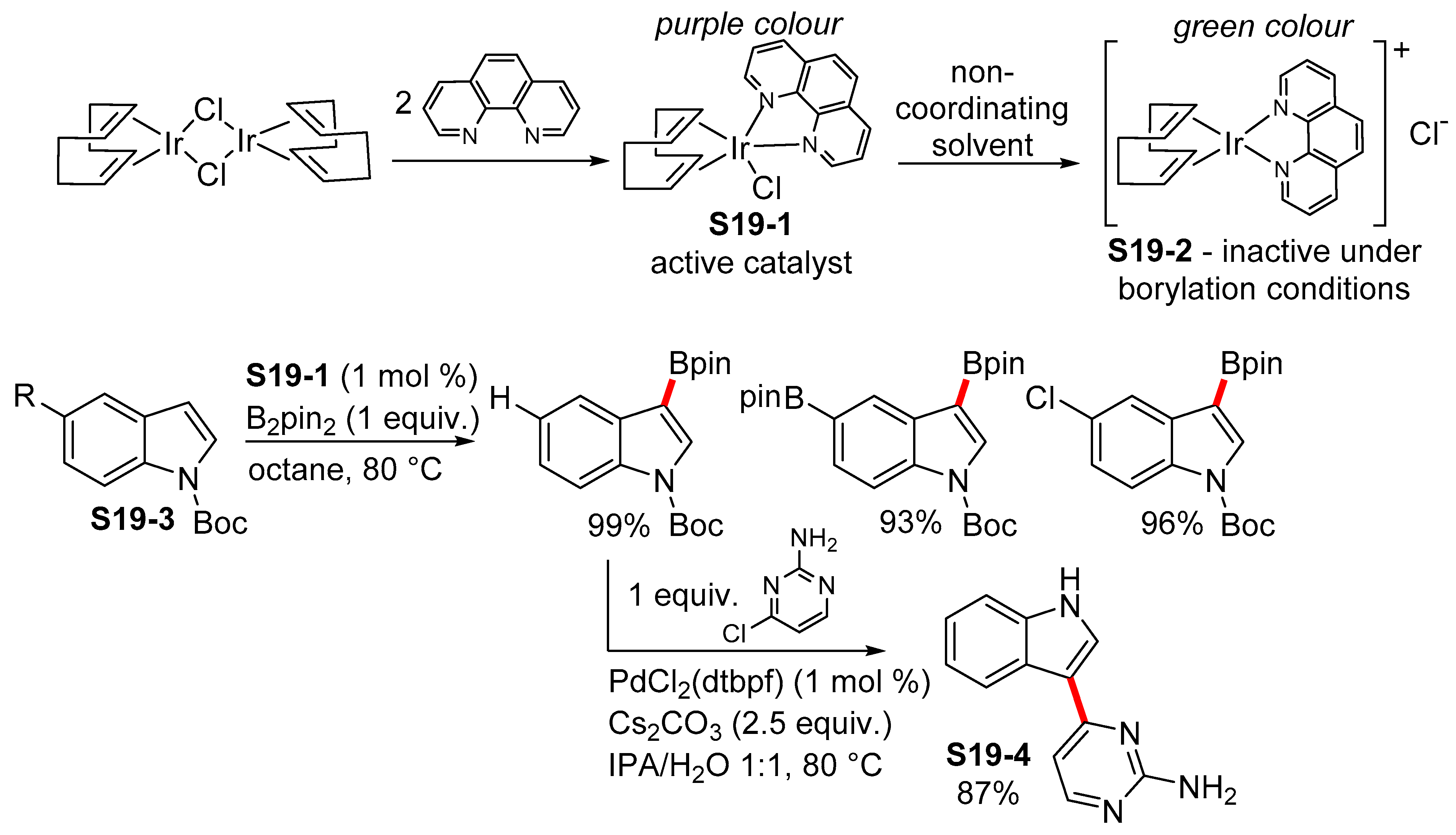
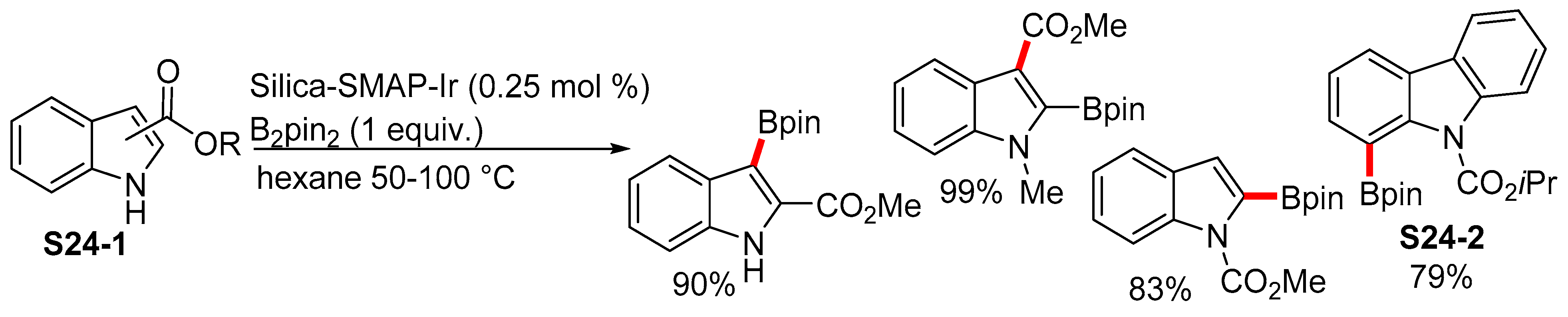
2.3.1. C2-Selective Borylation
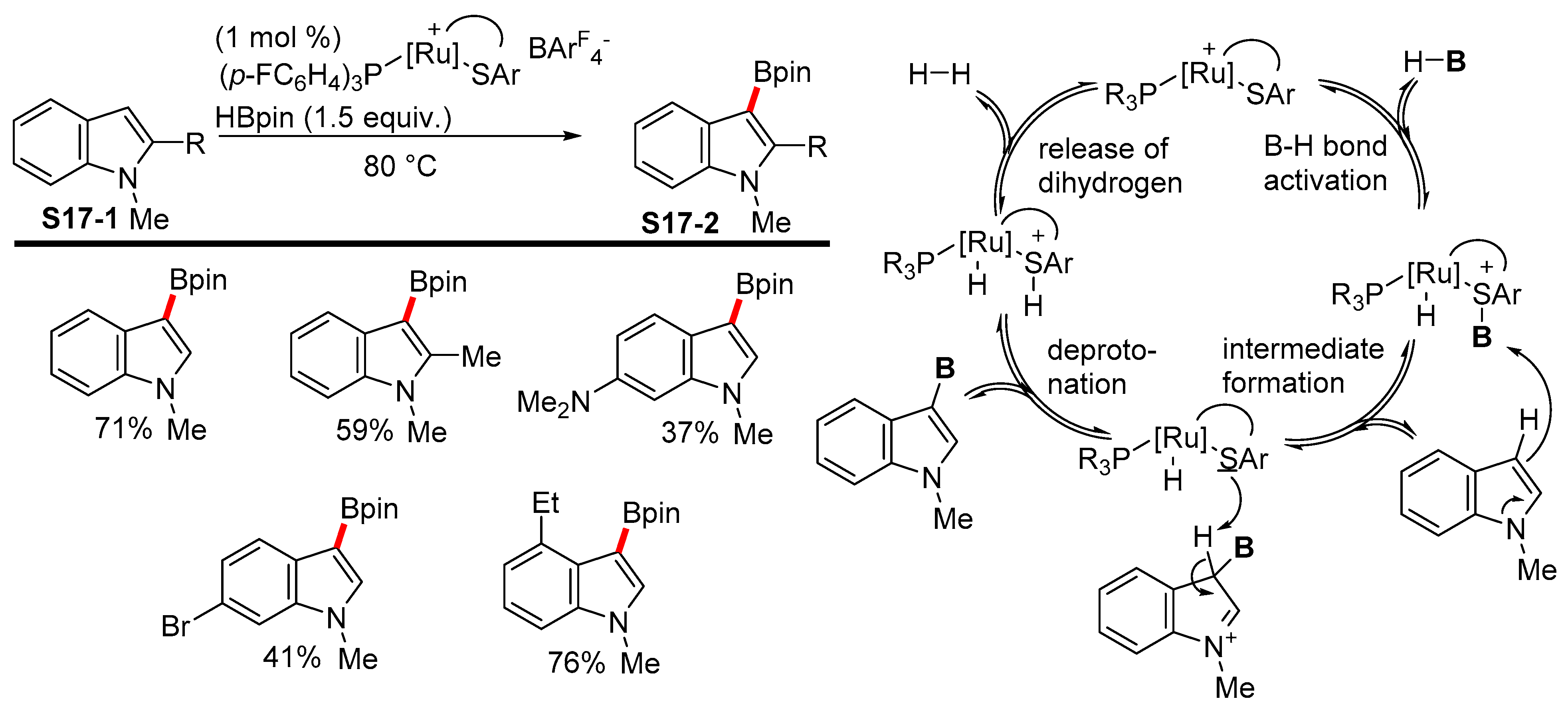
2.3.2. C3-Selective Borylation
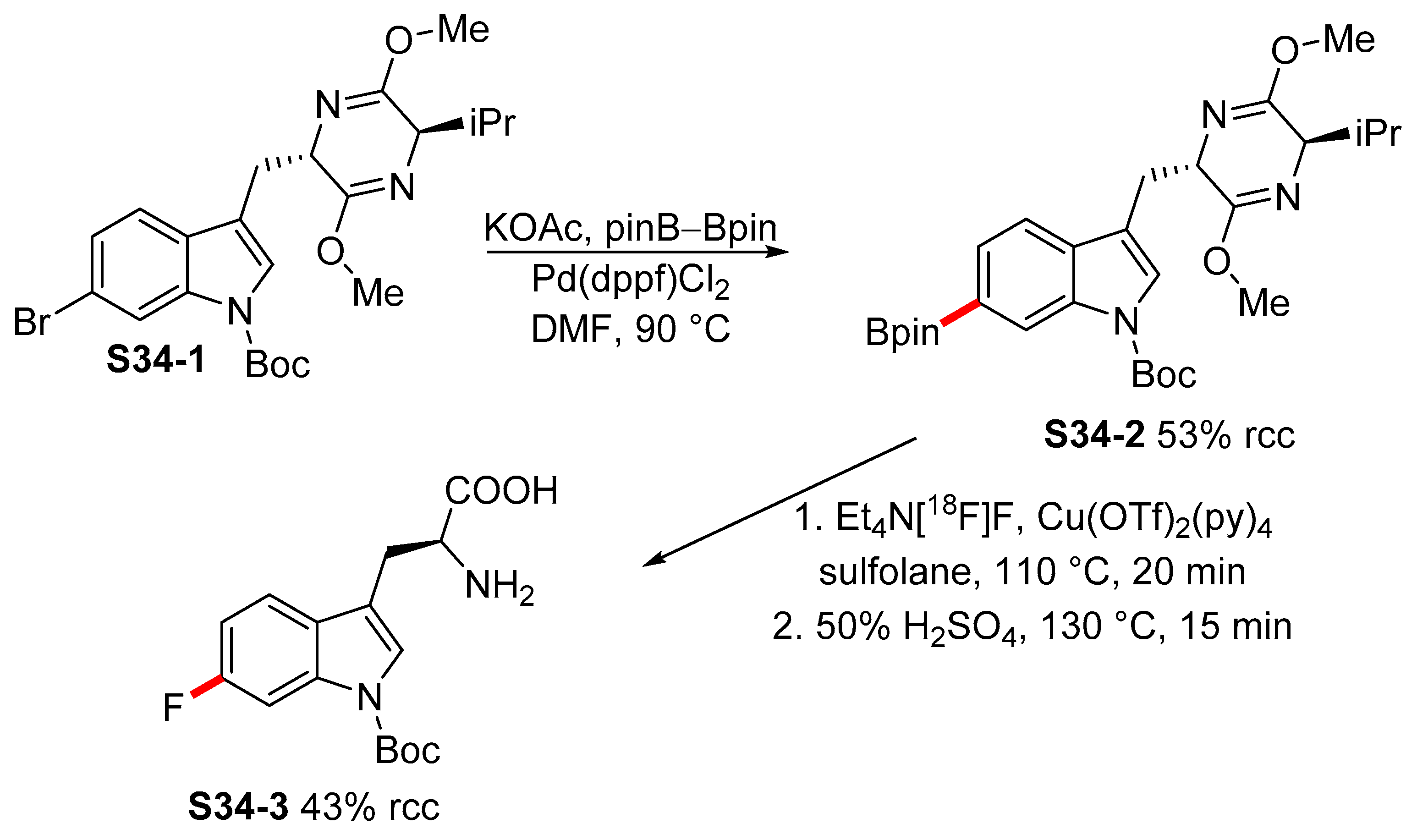
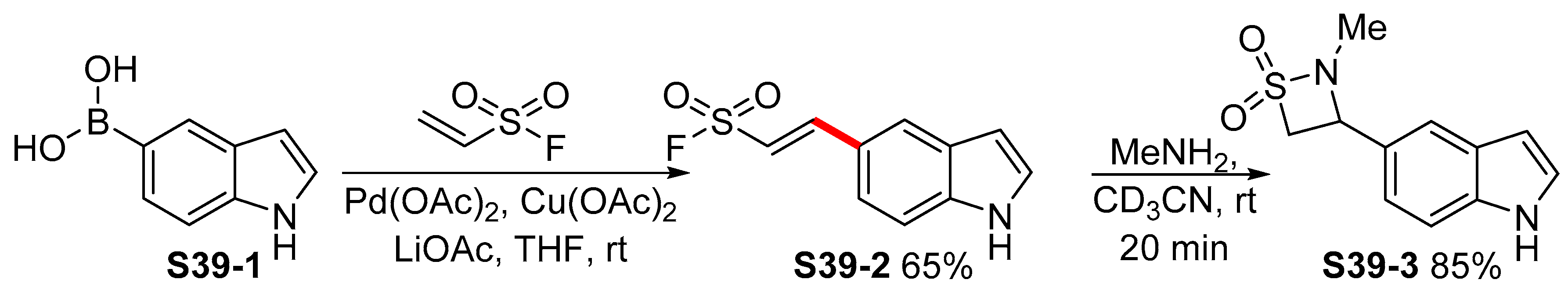
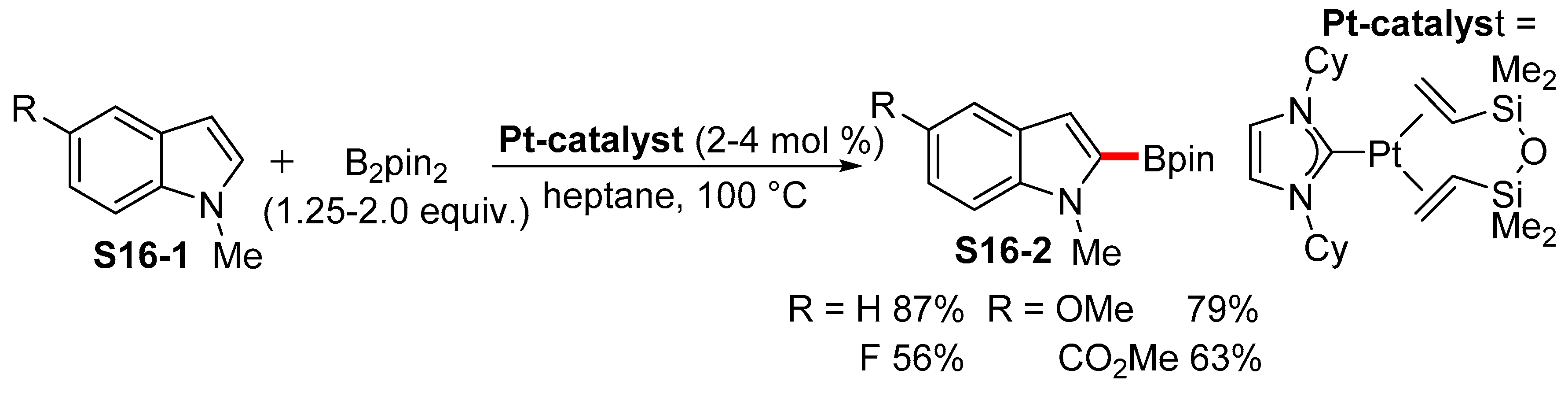
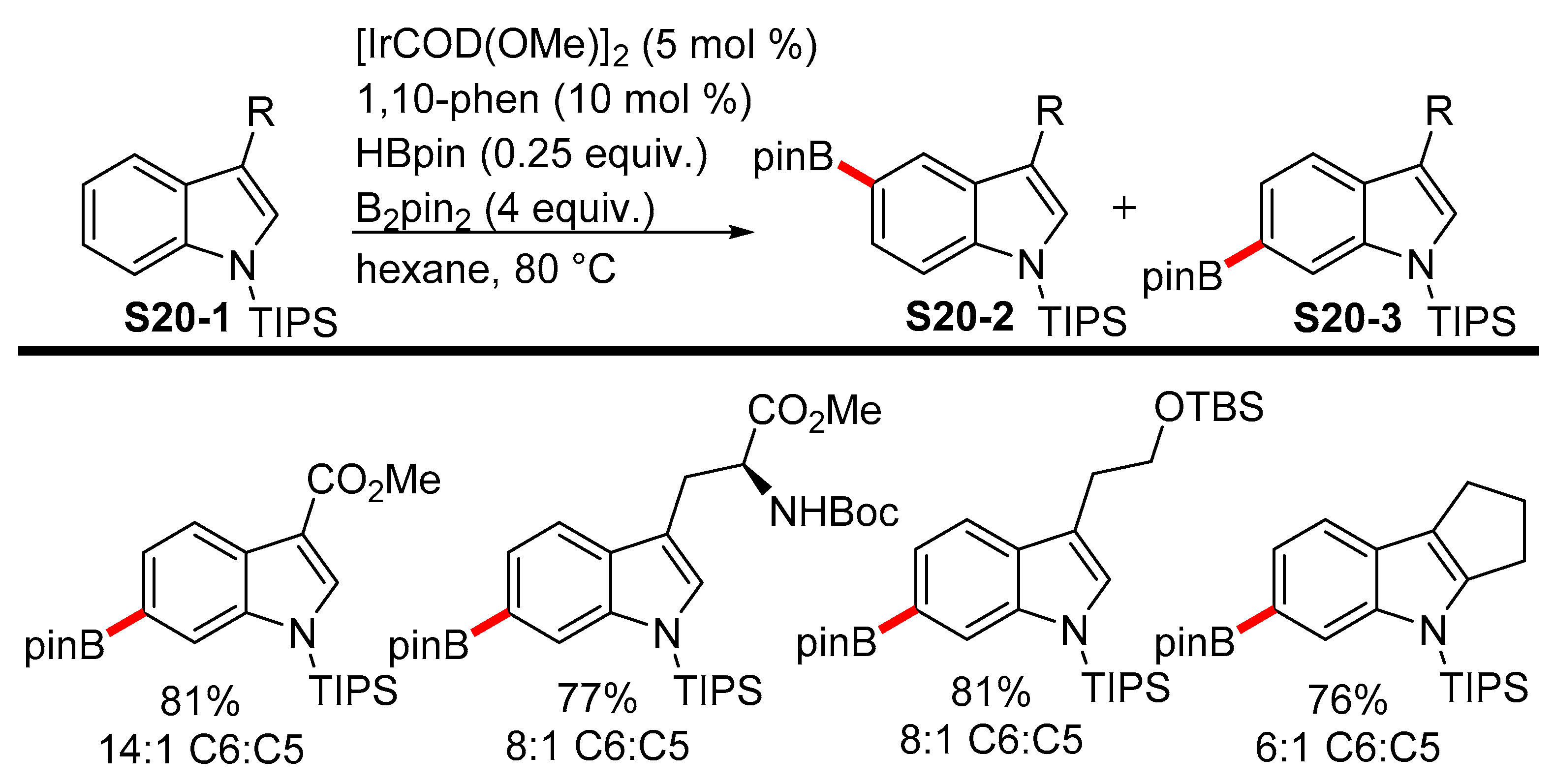
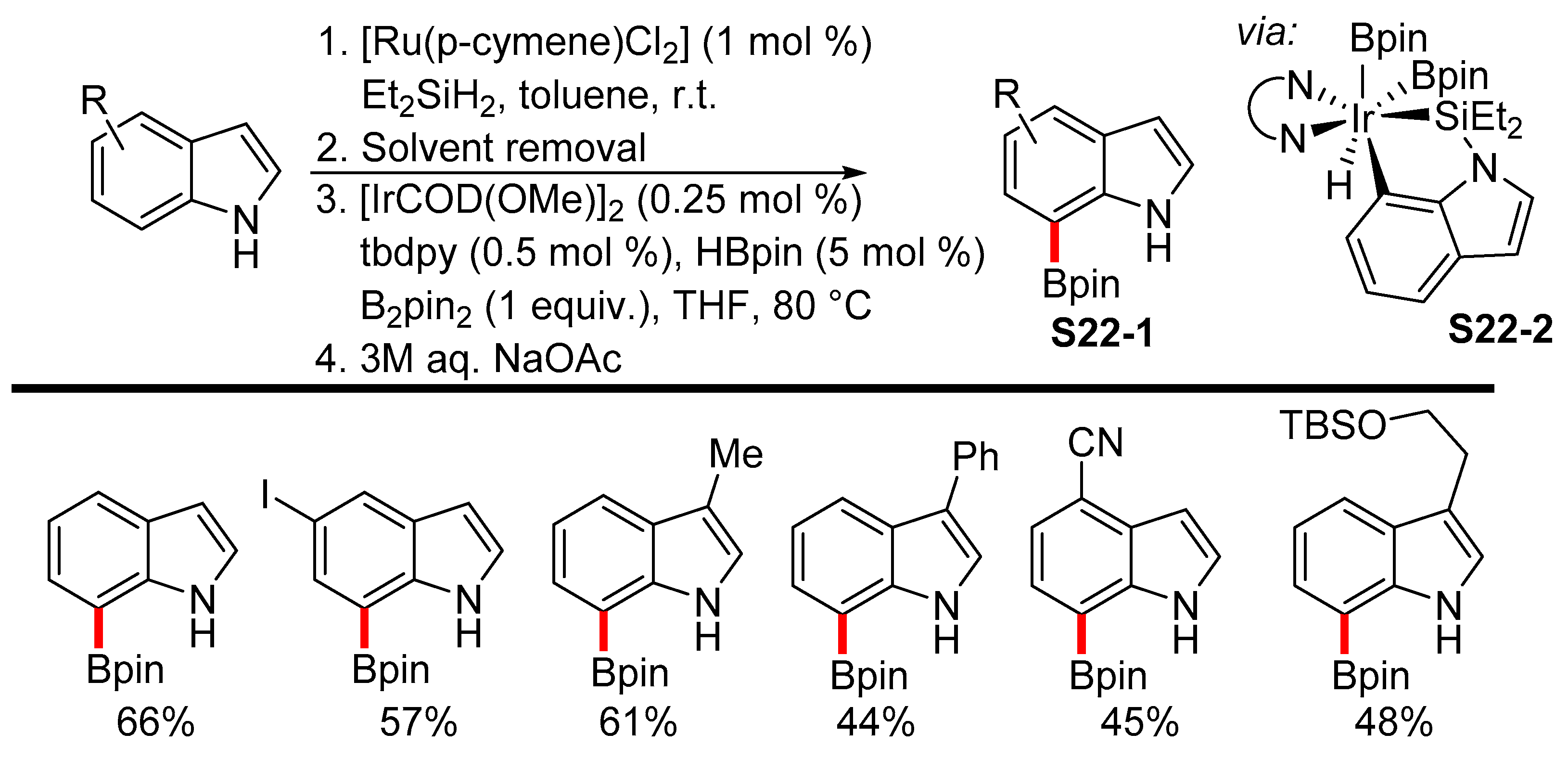
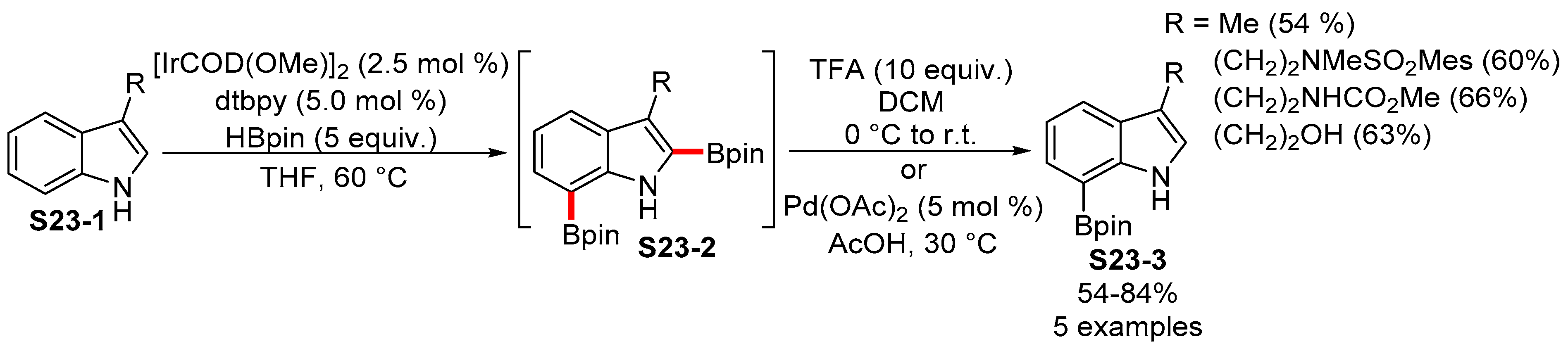
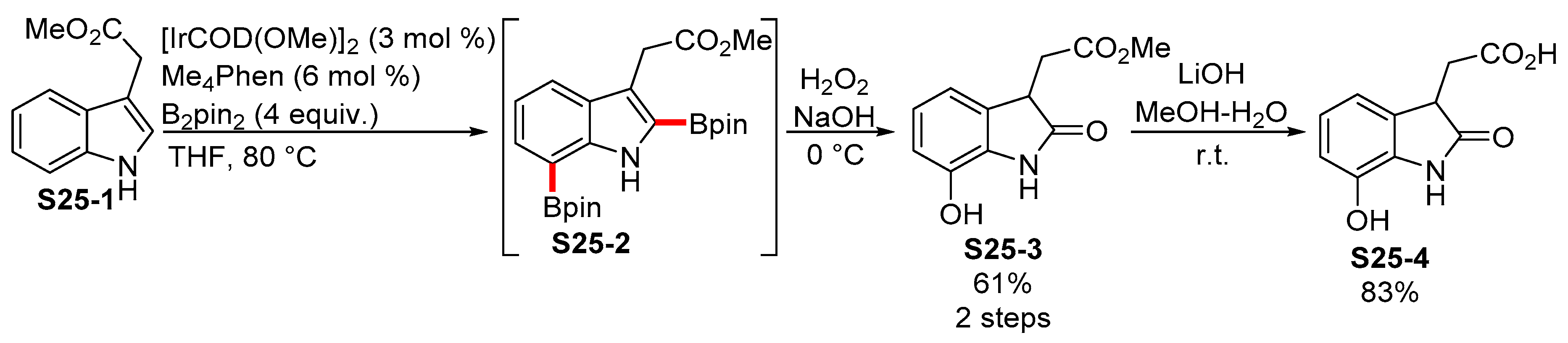
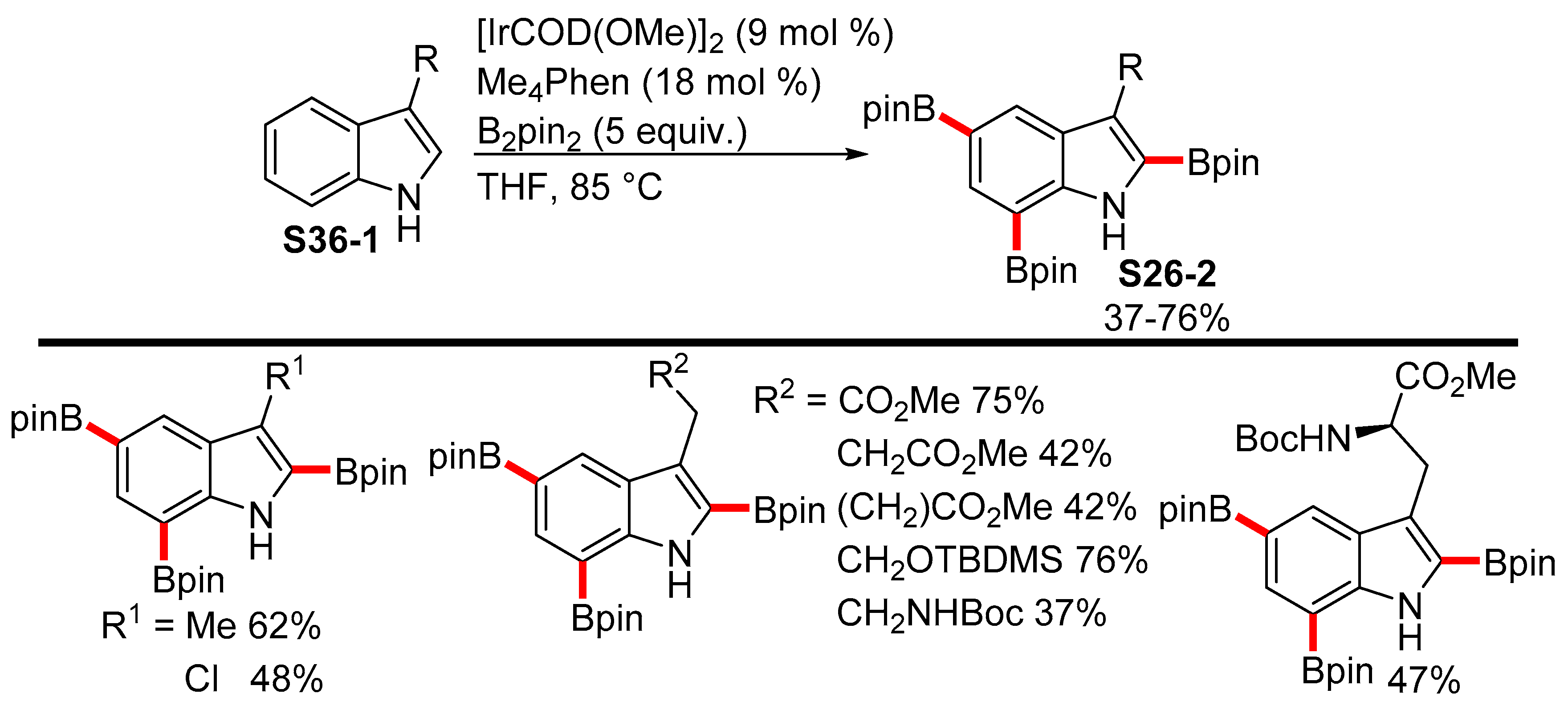
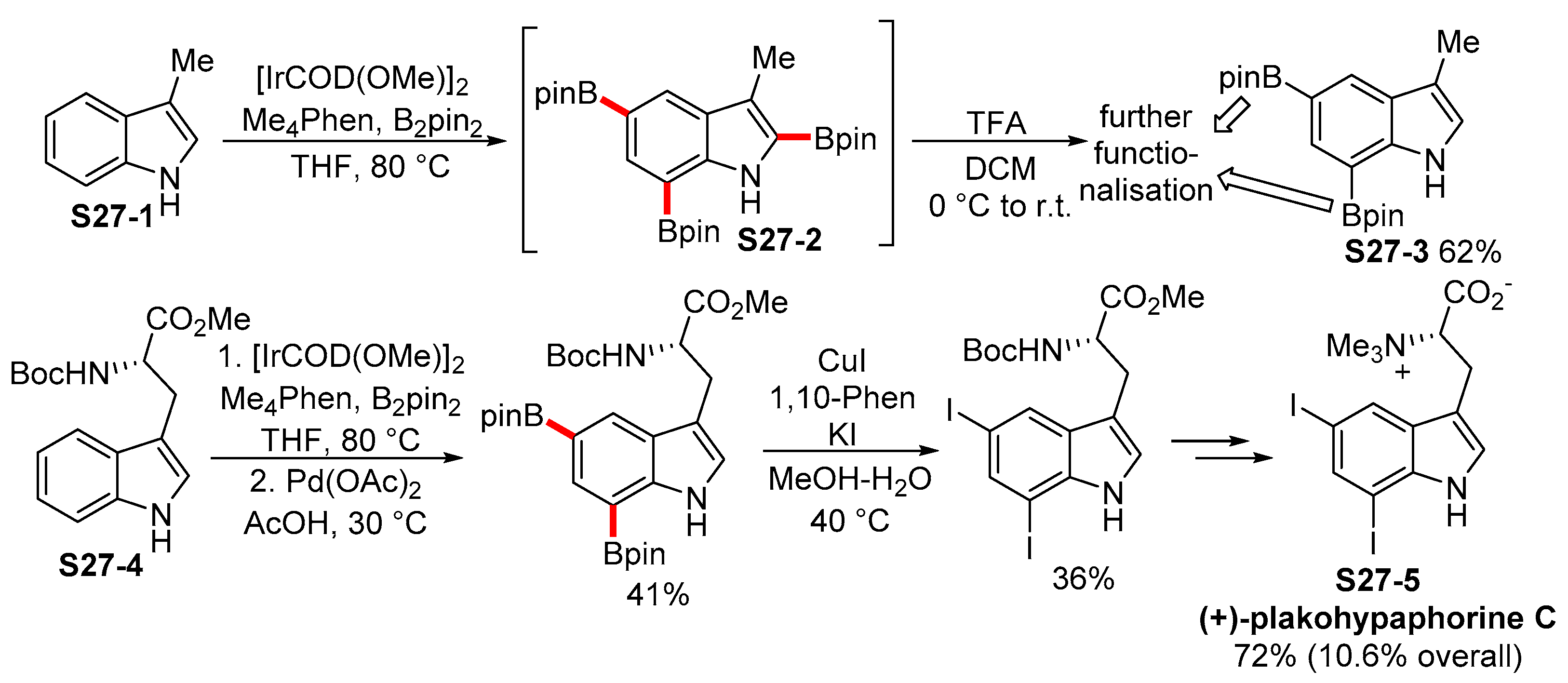
2.3.3. C6-, C7-Selective Borylation
2.3.4. Non-Selective Borylations
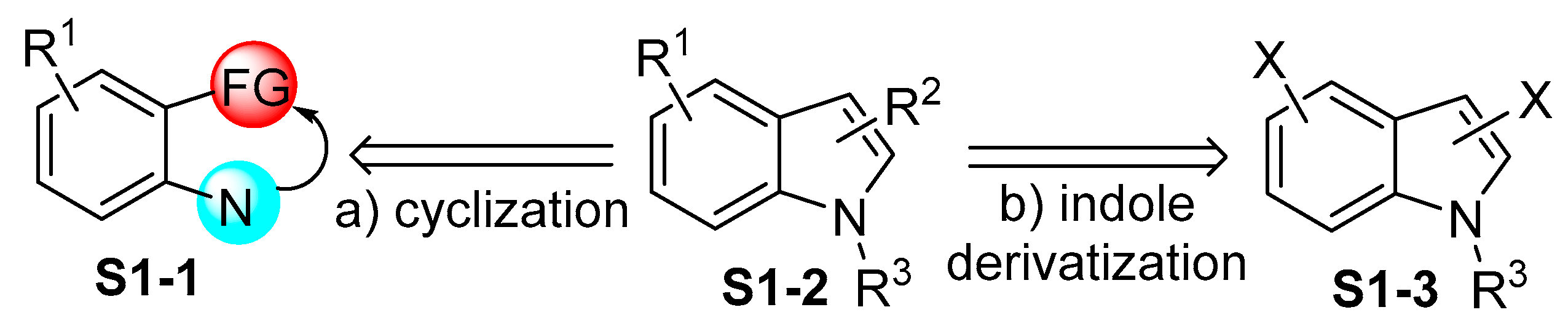
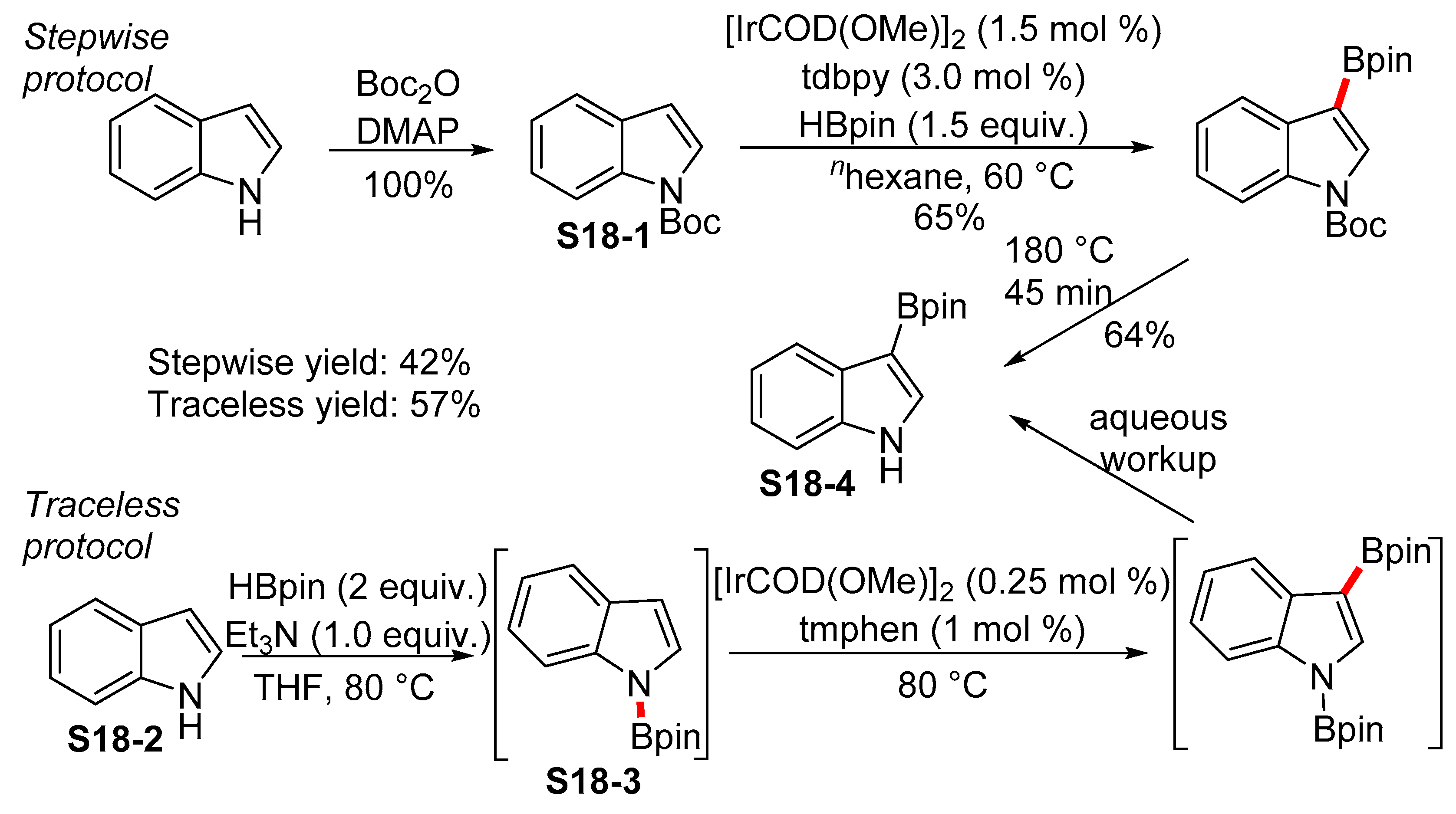
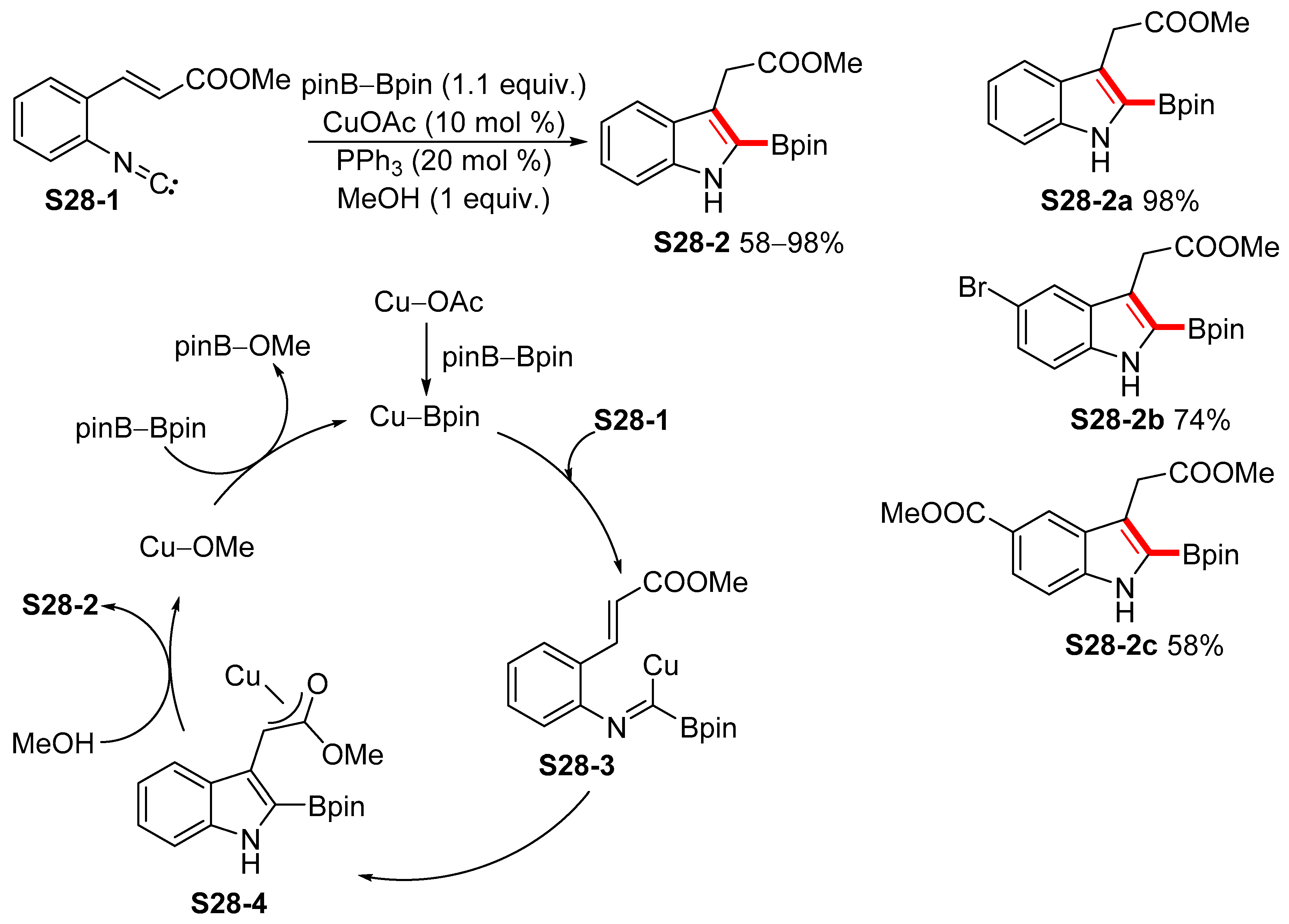
2.4. Cyclization Protocols for the Synthesis of Indolylboronic Acid Derivatives
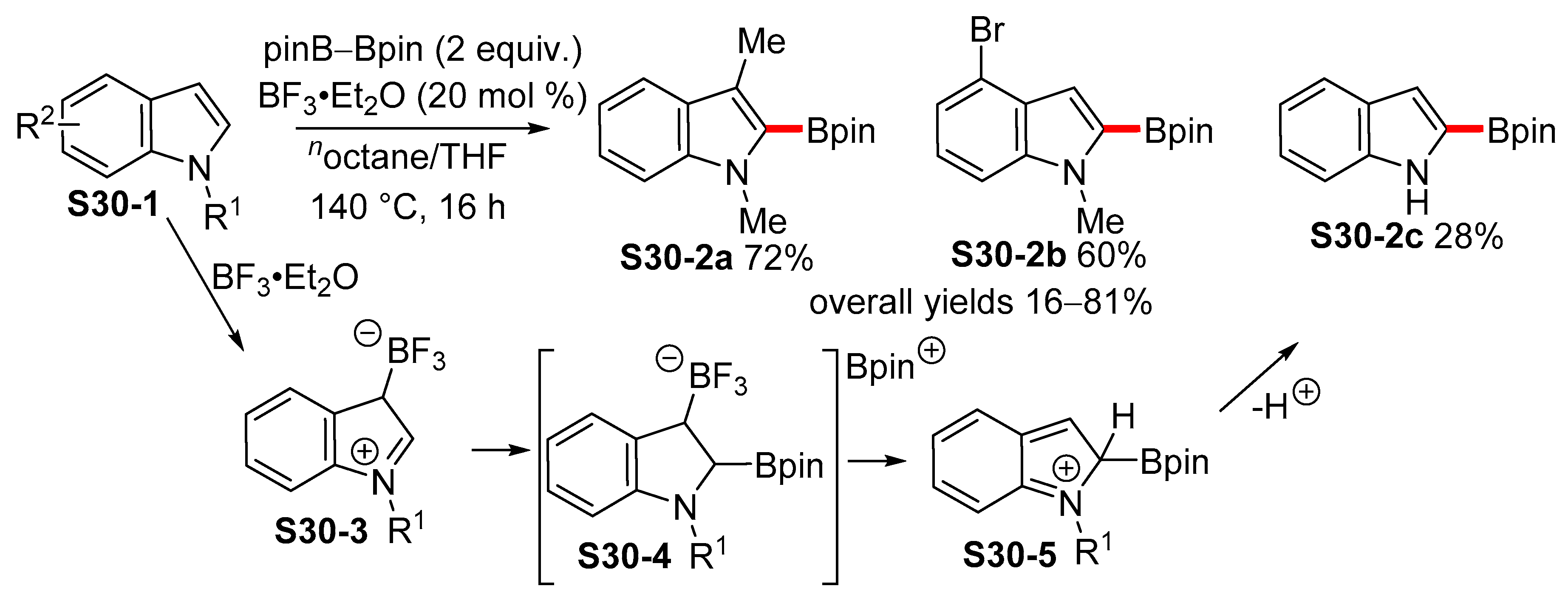
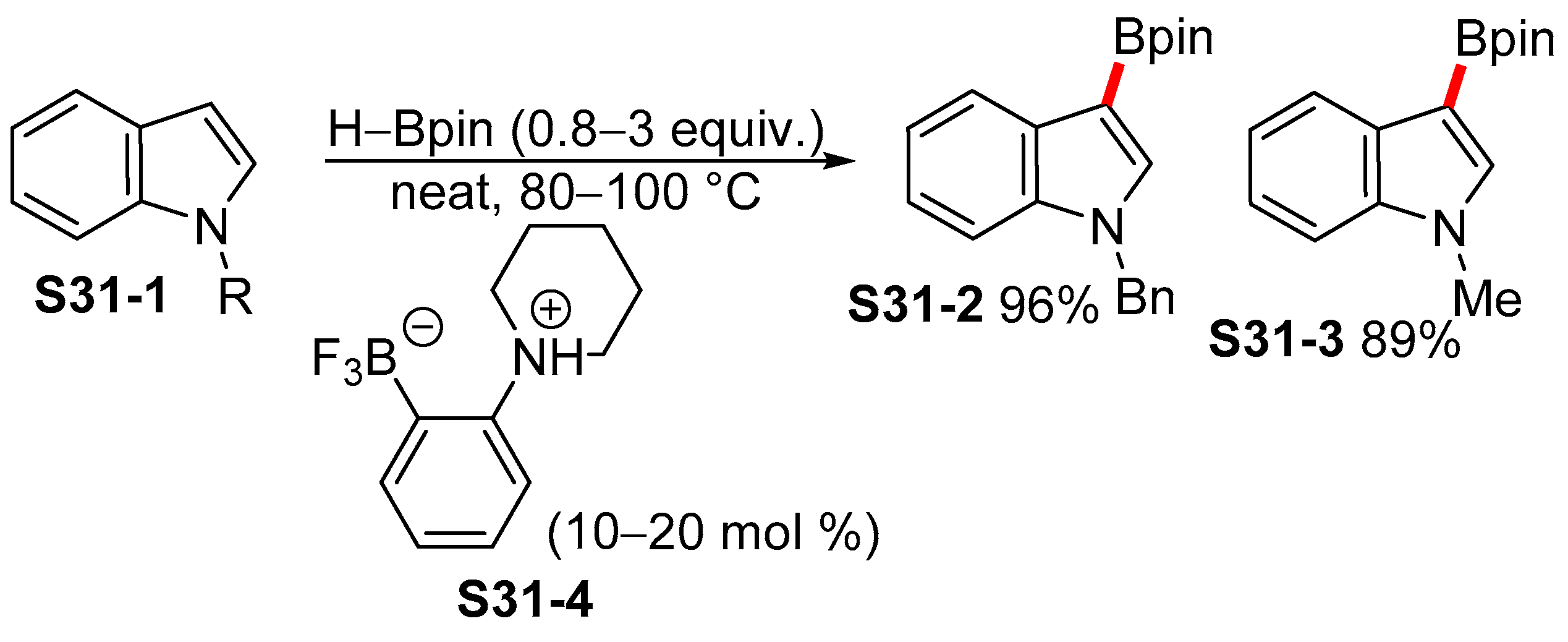
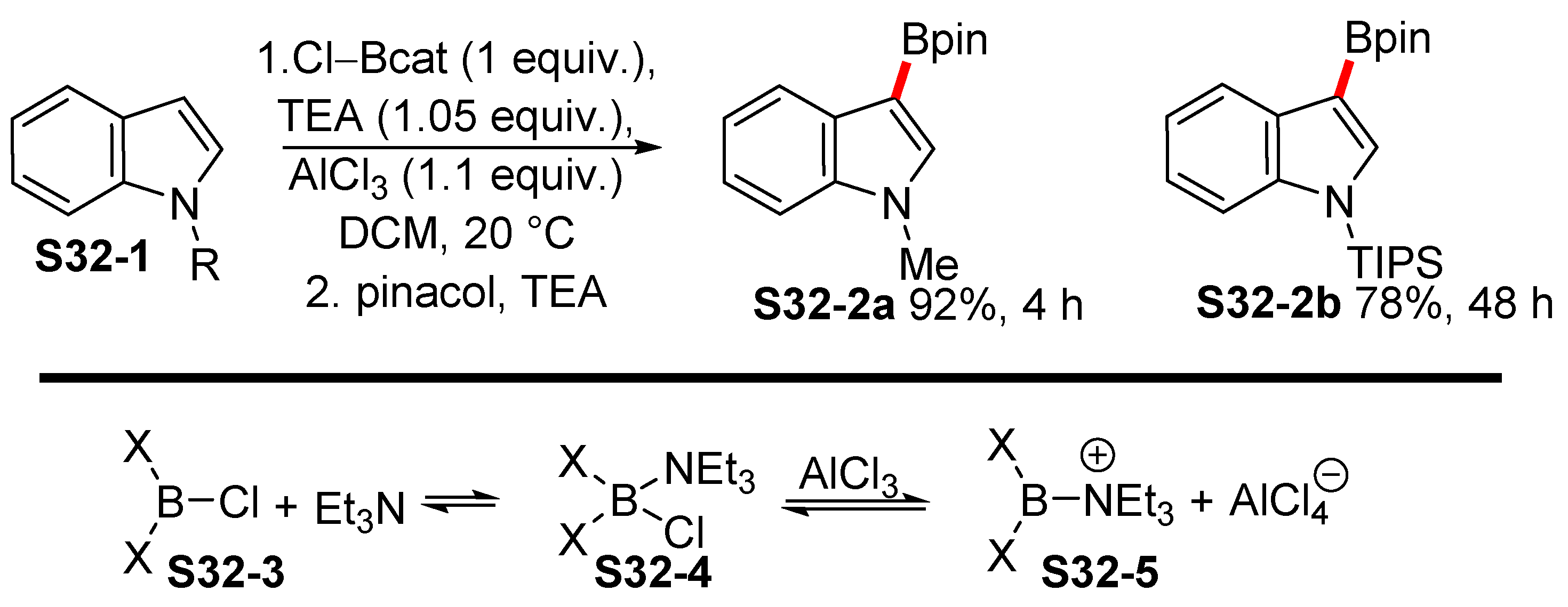
2.5. Lewis-acid-catalyzed Borylation of Indoles
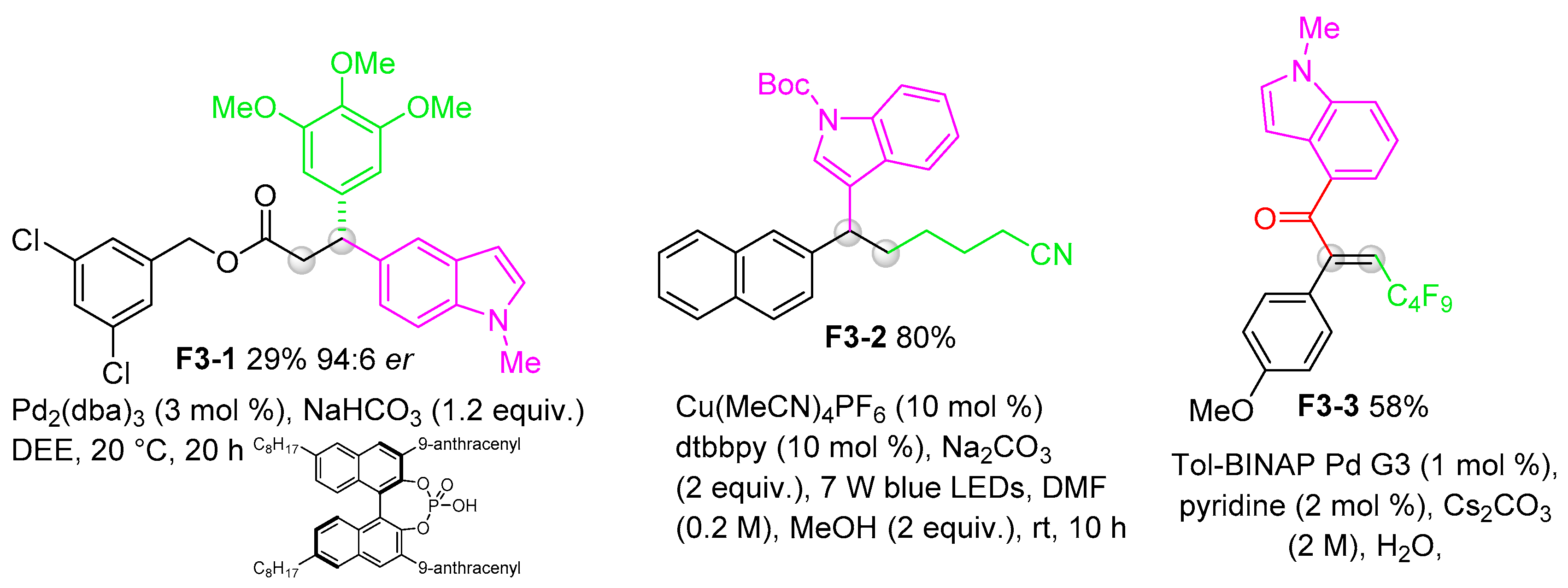
3. Chemical Transformations of Indolylboronic Acid Derivatives
3.1. Formal Substitution of Boron Moieties by Transition-Metal-Catalyzed or Transition-Metal-Free Reactions
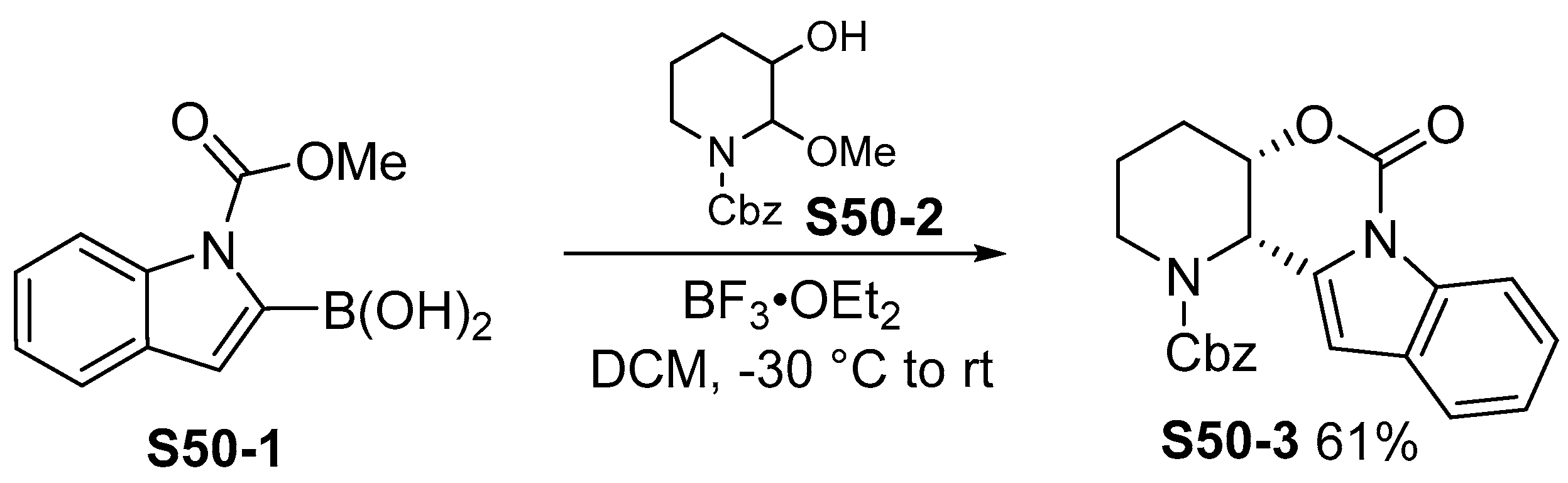
3.2. Cyclization Reactions of Indolylboronic Acid Derivatives
3.3. Multicomponent Reactions of Indolylboronic Acid Derivatives
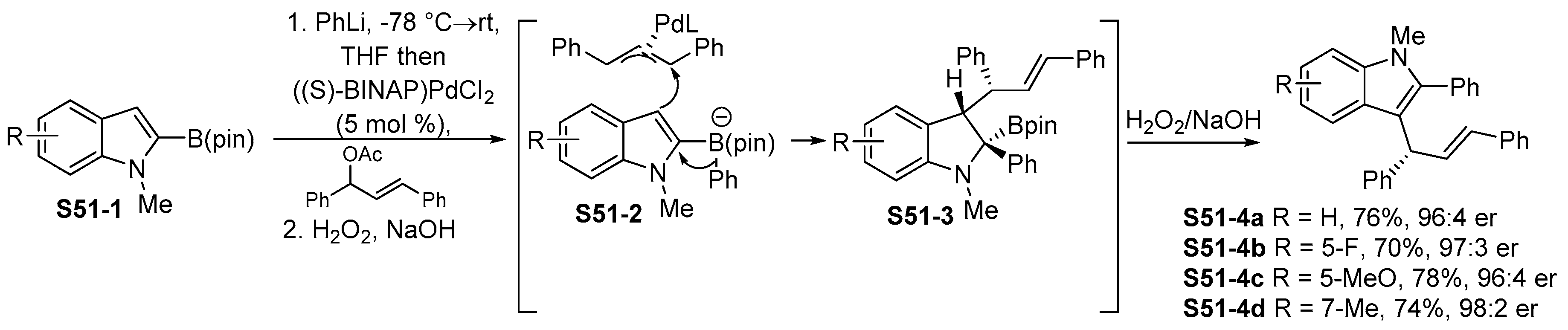
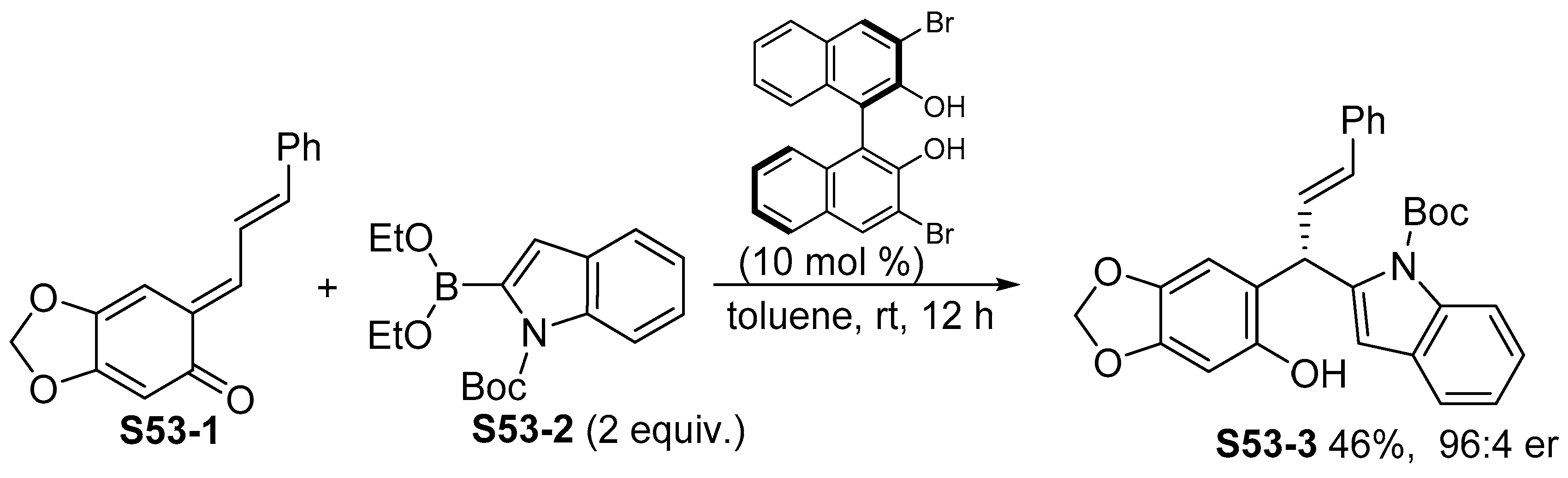
3.4. Addition of Indolylboronic Acids or Boronates to Multiple Bonds
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Foley, C.A.; Al-Issa, Y.A.; Hiller, K.P.; Mulcahy, S.P. Synthesis and Structure–Activity Relationships of 1-Aryl-β-carbolines as Affinity Probes for the 5-Hydroxytryptamine Receptor. ACS Omega 2019, 4, 9807–9812. [Google Scholar] [CrossRef]
- Cornelio, B.; Laronze-Cochard, M.; Miambo, R.; De Grandis, M.; Riccioni, R.; Borisova, B.; Dontchev, D.; Machado, C.; Ceruso, M.; Fontana, A.; et al. 5-Arylisothiazol-3(2H)-one-1,(1)-(di)oxides: A new class of selective tumor-associated carbonic anhydrases (hCA IX and XII) inhibitors. Eur. J. Med. Chem. 2019, 175, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.M.; Altman, M.D.; Cash, B.; Haidle, A.M.; Kubiak, R.L.; Maddess, M.L.; Yan, Y.; Northrup, A.B. Carboxamide Spleen Tyrosine Kinase (Syk) Inhibitors: Leveraging Ground State Interactions To Accelerate Optimization. ACS Med. Chem. Lett. 2016, 7, 1151–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Turpoff, A.; Zhang, X.; Huang, S.; Liu, Y.; Almstead, N.; Njoroge, F.G.; Gu, Z.; Graci, J.; Jung, S.P.; et al. Discovery of 2-(4-sulfonamidophenyl)-indole 3-carboxamides as potent and selective inhibitors with broad hepatitis C virus genotype activity targeting HCV NS4B. Bioorg. Med. Chem. Lett. 2016, 26, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; Venables, B.; Guernon, J.; Chen, J.; Sit, S.-Y.; Rajamani, R.; Knox, R.J.; Matchett, M.; Pieschl, R.L.; Herrington, J.; et al. Discovery of new indole-based acylsulfonamide Nav1.7 inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 659–663. [Google Scholar] [CrossRef]
- Kanada, R.; Tanabe, M.; Muromoto, R.; Sato, Y.; Kuwahara, T.; Fukuda, H.; Arisawa, M.; Matsuda, T.; Watanabe, M.; Shuto, S. Synthesis of Chiral cis-Cyclopropane Bearing Indole and Chromone as Potential TNFα Inhibitors. J. Org. Chem. 2018, 83, 7672–7682. [Google Scholar] [CrossRef]
- Bartoccini, F.; Venturi, S.; Retini, M.; Mari, M.; Piersanti, G. Total Synthesis of (−)-Clavicipitic Acid via γ,γ-Dimethylallyltryptophan (DMAT) and Chemoselective C–H Hydroxylation. J. Org. Chem. 2019, 84, 8027–8034. [Google Scholar] [CrossRef]
- Nabi, A.A.; Liyu, J.; Lindsay, A.C.; Sperry, J. C4−H alkoxylation of 6-bromoindole and its application to the synthesis of breitfussin B. Tetrahedron 2018, 74, 1199–1202. [Google Scholar] [CrossRef]
- Hu, L.; Li, Q.; Yao, L.; Xu, B.; Wang, X.; Liao, X. Enantioselective and Divergent Syntheses of Alstoscholarisines A, E and Their Enantiomers. Org. Lett. 2018, 20, 6202–6205. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, S.; Jia, Y. Formal Synthesis of (+)-Kopsihainanine A and Synthetic Study toward (+)-Limaspermidine. J. Org. Chem. 2015, 80, 12545–12551. [Google Scholar] [CrossRef]
- Zhou, S.; Jia, Y. Total Synthesis of (−)-Goniomitine. Org. Lett. 2014, 16, 3416–3418. [Google Scholar] [CrossRef] [PubMed]
- Baeyer, A.; Emmerling, A. Synthese des Indols. Ber. Dtsch. Chem. Ges. 1869, 2, 679–682. [Google Scholar] [CrossRef]
- Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. The reaction of vinyl grignard reagents with 2-substituted nitroarenes: A new approach to the synthesis of 7-substituted indoles. Tetrahedron Lett. 1989, 30, 2129–2132. [Google Scholar] [CrossRef]
- Fischer, E.; Hess, O. Synthese von Indolderivaten. Ber. Dtsch. Chem. Ges. 1884, 17, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, H.; Yamashita, T.; Reding, M.T.; Kaburagi, Y.; Fukuyama, T. Radical Cyclization of 2-Alkenylthioanilides: A Novel Synthesis of 2,3-Disubstituted Indoles. J. Am. Chem. Soc. 1999, 121, 3791–3792. [Google Scholar] [CrossRef]
- Gassman, P.G.; Van Bergen, T.J.; Gruetzmacher, G. Use of halogen-sulfide complexes in the synthesis of indoles, oxindoles, and alkylated aromatic amines. J. Am. Chem. Soc. 1973, 95, 6508–6509. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K. Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc. 1991, 113, 6689–6690. [Google Scholar] [CrossRef]
- Tanner, M.E. Mechanistic studies on the indole prenyltransferases. Nat. Prod. Rep. 2015, 32, 88–101. [Google Scholar] [CrossRef]
- Bandini, M.; Melloni, A.; Tommasi, S.; Umani-Ronchi, A. A Journey Across Recent Advances in Catalytic and Stereoselective Alkylation of Indoles. Synlett 2005, 1199–1222. [Google Scholar] [CrossRef]
- Makosza, M. Vicarious Nucleophilic Substitution of Hydrogen in the Chemistry of Heterocyclic Compounds. Synthesis 1991, 103–111. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Osipov, S.N. Selective Synthesis of 2- and 7-Substituted Indole Derivatives via Chelation-Assisted Metallocarbenoid C–H Bond Functionalization. Synthesis 2018, 50, 227–240. [Google Scholar]
- Petrini, M. Regioselective Direct C-Alkenylation of Indoles. Chem. Eur. J. 2017, 23, 16115–16151. [Google Scholar] [CrossRef] [PubMed]
- Bheeter, C.B.; Chen, L.; Soulé, J.-F.; Doucet, H. Regioselectivity in palladium-catalysed direct arylation of 5-membered ring heteroaromatics. Catal. Sci. Technol. 2016, 6, 2005–2049. [Google Scholar] [CrossRef]
- Sandtorv, A.H. Transition Metal-Catalyzed C−H Activation of Indoles. Adv. Synth. Catal. 2015, 357, 2403–2435. [Google Scholar] [CrossRef]
- Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Cross-Coupling of Heteroarenes by C−H Functionalization: Recent Progress towards Direct Arylation and Heteroarylation Reactions Involving Heteroarenes Containing One Heteroatom. Adv. Synth. Catal. 2014, 356, 17–117. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef] [Green Version]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc., Chem. Commun. 1979, 866–867. [Google Scholar] [CrossRef]
- Amatore, C.; Le Duc, G.; Jutand, A. Mechanism of Palladium-Catalyzed Suzuki–Miyaura Reactions: Multiple and Antagonistic Roles of Anionic “Bases” and Their Countercations. Chem. Eur. J. 2013, 19, 10082–10093. [Google Scholar] [CrossRef]
- Düfert, M.A.; Billingsley, K.L.; Buchwald, S.L. Suzuki-Miyaura Cross-Coupling of Unprotected, Nitrogen-Rich Heterocycles: Substrate Scope and Mechanistic Investigation. J. Am. Chem. Soc. 2013, 135, 12877–12885. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.L.; Murray, J.; Sneddon, H.F.; Jamieson, C.; Watson, A.J.B. Dimethylisosorbide (DMI) as a Bio-Derived Solvent for Pd-Catalyzed Cross-Coupling Reactions. Synlett 2018, 29, 2293–2297. [Google Scholar] [Green Version]
- Polák, P.; Tobrman, T. The synthesis of polysubstituted indoles from 3-bromo-2-indolyl phosphates. Org. Biomol. Chem. 2017, 15, 6233–6241. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Abbiati, G.; Canevari, V.; Celentano, G.; Magri, E. 2-Trifluoromethanesulfonyloxyindole-1-carboxylic Acid Ethyl Ester: A Practical Intermediate for the Synthesis of 2-Carbosubstituted Indoles. Synthesis 2006, 299–304. [Google Scholar] [CrossRef]
- Nazari, S.H.; Bourdeau, J.E.; Talley, M.R.; Valdivia-Berroeta, G.A.; Smith, S.J.; Michaelis, D.J. Nickel-Catalyzed Suzuki Cross Couplings with Unprotected Allylic Alcohols Enabled by Bidentate N-Heterocyclic Carbene (NHC)/Phosphine Ligands. ACS Catal. 2018, 8, 86–89. [Google Scholar] [CrossRef]
- Tan, J.; Chen, Y.; Li, H.; Yasuda, N. Suzuki-Miyaura Cross-Coupling Reactions of Unprotected Haloimidazoles. J. Org. Chem. 2014, 79, 8871–8876. [Google Scholar] [CrossRef]
- Kotek, V.; Polák, P.; Dvořáková, H.; Tobrman, T. Aluminum Chloride Promoted Cross-Coupling of Trisubstituted Enol Phosphates with Organozinc Reagents En Route to the Stereoselective Synthesis of Tamoxifen and Its Analogues. Eur. J. Org. Chem. 2016, 5037–5044. [Google Scholar] [CrossRef]
- Nambo, M.; Keske, E.C.; Rygus, J.P.G.; Yim, J.C.H.; Crudden, C.M. Development of Versatile Sulfone Electrophiles for Suzuki–Miyaura Cross-Coupling Reactions. ACS Catal. 2017, 7, 1108–1112. [Google Scholar] [CrossRef]
- Chang, S.; Sun, Y.B.; Zhang, X.R.; Dong, L.L.; Zhu, H.Y.; Lai, H.W.; Wang, D. Pd-catalyzed Desulfitative reaction of Aryltrifluoroborates with sodium Arenesulfinates in water. Appl. Organomet. Chem. 2018, 32, e3970. [Google Scholar] [CrossRef]
- Landstrom, E.B.; Handa, S.; Aue, D.H.; Gallou, F.; Lipshutz, B.H. EvanPhos: A ligand for ppm level Pd-catalyzed Suzuki–Miyaura couplings in either organic solvent or water. Green Chem. 2018, 20, 3436–3443. [Google Scholar] [CrossRef]
- Zou, Y.; Yue, G.; Xu, J.; Zhou, J. General Suzuki Coupling of Heteroaryl Bromides by Using Tri-tert-butylphosphine as a Supporting Ligand. Eur. J. Org. Chem. 2014, 5901–5905. [Google Scholar] [CrossRef]
- Boit, T.B.; Weires, N.A.; Kim, J.; Garg, N.K. Nickel-Catalyzed Suzuki–Miyaura Coupling of Aliphatic Amides. ACS Catal. 2018, 8, 1003–1008. [Google Scholar] [CrossRef]
- Schäfer, P.; Palacin, T.; Sidera, M.; Fletcher, S.P. Asymmetric Suzuki-Miyaura coupling of heterocycles via Rhodium-catalysed allylic arylation of racemates. Nat. Commun. 2017, 8, 15762. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Okuzono, C.; Adak, L.; Jin, M.; Nakamura, M. Iron-catalysed enantioselective Suzuki–Miyaura coupling of racemic alkyl bromides. Chem. Commun. 2019, 55, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Cobb, K.M.; Rabb-Lynch, J.M.; Hoerrner, M.E.; Manders, A.; Zhou, Q.; Watson, M.P. Stereospecific, Nickel-Catalyzed Suzuki–Miyaura Cross-Coupling of Allylic Pivalates To Deliver Quaternary Stereocenters. Org. Lett. 2017, 19, 4355–4358. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, E.; Chen, H.; Li, H.; Liu, Y.; Wang, P.G. A general and efficient synthesis of substituted fluorenes and heterocycle-fused indenes containing thiophene or indole rings utilizing a Suzuki–Miyaura coupling and acid-catalyzed Friedel–Crafts reactions as key steps. Tetrahedron 2008, 64, 9033–9043. [Google Scholar] [CrossRef]
- Vazquez, E.; Davies, I.W.; Payack, J.F. A Non-cryogenic Method for the Preparation of 2-(Indolyl) Borates, Silanes, and Silanols. J. Org. Chem. 2002, 67, 7551–7552. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Broda, E.; Snieckus, V. Directed ortho-Metalation–Cross-Coupling Strategies. One-Pot Suzuki Reaction to Biaryl and Heterobiaryl Sulfonamides. Org. Lett. 2011, 13, 3588–3591. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron 2007, 63, 1568–1576. [Google Scholar] [CrossRef]
- Mesganaw, T.; Fine Nathel, N.F.; Garg, N.K. Cine Substitution of Arenes Using the Aryl Carbamate as a Removable Directing Group. Org. Lett. 2012, 14, 2918–2921. [Google Scholar] [CrossRef]
- Al-Saedy, M.A.E.; Harrity, J.P.A. Synthesis and Stabilities of 3-Borylated Indoles. Synlett 2016, 27, 1674–1676. [Google Scholar]
- Guerrand, H.D.S.; Marciasini, L.D.; Jousseaume, M.; Vaultier, M.; Pucheault, M. Borylation of Unactivated Aryl Chlorides under Mild Conditions by Using Diisopropylaminoborane as a Borylating Reagent. Chem. Eur. J. 2014, 20, 5573–5579. [Google Scholar] [CrossRef]
- Nitelet, A.; Thevenet, D.; Schiavi, B.; Hardouin, C.; Fournier, J.; Tamion, R.; Pannecoucke, X.; Jubault, P.; Poisson, T. Copper-Photocatalyzed Borylation of Organic Halides under Batch and Continuous-Flow Conditions. Chem. Eur. J. 2019, 25, 3262–3266. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.K.; Mandal, S.; Geetharani, K. Efficient Synthesis of Aryl Boronates via Cobalt-Catalyzed Borylation of Aryl Chlorides and Bromides. ACS Catal. 2018, 8, 4049–4054. [Google Scholar] [CrossRef]
- Labre, F.; Gimbert, Y.; Bannwarth, P.; Olivero, S.; Duñach, E.; Chavant, P.Y. Application of Cooperative Iron/Copper Catalysis to a Palladium-Free Borylation of Aryl Bromides with Pinacolborane. Org. Lett. 2014, 16, 2366–2369. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Song, D.; Jeon, S.; Kim, Y.; Kim, H.; Lee, S.; Cho, H.; Lee, B.C.; Kim, S.E.; Kim, K.; et al. Cobalt-Catalyzed C–F Bond Borylation of Aryl Fluorides. Org. Lett. 2018, 20, 7249–7252. [Google Scholar] [CrossRef]
- Liu, X.-W.; Echavarren, J.; Zarate, C.; Martin, R. Ni-Catalyzed Borylation of Aryl Fluorides via C–F Cleavage. J. Am. Chem. Soc. 2015, 137, 12470–12473. [Google Scholar] [CrossRef]
- Tobisu, M.; Kinuta, H.; Kita, Y.; Rémond, E.; Chatani, N. Rhodium(I)-Catalyzed Borylation of Nitriles through the Cleavage of Carbon–Cyano Bonds. J. Am. Chem. Soc. 2012, 134, 115–118. [Google Scholar] [CrossRef]
- Kinuta, H.; Kita, Y.; Rémond, E.; Tobisu, M.; Chatani, N. Novel Synthetic Approach to Arylboronates via Rhodium-Catalyzed Carbon–Cyano Bond Cleavage of Nitriles. Synthesis 2012, 44, 2999–3002. [Google Scholar] [CrossRef]
- Chen, K.; Cheung, M.S.; Lin, Z.; Li, P. Metal-free borylation of electron-rich aryl (pseudo)halides under continuous-flow photolytic conditions. Org. Chem. Front. 2016, 3, 875–879. [Google Scholar] [CrossRef]
- Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. Anomalous Reactivity of Silylborane: Transition-Metal-Free Boryl Substitution of Aryl, Alkenyl, and Alkyl Halides with Silylborane/Alkoxy Base Systems. J. Am. Chem. Soc. 2012, 134, 19997–20000. [Google Scholar] [CrossRef]
- Yamamoto, E.; Izumi, K.; Horita, Y.; Ukigai, S.; Ito, H. Formal Nucleophilic Boryl Substitution of Organic Halides with Silylborane/Alkoxy Base System. Top. Catal. 2014, 57, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Erb, W.; Hellal, A.; Albini, M.; Rouden, J.; Blanchet, J. An Easy Route to (Hetero)arylboronic Acids. Chem. Eur. J. 2014, 20, 6608–6612. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ji, C.-L.; Hong, X.; Szostak, M. Palladium-Catalyzed Decarbonylative Borylation of Carboxylic Acids: Tuning Reaction Selectivity by Computation. Angew. Chem. 2018, 57, 16721–16726. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Uetake, Y.; Niwa, T.; Hosoya, T. Rhodium-Catalyzed Decarbonylative Borylation of Aromatic Thioesters for Facile Diversification of Aromatic Carboxylic Acids. Angew. Chem. Int. Ed. 2017, 56, 2482–2486. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-M.; Shang, R.; Zhao, B.; Xing, W.-L.; Fu, Y. Isonicotinate Ester Catalyzed Decarboxylative Borylation of (Hetero)Aryl and Alkenyl Carboxylic Acids through N-Hydroxyphthalimide Esters. Org. Lett. 2017, 19, 4291–4294. [Google Scholar] [CrossRef] [PubMed]
- Candish, L.; Teders, M.; Glorius, F. Transition-Metal-Free, Visible-Light-Enabled Decarboxylative Borylation of Aryl N-Hydroxyphthalimide Esters. J. Am. Chem. Soc. 2017, 139, 7440–7443. [Google Scholar] [CrossRef]
- Iverson, C.N.; Smith, M.R. Stoichiometric and Catalytic B−C Bond Formation from Unactivated Hydrocarbons and Boranes. J. Am. Chem. Soc. 1999, 121, 7696–7697. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N.R.; Hartwig, J.F. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions, and isolation of a potential intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef]
- Ishiyama, T.; Nobuta, Y.; Hartwig, J.F.; Miyaura, N. Room temperature borylation of arenes and heteroarenes using stoichiometric amounts of pinacolborane catalyzed by iridium complexes in an inert solvent. Chem. Commun. 2003, 2924–2925. [Google Scholar] [CrossRef]
- Liskey, C.W.; Hartwig, J.F. Borylation of Arenes with Bis(hexylene glycolato)diboron. Synthesis 2013, 45, 1837–1842. [Google Scholar]
- Tobisu, M.; Igarashi, T.; Chatani, N. Iridium/N-heterocyclic carbene-catalyzed C–H borylation of arenes by diisopropylaminoborane. Beilstein J. Org. Chem. 2016, 12, 654–661. [Google Scholar] [CrossRef]
- Hoque, M.E.; Bisht, R.; Haldar, C.; Chattopadhyay, B. Noncovalent Interactions in Ir-Catalyzed C–H Activation: L-Shaped Ligand for Para-Selective Borylation of Aromatic Esters. J. Am. Chem. Soc. 2017, 139, 7745–7748. [Google Scholar] [CrossRef] [PubMed]
- Bisht, R.; Hoque, M.E.; Chattopadhyay, B. Amide Effects in C−H Activation: Noncovalent Interactions with L-Shaped Ligand for meta Borylation of Aromatic Amides. Angew. Chem. Int. Ed. 2018, 57, 15762–15766. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, I.; Ikeda, T.; Amou, T.; Taguchi, J.; Ito, H.; Ishiyama, T. Regioselective C–H Borylation of Heteroaromatic Aldimines with Iridium Complexes. Synlett 2016, 27, 1582–1586. [Google Scholar]
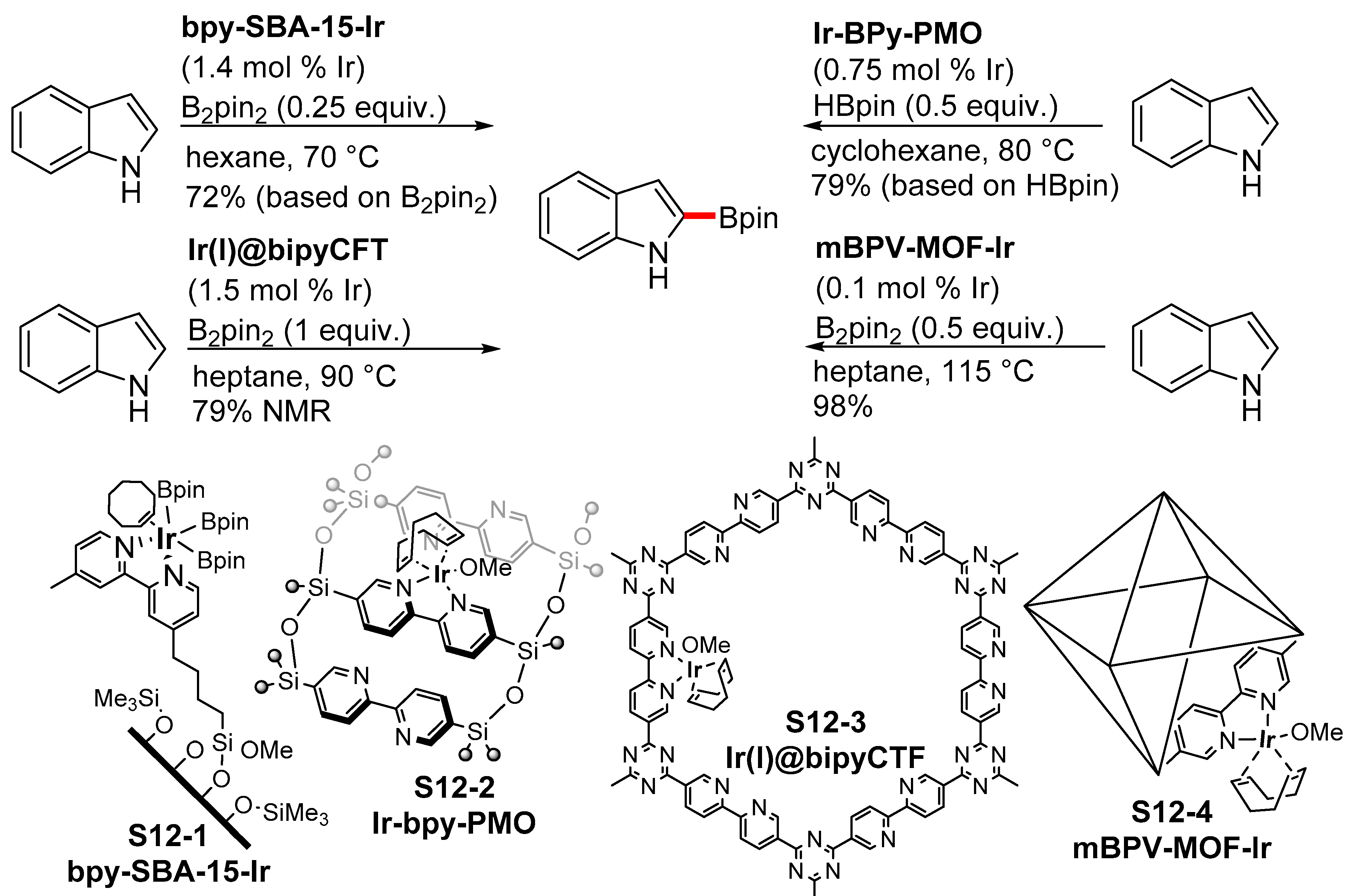
- Wu, F.; Feng, Y.; Jones, C.W. Recyclable Silica-Supported Iridium Bipyridine Catalyst for Aromatic C–H Borylation. ACS Catal. 2014, 4, 1365–1375. [Google Scholar] [CrossRef]
- Maegawa, Y.; Inagaki, S. Iridium–bipyridine periodic mesoporous organosilica catalyzed direct C–H borylation using a pinacolborane. Dalton Trans. 2015, 44, 13007–13016. [Google Scholar] [CrossRef] [PubMed]
- Tahir, N.; Muniz-Miranda, F.; Everaert, J.; Tack, P.; Heugebaert, T.; Leus, K.; Vincze, L.; Stevens, C.V.; Van Speybroeck, V.; Van Der Voort, P. Immobilization of Ir(I) complex on covalent triazine frameworks for CH borylation reactions: A combined experimental and computational study. J. Catal. 2019, 371, 135–143. [Google Scholar] [CrossRef]
- Manna, K.; Zhang, T.; Greene, F.X.; Lin, W. Bipyridine- and phenanthroline-based metal-organic frameworks for highly efficient and tandem catalytic organic transformations via directed C–H activation. J. Am. Chem. Soc. 2015, 137, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
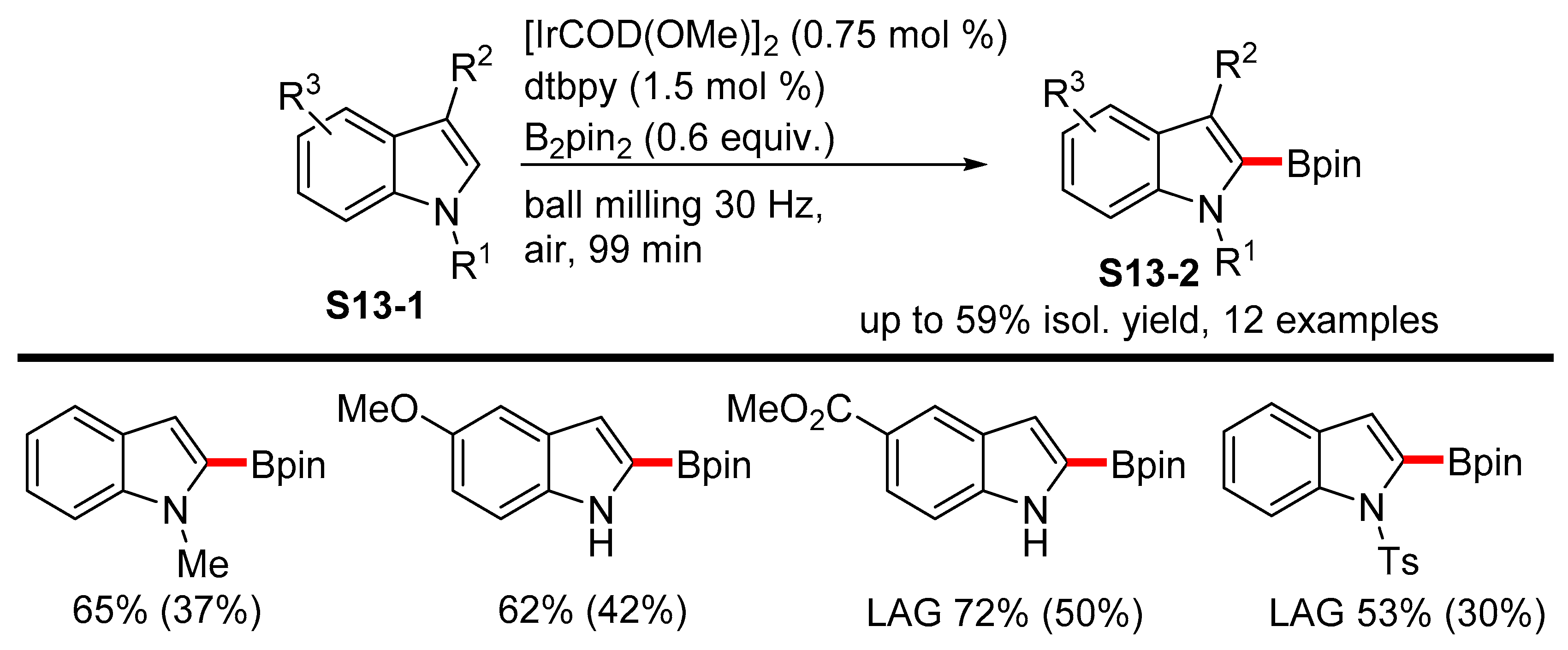
- Pang, Y.; Ishiyama, T.; Kubota, K.; Ito, H. Iridium(I)-Catalyzed C−H Borylation in Air by Using Mechanochemistry. Chem. Eur. J. 2019, 25, 4654–4659. [Google Scholar] [CrossRef]
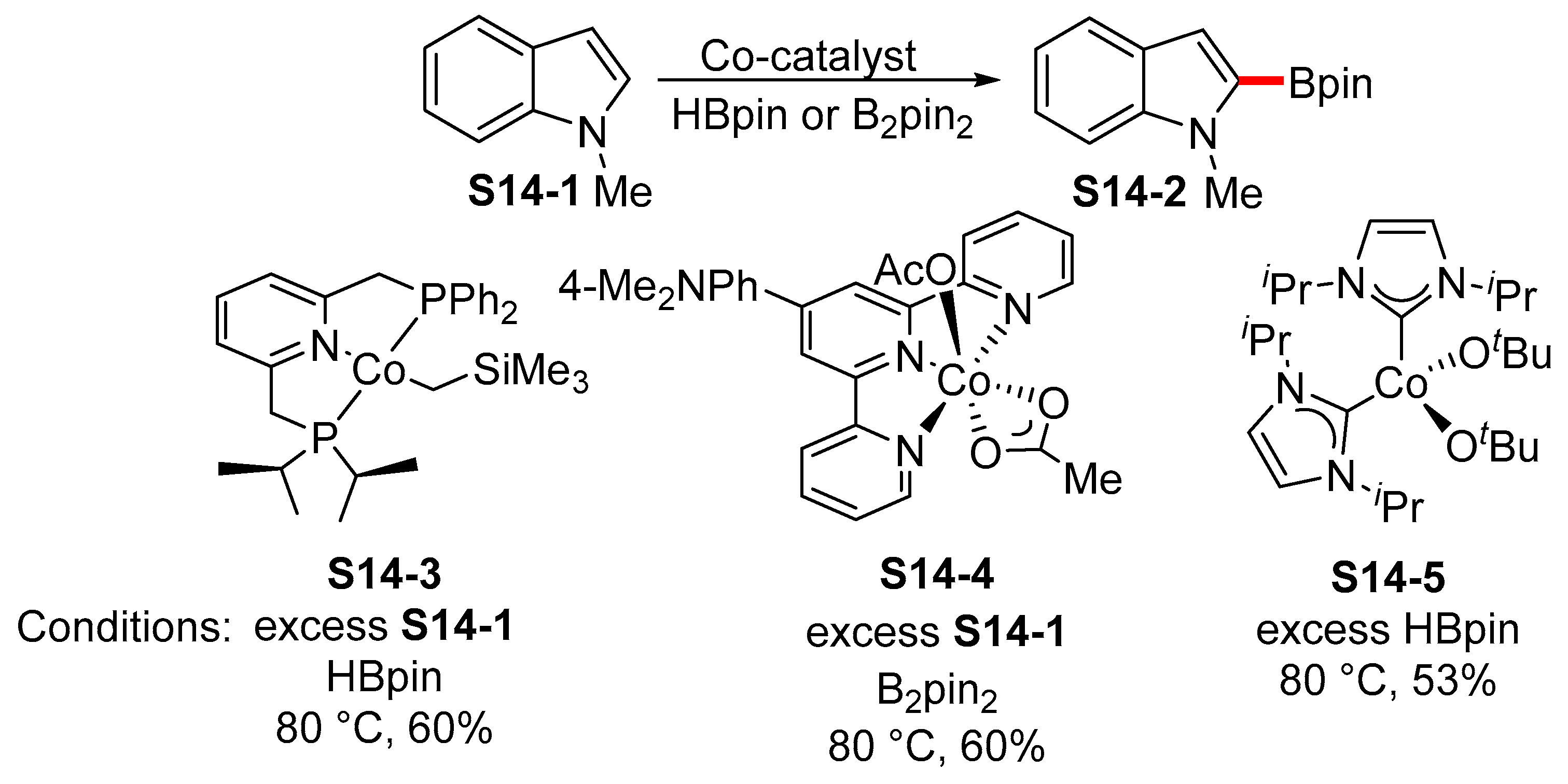
- Obligacion, J.V.; Semproni, S.P.; Chirik, P.J. Cobalt-catalyzed C-H borylation. J. Am. Chem. Soc. 2014, 136, 4133–4136. [Google Scholar] [CrossRef]
- Léonard, N.G.; Bezdek, M.J.; Chirik, P.J. Cobalt-Catalyzed C(sp2)–H Borylation with an Air-Stable, Readily Prepared Terpyridine Cobalt(II) Bis(acetate) Precatalyst. Organometallics 2017, 36, 142–150. [Google Scholar] [CrossRef]
- Jayasundara, C.R.K.; Sabasovs, D.; Staples, R.J.; Oppenheimer, J.; Smith, M.R.; Maleczka, R.E. Cobalt-Catalyzed C–H Borylation of Alkyl Arenes and Heteroarenes Including the First Selective Borylations of Secondary Benzylic C–H Bonds. Organometallics 2018, 37, 1567–1574. [Google Scholar] [CrossRef]
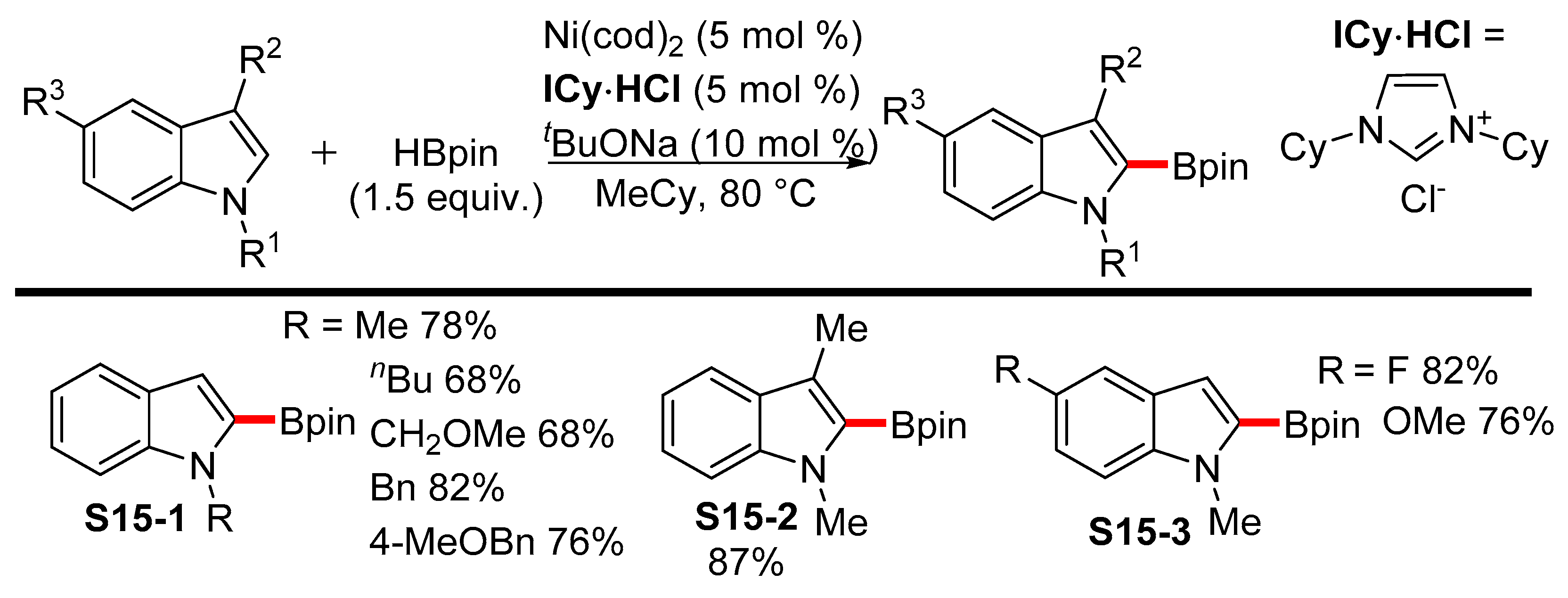
- Furukawa, T.; Tobisu, M.; Chatani, N. Nickel-catalyzed borylation of arenes and indoles via C–H bond cleavage. Chem. Commun. 2015, 51, 6508–6511. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Tobisu, M.; Chatani, N. C–H Functionalization at Sterically Congested Positions by the Platinum-Catalyzed Borylation of Arenes. J. Am. Chem. Soc. 2015, 137, 12211–12214. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Ito, S.; Nomoto, H.; Saito, N.; Kirai, N.; Iwasawa, N. Fluorine-controlled C–H borylation of arenes catalyzed by a PSiN-pincer platinum complex. Chem. Commun. 2015, 51, 17662–17665. [Google Scholar] [CrossRef] [PubMed]
- Stahl, T.; Müther, K.; Ohki, Y.; Tatsumi, K.; Oestreich, M. Catalytic Generation of Borenium Ions by Cooperative B–H Bond Activation: The Elusive Direct Electrophilic Borylation of Nitrogen Heterocycles with Pinacolborane. J. Am. Chem. Soc. 2013, 135, 10978–10981. [Google Scholar] [CrossRef] [PubMed]
- Kallepalli, V.A.; Shi, F.; Paul, S.; Onyeozili, E.N.; Maleczka, R.E., Jr.; Smith, M.R., III. Boc groups as protectors and directors for Ir-catalyzed C-H borylation of heterocycles. J. Org. Chem. 2009, 74, 9199–9201. [Google Scholar] [CrossRef] [PubMed]
- Preshlock, S.M.; Plattner, D.L.; Maligres, P.E.; Krska, S.W.; Maleczka, R.E., Jr.; Smith, M.R., III. A Traceless Directing Group for C−H Borylation. Angew. Chem. Int. Ed. 2013, 52, 12915–12919. [Google Scholar] [CrossRef] [PubMed]
- Seechurn, C.C.C.J.; Sivakumar, V.; Satoskar, D.; Colacot, T.J. Iridium-Catalyzed C–H Borylation of Heterocycles Using an Overlooked 1,10-Phenanthroline Ligand: Reinventing the Catalytic Activity by Understanding the Solvent-Assisted Neutral to Cationic Switch. Organometallics 2014, 33, 3514–3522. [Google Scholar] [CrossRef]
- Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, L.R.; Baran, P.S. Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C–H Borylation. J. Am. Chem. Soc. 2015, 137, 10160–10163. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.-M.; Liras, S.; Guzman-Perez, A.; Perreault, C.; Bian, J.; James, K. Functionalization of Aromatic Amino Acids via Direct C−H Activation: Generation of Versatile Building Blocks for Accessing Novel Peptide Space. Org. Lett. 2010, 12, 3870–3873. [Google Scholar] [CrossRef] [PubMed]
- Eastabrook, A.S.; Wang, C.; Davison, E.K.; Sperry, J. A Procedure for Transforming Indoles into Indolequinones. J. Org. Chem. 2015, 80, 1006–1017. [Google Scholar] [CrossRef]
- Batool, F.; Parveen, S.; Emwas, A.-H.; Sioud, S.; Gao, X.; Munawar, M.A.; Chotana, G.A. Synthesis of Fluoroalkoxy Substituted Arylboronic Esters by Iridium-Catalyzed Aromatic C–H Borylation. Org. Lett. 2015, 17, 4256–4259. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.W.; Boebel, T.A.; Hartwig, J.F. Iridium-Catalyzed, Silyl-Directed Borylation of Nitrogen-Containing Heterocycles. J. Am. Chem. Soc. 2010, 132, 4068–4069. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sperry, J. Iridium-Catalyzed C–H Borylation-Based Synthesis of Natural Indolequinones. J. Org. Chem. 2012, 77, 2584–2587. [Google Scholar] [CrossRef] [PubMed]
- Loach, R.P.; Fenton, O.S.; Amaike, K.; Siegel, D.S.; Ozkal, E.; Movassaghi, M. C7-Derivatization of C3-Alkylindoles Including Tryptophans and Tryptamines. J. Org. Chem. 2014, 79, 11254–11263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallepalli, V.A.; Gore, K.A.; Shi, F.; Sanchez, L.; Chotana, G.A.; Miller, S.L.; Maleczka, R.E.; Smith, M.R. Harnessing C–H Borylation/Deborylation for Selective Deuteration, Synthesis of Boronate Esters, and Late Stage Functionalization. J. Org. Chem. 2015, 80, 8341–8353. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Tyagarajan, S.; Perera, D.; Krska, S.W.; Maligres, P.E.; Smith, M.R.; Maleczka, R.E. Bismuth Acetate as a Catalyst for the Sequential Protodeboronation of di-and Triborylated Indoles. Org. Lett. 2016, 18, 1554–1557. [Google Scholar] [CrossRef]
- Smith, M.W.; Falk, I.D.; Ikemoto, H.; Burns, N.Z. A convenient C–H functionalization platform for pyrroloiminoquinone alkaloid synthesis. Tetrahedron 2019, 75, 3366–3370. [Google Scholar] [CrossRef]
- Wang, G.; Xu, L.; Li, P. Double N,B-Type Bidentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation. J. Am. Chem. Soc. 2015, 137, 8058–8061. [Google Scholar] [CrossRef]
- Kawamorita, S.; Ohmiya, H.; Sawamura, M. Ester-Directed Regioselective Borylation of Heteroarenes Catalyzed by a Silica-Supported Iridium Complex. J. Org. Chem. 2010, 75, 3855–3858. [Google Scholar] [CrossRef]
- Demory, E.; Devaraj, K.; Orthaber, A.; Gates, P.J.; Pilarski, L.T. Boryl (Hetero)aryne Precursors as Versatile Arylation Reagents: Synthesis through C−H Activation and Orthogonal Reactivity. Angew. Chem. Int. Ed. 2015, 54, 11765–11769. [Google Scholar] [CrossRef] [PubMed]
- Homer, J.A.; Sperry, J. A short synthesis of the endogenous plant metabolite 7-hydroxyoxindole-3-acetic acid (7-OH-OxIAA) using simultaneous C–H borylations. Tetrahedron Lett. 2014, 55, 5798–5800. [Google Scholar] [CrossRef]
- Eastabrook, A.S.; Sperry, J. Iridium-Catalyzed Triborylation of 3-Substituted Indoles. Australian J. Chem. 2015, 68, 1810–1814. [Google Scholar] [CrossRef]
- Eastabrook, A.S.; Sperry, J. Synthetic Access to 3,5,7-Trisubstituted Indoles Enabled by Iridium-Catalyzed C–H Borylation. Synthesis 2017, 49, 4731–4737. [Google Scholar]
- Lv, J.; Zhao, B.; Liu, L.; Han, Y.; Yuan, Y.; Shi, Z. Boron Trichloride-Mediated Synthesis of Indoles via the Aminoboration of Alkynes. Adv. Synth. Catal. 2018, 360, 4054–4059. [Google Scholar] [CrossRef]
- Yuan, K.; Wang, S. trans-Aminoboration across Internal Alkynes Catalyzed by B (C6F5)3 for the Synthesis of Borylated Indoles. Org. Lett. 2017, 19, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Macdonald, S.J.F.; Harrity, J.P.A. A borylative cyclisation towards indole boronic esters. Chem. Commun. 2010, 46, 8770–8772. [Google Scholar] [CrossRef]
- Chong, E.; Blum, S.A. Aminoboration: Addition of B–N σ Bonds across C–C π Bonds. J. Am. Chem. Soc. 2015, 137, 10144–10147. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, Z.; Fu, Y. Theoretical Investigation on the ClBcat-Promoted Synthesis of Heterocyclic Boronic Esters. Asian, J. Org. Chem. 2017, 6, 282–289. [Google Scholar] [CrossRef]
- Seath, C.P.; Wilson, K.L.; Campbell, A.; Mowat, J.M.; Watson, A.J.B. Synthesis of 2-BMIDA 6,5-bicyclic heterocycles by Cu(i)/Pd(0)/Cu(ii) cascade catalysis of 2-iodoaniline/phenols. Chem. Commun. 2016, 52, 8703–8706. [Google Scholar] [CrossRef] [Green Version]
- Tobisu, M.; Fujihara, H.; Koh, K.; Chatani, N. Synthesis of 2-Boryl- and Silylindoles by Copper-Catalyzed Borylative and Silylative Cyclization of 2-Alkenylaryl Isocyanides. J. Org. Chem. 2010, 75, 4841–4847. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Jin, L.; Zheng, Z.; Meng, H.; Mo, F.; Wang, X.; Zhang, Y.; Wang, J. Synthesis of Pinacol Arylboronates from Aromatic Amines: A Metal-Free Transformation. J. Org. Chem. 2013, 78, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Zhang, Y.; Wang, J. Direct synthesis of arylboronic pinacol esters from arylamines. Org. Chem. Front. 2014, 1, 422–425. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Qin, S.; Yin, Y.; Hu, J.; Zhang, H. Boron(III)-Catalyzed C2-Selective C−H Borylation of Heteroarenes. Angew. Chem. Int. Ed. 2018, 57, 14891–14895. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Han, Y.; He, J.; Zhang, Y. B (C6F5)3-Catalyzed C3-Selective C–H Borylation of Indoles: Synthesis, Intermediates, and Reaction Mechanism. J. Org. Chem. 2018, 83, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Klare, H.F.T.; Oestreich, M. Catalytic Friedel–Crafts C−H Borylation of Electron-Rich Arenes: Dramatic Rate Acceleration by Added Alkenes. Angew. Chem. Int. Ed. 2017, 56, 3712–3717. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Kehr, G.; Daniliuc, C.G.; Erker, G. Metal-Free Arene and Heteroarene Borylation Catalyzed by Strongly Electrophilic Bis-boranes. Chem. Eur. J. 2017, 23, 12141–12144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Légaré Lavergne, J.; Jayaraman, A.; Misal Castro, L.C.; Rochette, É.; Fontaine, F.-G. Metal-Free Borylation of Heteroarenes Using Ambiphilic Aminoboranes: On the Importance of Sterics in Frustrated Lewis Pair C–H Bond Activation. J. Am. Chem. Soc. 2017, 139, 14714–14723. [Google Scholar] [CrossRef]
- Légaré, M.-A.; Courtemanche, M.-A.; Rochette, É.; Fontaine, F.-G. Metal-free catalytic C-H bond activation and borylation of heteroarenes. Science 2015, 349, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Légaré, M.-A.; Rochette, É.; Lavergne, J.L.; Bouchard, N.; Fontaine, F.-G. Bench-stable frustrated Lewis pair chemistry: Fluoroborate salts as precatalysts for the C–H borylation of heteroarenes. Chem. Commun. 2016, 52, 5387–5390. [Google Scholar] [CrossRef]
- Jayaraman, A.; Misal Castro, L.C.; Fontaine, F.-G. Practical and Scalable Synthesis of Borylated Heterocycles Using Bench-Stable Precursors of Metal-Free Lewis Pair Catalysts. Org. Process. Res. Dev. 2018, 22, 1489–1499. [Google Scholar] [CrossRef]
- Bagutski, V.; Del Grosso, A.; Carrillo, J.A.; Cade, I.A.; Helm, M.D.; Lawson, J.R.; Singleton, P.J.; Solomon, S.A.; Marcelli, T.; Ingleson, M.J. Mechanistic Studies into Amine-Mediated Electrophilic Arene Borylation and Its Application in MIDA Boronate Synthesis. J. Am. Chem. Soc. 2013, 135, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, A.; Singleton, P.J.; Muryn, C.A.; Ingleson, M.J. Pinacol Boronates by Direct Arene Borylation with Borenium Cations. Angew. Chem. Int. Ed. 2011, 50, 2102–2106. [Google Scholar] [CrossRef]
- Grosso, A.D.; Helm, M.D.; Solomon, S.A.; Caras-Quintero, D.; Ingleson, M.J. Simple inexpensive boron electrophiles for direct arene borylation. Chem. Commun. 2011, 47, 12459–12461. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.A.; Del Grosso, A.; Clark, E.R.; Bagutski, V.; McDouall, J.J.W.; Ingleson, M.J. Reactivity of Lewis Acid Activated Diaza- and Dithiaboroles in Electrophilic Arene Borylation. Organometallics 2012, 31, 1908–1916. [Google Scholar] [CrossRef]
- Tanaka, S.; Saito, Y.; Yamamoto, T.; Hattori, T. Electrophilic Borylation of Terminal Alkenes with BBr3/2,6-Disubstituted Pyridines. Org. Lett. 2018, 20, 1828–1831. [Google Scholar] [CrossRef]
- Bartolucci, S.; Bartoccini, F.; Righi, M.; Piersanti, G. Direct, Regioselective, and Chemoselective Preparation of Novel Boronated Tryptophans by Friedel–Crafts Alkylation. Org. Lett. 2012, 14, 600–603. [Google Scholar] [CrossRef]
- Churches, Q.I.; Hooper, J.F.; Hutton, C.A. A General Method for Interconversion of Boronic Acid Protecting Groups: Trifluoroborates as Common Intermediates. J. Org. Chem. 2015, 80, 5428–5435. [Google Scholar] [CrossRef] [Green Version]
- Borah, A.J.; Shi, Z. Palladium-catalyzed regioselective C–H fluoroalkylation of indoles at the C4-position. Chem. Commun. 2017, 53, 3945–3948. [Google Scholar] [CrossRef]
- Fei, X.; Li, C.; Yu, X.; Liu, H. Rh(III)-Catalyzed Hydroarylation of Alkyne MIDA Boronates via C–H Activation of Indole Derivatives. J. Org. Chem. 2019, 84, 6840–6850. [Google Scholar] [CrossRef]
- Caramenti, P.; Nandi, R.K.; Waser, J. Metal-Free Oxidative Cross Coupling of Indoles with Electron-Rich (Hetero) arenes. Chem. Eur. J. 2018, 24, 10049–10053. [Google Scholar] [CrossRef] [PubMed]
- Caramenti, P.; Nicolai, S.; Waser, J. Indole- and Pyrrole-BX: Bench-Stable Hypervalent Iodine Reagents for Heterocycle Umpolung. Chem. Eur. J. 2017, 23, 14702–14706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wu, J. Catalytic vinylogous cross-coupling reactions of rhenium vinylcarbenoids. Chem. Sci. 2018, 9, 2489–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, G.; Webster, S.; Johnson, D.G.; Curley, R.; Andrews, M.; Young, P.C.; Macgregor, S.A.; Lee, A.-L. Gold-Catalyzed Proto- and Deuterodeboronation. J. Org. Chem. 2015, 80, 9807–9816. [Google Scholar] [CrossRef] [PubMed]
- Grimes, K.D.; Gupte, A.; Aldrich, C.C. Copper(II)-Catalyzed Conversion of Aryl/Heteroaryl Boronic Acids, Boronates, and Trifluoroborates into the Corresponding Azides: Substrate Scope and Limitations. Synthesis 2010, 1441–1448. [Google Scholar] [CrossRef]
- Zhang, Z.; Niwa, T.; Watanabe, Y.; Hosoya, T. Palladium(ii)-mediated rapid 11C-cyanation of (hetero)arylborons. Org. Biomol. Chem. 2018, 16, 7711–7716. [Google Scholar] [CrossRef]
- Tramutola, F.; Chiummiento, L.; Funicello, M.; Lupattelli, P. Practical and efficient ipso-iodination of arylboronic acids via KF/I2 system. Tetrahedron Lett. 2015, 56, 1122–1123. [Google Scholar] [CrossRef]
- Matsuzono, M.; Fukuda, T.; Iwao, M. Direct C-3 lithiation of 1-(triisopropylsilyl) indole. Tetrahedron Lett. 2001, 42, 7621–7623. [Google Scholar] [CrossRef]
- Fier, P.S.; Luo, J.; Hartwig, J.F. Copper-Mediated Fluorination of Arylboronate Esters. Identification of a Copper(III) Fluoride Complex. J. Am. Chem. Soc. 2013, 135, 2552–2559. [Google Scholar] [CrossRef] [Green Version]
- Antuganov, D.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Fedorova, O.; Krasikova, R. Copper-Mediated Radiofluorination of Aryl Pinacolboronate Esters: A Straightforward Protocol by Using Pyridinium Sulfonates. Eur. J. Org. Chem. 2019, 918–922. [Google Scholar] [CrossRef]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, D.; Weiß, P.; Ermert, J.; Castillo Meleán, J.; Zarrad, F.; Neumaier, B. Preparation of No-Carrier-Added 6-[18F]Fluoro-l-tryptophan via Cu-Mediated Radiofluorination. Eur. J. Org. Chem. 2016, 4621–4628. [Google Scholar]
- Konas, D.W.; Seci, D.; Tamimi, S. Synthesis of (l)-4-Fluorotryptophan. Synthetic Commun. 2012, 42, 144–152. [Google Scholar] [CrossRef]
- Senecal, T.D.; Parsons, A.T.; Buchwald, S.L. Room temperature aryl trifluoromethylation via copper-mediated oxidative cross-coupling. J. Org. Chem. 2011, 76, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.A.; Buba, A.E.; Gooßen, L.J. Oxidative trifluoromethylation of arylboronates with shelf-stable potassium (trifluoromethyl) trimethoxyborate. Chem. Eur. J. 2012, 18, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Luo, D.F.; Xiao, B.; Liu, Z.J.; Gong, T.J.; Fu, Y.; Liu, L. Copper-catalyzed trifluoromethylation of aryl boronic acids using a CF3+ reagent. Chem. Commun. 2011, 47, 4300–4302. [Google Scholar] [CrossRef]
- Liu, T.; Shen, Q. Copper-catalyzed trifluoromethylation of aryl and vinyl boronic acids with an electrophilic trifluoromethylating reagent. Org. Lett. 2011, 13, 2342–2345. [Google Scholar] [CrossRef]
- Eisenberger, P.; Gischig, S.; Togni, A. Novel 10-I-3 Hypervalent Iodine-Based Compounds for Electrophilic Trifluoromethylation. Chem. Eur. J. 2006, 12, 2579–2586. [Google Scholar] [CrossRef]
- Presset, M.; Oehlrich, D.; Rombouts, F.; Molander, G.A. Copper-mediated radical trifluoromethylation of unsaturated potassium organotrifluoroborates. J. Org. Chem. 2013, 78, 12837–12843. [Google Scholar] [CrossRef]
- Huang, C.; Liang, T.; Harada, S.; Lee, E.; Ritter, T. Silver-mediated trifluoromethoxylation of aryl stannanes and arylboronic acids. J. Am. Chem. Soc. 2011, 133, 13308–13310. [Google Scholar] [CrossRef]
- Nakajima, S.; Takaya, H.; Nakamura, M. Iron-catalyzed methylation of arylboron compounds with iodomethane. Chem. Lett. 2017, 46, 711–714. [Google Scholar] [CrossRef]
- Haydl, A.M.; Hartwig, J.F. Palladium-Catalyzed Methylation of Aryl, Heteroaryl, and Vinyl Boronate Esters. Org. Lett. 2019, 21, 1337–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.T.; Li, H.; Haydl, A.M.; Whiteker, G.T.; Hartwig, J.F. Trimethylphosphate as a Methylating Agent for Cross Coupling: A Slow-Release Mechanism for the Methylation of Arylboronic Esters. J. Am. Chem. Soc. 2018, 140, 17197–17202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, A.M.; Landry-Bayle, A.; Gomez, L. Palladium-catalyzed benzylation of N-Boc indole boronic acids. Tetrahedron Lett. 2010, 51, 2281–2283. [Google Scholar] [CrossRef]
- Liao, J.; Guan, W.; Boscoe, B.P.; Tucker, J.W.; Tomlin, J.W.; Garnsey, M.R.; Watson, M.P. Transforming Benzylic Amines into Diarylmethanes: Cross-Couplings of Benzylic Pyridinium Salts via C–N Bond Activation. Org. Lett. 2018, 20, 3030–3033. [Google Scholar] [CrossRef] [PubMed]
- Tobisu, M.; Yasutome, A.; Kinuta, H.; Nakamura, K.; Chatani, N. 1,3-Dicyclohexylimidazol-2-ylidene as a Superior Ligand for the Nickel-Catalyzed Cross-Couplings of Aryl and Benzyl Methyl Ethers with Organoboron Reagents. Org. Lett. 2014, 16, 5572–5575. [Google Scholar] [CrossRef]
- Takeda, M.; Takatsu, K.; Shintani, R.; Hayashi, T. Synthesis of quaternary carbon stereocenters by copper-catalyzed asymmetric allylic substitution of allyl phosphates with arylboronates. J. Org. Chem. 2014, 79, 2354–2367. [Google Scholar] [CrossRef] [PubMed]
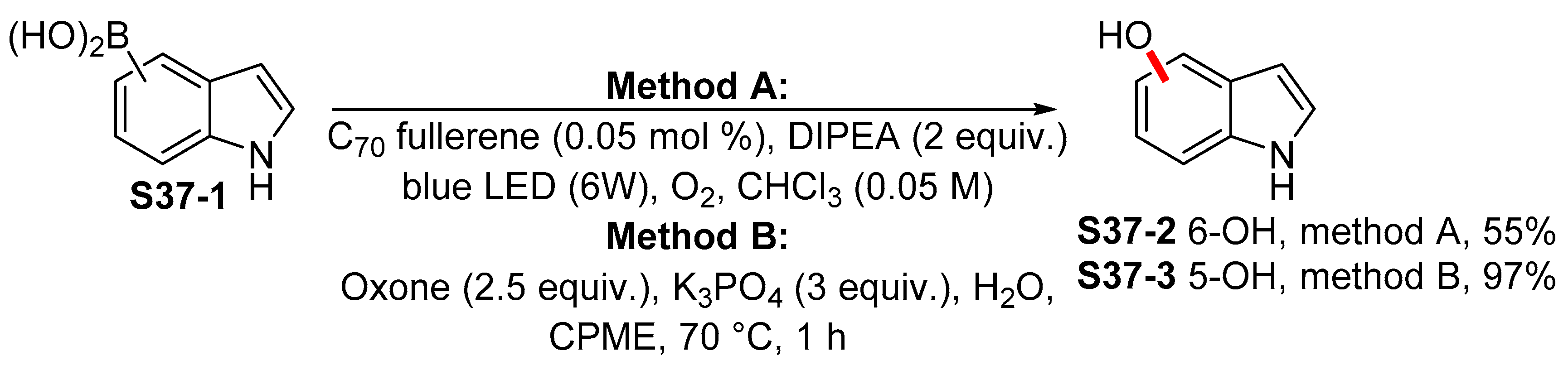
- Kumar, I.; Sharma, R.; Kumar, R.; Kumar, R.; Sharma, U. C70 Fullerene-Catalyzed Metal-Free Photocatalytic ipso-Hydroxylation of Aryl Boronic Acids: Synthesis of Phenols. Adv. Synth. Catal. 2018, 360, 2013–2019. [Google Scholar] [CrossRef]
- Molloy, J.J.; Clohessy, T.A.; Irving, C.; Anderson, N.A.; Lloyd-Jones, G.C.; Watson, A.J.B. Chemoselective oxidation of aryl organoboron systems enabled by boronic acid-selective phase transfer. Chem. Sci. 2017, 8, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.L.; Frederiksen, M.W.; Domino, K.; Skrydstrup, T. Direct Access to α,α-Difluoroacylated Arenes by Palladium-Catalyzed Carbonylation of (Hetero) Aryl Boronic Acid Derivatives. Angew. Chem. Int. Ed. 2016, 55, 10396–10400. [Google Scholar] [CrossRef]
- Meng, G.; Szostak, M. Palladium-catalyzed Suzuki–Miyaura coupling of amides by carbon–nitrogen cleavage: General strategy for amide N–C bond activation. Org. Biomol. Chem. 2016, 14, 5690–5707. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y. The First General and Selective Palladium(II)-Catalyzed Alkoxycarbonylation of Arylboronates: Interplay among Benzoquinone-Ligated Palladium(0) Complex, Organoboron, and Alcohol Solvent. Adv. Synth. Catal. 2010, 352, 478–492. [Google Scholar] [CrossRef]
- Nahm, S.; Weinreb, S.M. N-methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Krishnamoorthy, R.; Lam, S.Q.; Manley, C.M.; Herr, R.J. Palladium-Catalyzed Preparation of Weinreb Amides from Boronic Acids and N-Methyl-N-methoxycarbamoyl Chloride. J. Org. Chem. 2010, 75, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yasui, K.; Tobisu, M.; Chatani, N. Rhodium-catalyzed cross-coupling of aryl carbamates with arylboron reagents. Tetrahedron 2015, 71, 4484–4489. [Google Scholar] [CrossRef] [Green Version]
- Betancourt-Mendiola, L.; Valois-Escamilla, I.; Arbeloa, T.; Bañuelos, J.; López Arbeloa, I.; Flores-Rizo, J.O.; Hu, R.; Lager, E.; Gómez-Durán, C.F.A.; Belmonte-Vázquez, J.L.; et al. Scope and Limitations of the Liebeskind–Srogl Cross-Coupling Reactions Involving the Biellmann BODIPY. J. Org. Chem. 2015, 80, 5771–5782. [Google Scholar] [CrossRef] [PubMed]
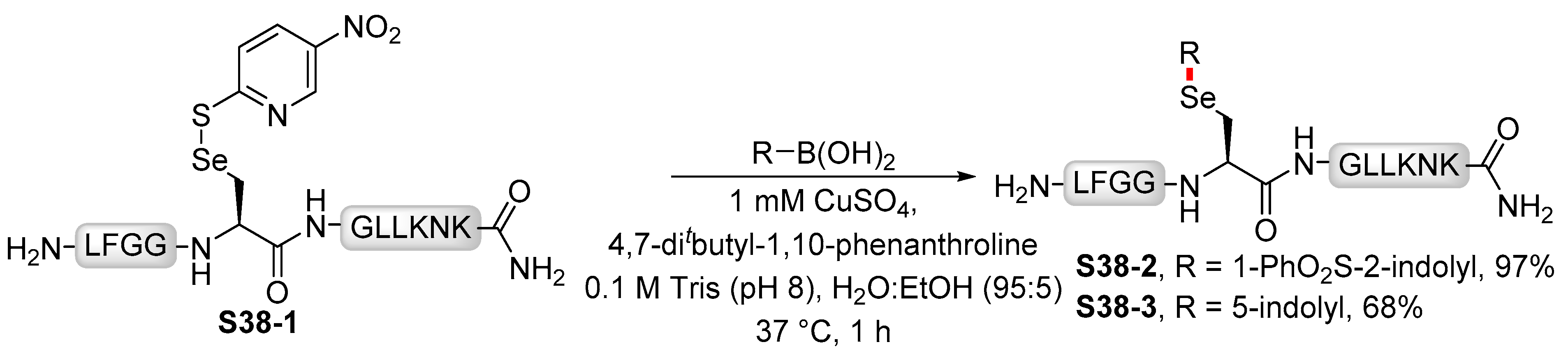
- Cohen, D.T.; Zhang, C.; Pentelute, B.L.; Buchwald, S.L. An Umpolung Approach for the Chemoselective Arylation of Selenocysteine in Unprotected Peptides. J. Am. Chem. Soc. 2015, 137, 9784–9787. [Google Scholar] [CrossRef] [Green Version]
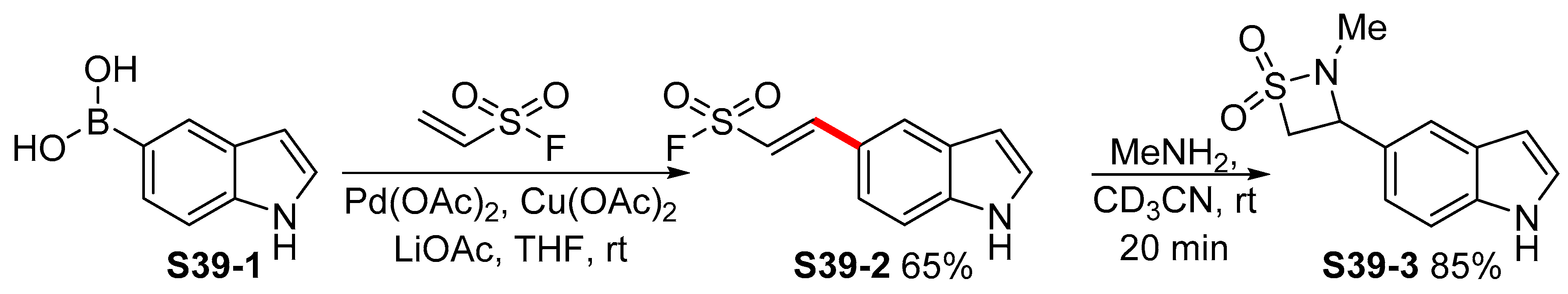
- Chinthakindi, P.K.; Govender, K.B.; Kumar, A.S.; Kruger, H.G.; Govender, T.; Naicker, T.; Arvidsson, P.I. A Synthesis of “Dual Warhead” β-Aryl Ethenesulfonyl Fluorides and One-Pot Reaction to β-Sultams. Org. Lett. 2017, 19, 480–483. [Google Scholar] [CrossRef]
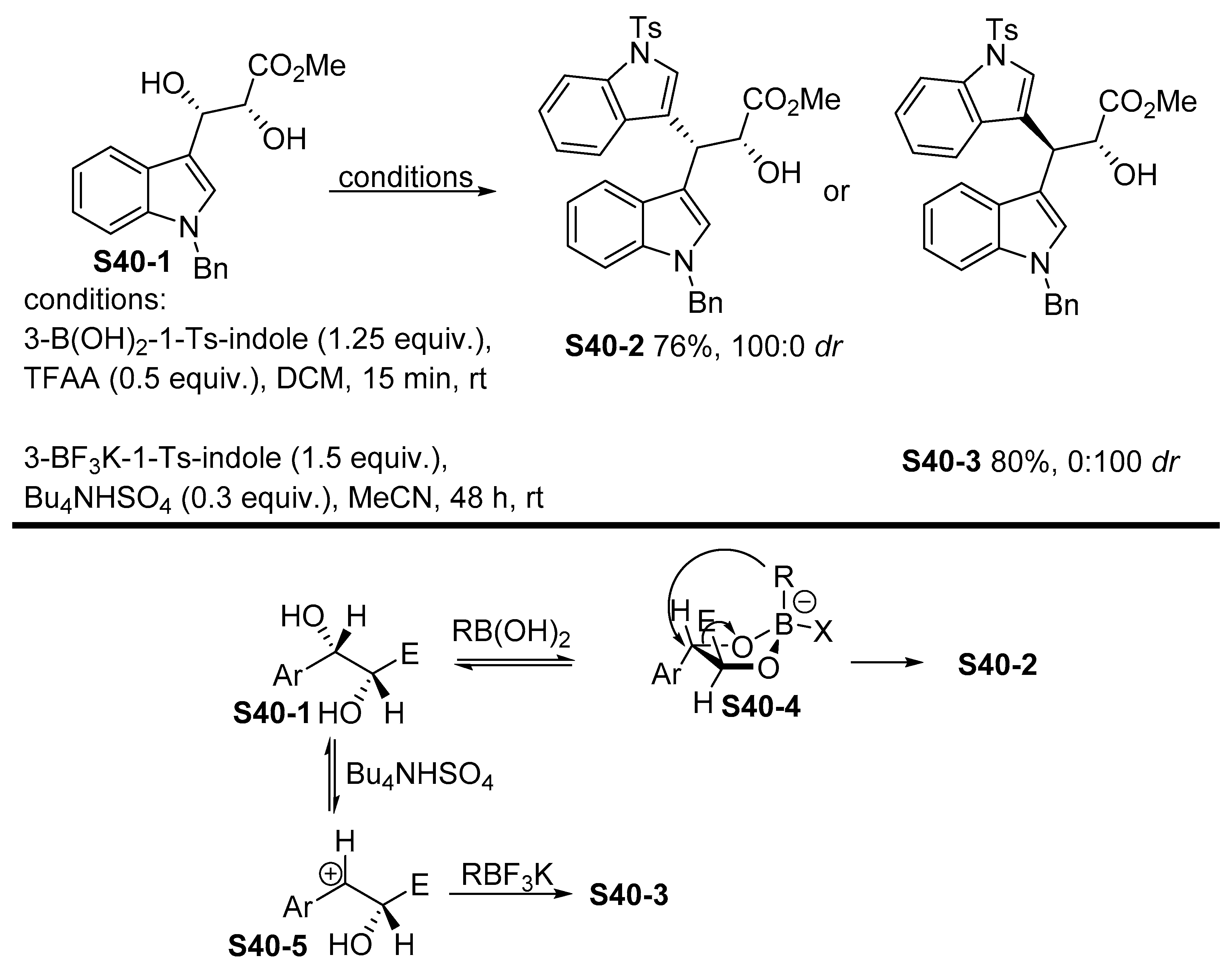
- Ortega, V.; del Castillo, E.; Csákÿ, A.G. Transition-Metal-Free Stereocomplementary Cross-Coupling of Diols with Boronic Acids as Nucleophiles. Org. Lett. 2017, 19, 6236–6239. [Google Scholar] [CrossRef]
- Armstrong, R.J.; Niwetmarin, W.; Aggarwal, V.K. Synthesis of Functionalized Alkenes by a Transition-Metal-Free Zweifel Coupling. Org. Lett. 2017, 19, 2762–2765. [Google Scholar] [CrossRef]
- Sun, H.-B.; Gong, L.; Tian, Y.-B.; Wu, J.-G.; Zhang, X.; Liu, J.; Fu, Z.; Niu, D. Metal- and Base-Free Room-Temperature Amination of Organoboronic Acids with N-Alkyl Hydroxylamines. Angew. Chem. Int. Ed. 2018, 57, 9456–9460. [Google Scholar] [CrossRef] [PubMed]
- Nageswar Rao, D.; Rasheed, S.; Vishwakarma, R.A.; Das, P. Copper-catalyzed sequential N-arylation of C-amino-NH-azoles. Chem. Commun. 2014, 50, 12911–12914. [Google Scholar] [CrossRef] [PubMed]
- Selmani, A.; Serpier, F.; Darses, S. From Tetrahydrofurans to Tetrahydrobenzo[d]oxepines via a Regioselective Control of Alkyne Insertion in Rhodium-Catalyzed Arylative Cyclization. J. Org. Chem. 2019, 84, 4566–4574. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, A.; Scott, J.S.; Bower, J.F. An Umpolung Approach to Alkene Carboamination: Palladium Catalyzed 1,2-Amino-Acylation, -Carboxylation, -Arylation, -Vinylation, and -Alkynylation. J. Am. Chem. Soc. 2015, 137, 7224–7230. [Google Scholar] [CrossRef] [PubMed]
- Hazelden, I.R.; Carmona, R.C.; Langer, T.; Pringle, P.G.; Bower, J.F. Pyrrolidines and Piperidines by Ligand-Enabled Aza-Heck Cyclizations and Cascades of N-(Pentafluorobenzoyloxy) carbamates. Angew. Chem. Int. Ed. 2018, 57, 5124–5128. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-B.; Pathipati, S.R.; Selander, N. Nickel-Catalyzed 1,2-Aminoarylation of Oxime Ester-Tethered Alkenes with Boronic Acids. ACS Catal. 2017, 7, 8441–8445. [Google Scholar] [CrossRef]
- Zhou, Y.; Rao, C.; Song, Q. Z-Selective Synthesis of γ,δ-Unsaturated Ketones via Pd-Catalyzed Ring Opening of 2-Alkylenecyclobutanones with Arylboronic Acids. Org. Lett. 2016, 18, 4000–4003. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Ouadoudi, O.; Andreadou, E.; Manolikakes, G. Sulfonamides as Amine Component in the Petasis-Borono Mannich Reaction: A Concise Synthesis of α-Aryl-and α-Alkenylglycine Derivatives. Synthesis 2018, 50, 3936–3946. [Google Scholar]
- Deeming, A.S.; Russell, C.J.; Willis, M.C. Palladium (II)-Catalyzed Synthesis of Sulfinates from Boronic Acids and DABSO: A Redox-Neutral, Phosphine-Free Transformation. Angew. Chem. Int. Ed. 2016, 55, 747–750. [Google Scholar] [CrossRef]
- Vedovato, V.; Talbot, E.P.A.; Willis, M.C. Copper-Catalyzed Synthesis of Activated Sulfonate Esters from Boronic Acids, DABSO, and Pentafluorophenol. Org. Lett. 2018, 20, 5493–5496. [Google Scholar] [CrossRef]
- Chen, Y.; Murray, P.R.D.; Davies, A.T.; Willis, M.C. Direct Copper-Catalyzed Three-Component Synthesis of Sulfonamides. J. Am. Chem. Soc. 2018, 140, 8781–8787. [Google Scholar] [CrossRef] [PubMed]
- Le Quement, S.T.; Flagstad, T.; Mikkelsen, R.J.T.; Hansen, M.R.; Givskov, M.C.; Nielsen, T.E. Petasis three-component coupling reactions of hydrazides for the synthesis of oxadiazolones and oxazolidinones. Org. Lett. 2012, 14, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Flagstad, T.; Petersen, M.T.; Nielsen, T.E. A Four-Component Reaction for the Synthesis of Dioxadiazaborocines. Angew. Chem. Int. Ed. 2015, 54, 8395–8397. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, S.; Onomura, O. Diastereoselective addition to N-acyliminium ions with aryl-and alkenyl boronic acids via a Petasis-type reaction. RSC Adv. 2012, 2, 2266–2269. [Google Scholar] [CrossRef]
- Panda, S.; Ready, J.M. Palladium Catalyzed Asymmetric Three-Component Coupling of Boronic Esters, Indoles, and Allylic Acetates. J. Am. Chem. Soc. 2017, 139, 6038–6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, E.; Hilton, M.J.; Orlandi, M.; Saini, V.; Toste, F.D.; Sigman, M.S. Development and Analysis of a Pd (0)-Catalyzed Enantioselective 1,1-Diarylation of Acrylates Enabled by Chiral Anion Phase Transfer. J. Am. Chem. Soc. 2016, 138, 15877–15880. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Zhao, Q.Q.; Chen, J.; Chen, J.R.; Xiao, W.J. Copper-Catalyzed Radical Cross-Coupling of Redox-Active Oxime Esters, Styrenes, and Boronic Acids. Angew. Chem. Int. Ed. 2018, 57, 15505–15509. [Google Scholar] [CrossRef]
- Lovinger, G.J.; Aparece, M.D.; Morken, J.P. Pd-Catalyzed Conjunctive Cross-Coupling between Grignard-Derived Boron “Ate” Complexes and C(sp2) Halides or Triflates: NaOTf as a Grignard Activator and Halide Scavenger. J. Am. Chem. Soc. 2017, 139, 3153–3160. [Google Scholar] [CrossRef]
- Domański, S.; Staszewska-Krajewska, O.; Chaładaj, W. Pd-Catalyzed Carbonylative Carboperfluoroalkylation of Alkynes. Through-Space 13C–19F Coupling as a Probe for Configuration Assignment of Fluoroalkyl-Substituted Olefins. J. Org. Chem. 2017, 82, 7998–8007. [Google Scholar] [CrossRef]
- Croix, C.; Prié, G.; Chaulet, C.; Viaud-Massuard, M.-C. Rhodium-Catalyzed 1,4-Addition of Arylboronic Acids to 3-Benzylidene-1H-pyrrolo[2,3-b]pyridin-2(3H)-one Derivatives. J. Org. Chem. 2015, 80, 3264–3269. [Google Scholar] [CrossRef]
- Malapit, C.A.; Luvaga, I.K.; Caldwell, D.R.; Schipper, N.K.; Howell, A.R. Rh-Catalyzed Conjugate Addition of Aryl and Alkenyl Boronic Acids to α-Methylene-β-lactones: Stereoselective Synthesis of trans-3,4-Disubstituted β-Lactones. Org. Lett. 2017, 19, 4460–4463. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, R.; Jankins, T.C.; Hill, D.E.; Yang, K.S.; Gallego, G.M.; Yang, S.; He, M.; Wang, F.; Marsters, R.P.; McAlpine, I.; et al. Palladium (ii)-catalyzed γ-selective hydroarylation of alkenyl carbonyl compounds with arylboronic acids. Chem. Sci. 2018, 9, 8363–8368. [Google Scholar] [CrossRef] [PubMed]
- Chiminazzo, A.; Sperni, L.; Damuzzo, M.; Strukul, G.; Scarso, A. Copper-mediated 1,4-Conjugate Addition of Boronic Acids and Indoles to Vinylidenebisphosphonate leading to gem-Bisphosphonates as Potential Antiresorption Bone Drugs. ChemCatChem 2014, 6, 2712–2718. [Google Scholar] [CrossRef]
- Hanna, L.E.; Konev, M.O.; Jarvo, E.R. Nickel-Catalyzed Directed Hydroarylation of Alkynes with Boronic Acids. Eur. J. Org. Chem. 2019, 184–187. [Google Scholar] [CrossRef]
- Yin, J.; Mekelburg, T.; Hyland, C. Unusual (Z)-selective palladium (ii)-catalysed addition of aryl boronic acids to vinylaziridines. Org. Biom. Chem. 2014, 12, 9113–9115. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Schaus, S.E. Enantioselective Addition of Boronates to o-Quinone Methides Catalyzed by Chiral Biphenols. J. Am. Chem. Soc. 2012, 134, 19965–19968. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.; Buttard, F.; Vallée, A.; Riguet, E. Organocatalytic Gram-Scale Synthesis and Alkylation of Heteroaryl and Electron-Rich Aryl α-Substituted γ-Lactones. Synthesis 2019, 51, 3151–3159. [Google Scholar] [CrossRef]
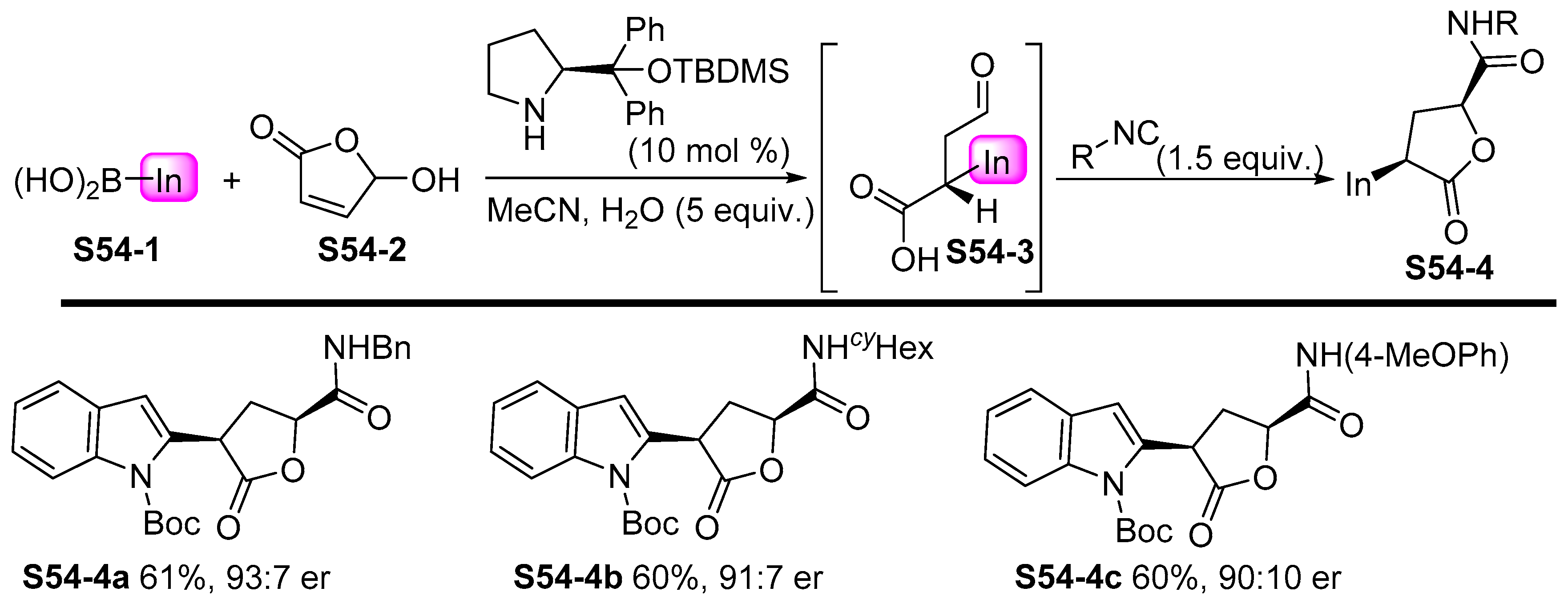
- Bos, M.; Riguet, E. Synthesis of Chiral γ-Lactones by One-Pot Sequential Enantioselective Organocatalytic Michael Addition of Boronic Acids and Diastereoselective Intramolecular Passerini Reaction. J. Org. Chem. 2014, 79, 10881–10889. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yu, C.; Li, X.; Zhang, Y.; Zhang, Y.; Chen, X.; Mariano, P.S.; Xie, H.; Wang, W. Synthesis of Aldehydes by Organocatalytic Formylation Reactions of Boronic Acids with Glyoxylic Acid. Angew. Chem. Int. Ed. 2017, 56, 8201–8205. [Google Scholar] [CrossRef]
- Kuriyama, M.; Ishiyama, N.; Shimazawa, R.; Onomura, O. Palladium-imidazolinium carbene-catalyzed arylation of aldehydes with arylboronic acids in water. Tetrahedron 2010, 66, 6814–6819. [Google Scholar] [CrossRef] [Green Version]
- Malapit, C.A.; Caldwell, D.R.; Luvaga, I.K.; Reeves, J.T.; Volchkov, I.; Gonnella, N.C.; Han, Z.S.; Busacca, C.A.; Howell, A.R.; Senanayake, C.H. Rhodium-Catalyzed Addition of Aryl Boronic Acids to 2,2-Disubstituted Malononitriles. Angew. Chem. Int. Ed. 2017, 56, 6999–7002. [Google Scholar] [CrossRef] [PubMed]
- Malapit, C.A.; Reeves, J.T.; Busacca, C.A.; Howell, A.R.; Senanayake, C.H. Rhodium-Catalyzed Transnitrilation of Aryl Boronic Acids with Dimethylmalononitrile. Angew. Chem. Int. Ed. 2016, 55, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, T.-S.; Xu, M.-H. Rhodium-catalyzed enantioselective 1,2-addition of arylboronic acids to heteroaryl α-ketoesters for synthesis of heteroaromatic α-hydroxy esters. Org. Biomol. Chem. 2012, 10, 9158–9164. [Google Scholar] [CrossRef] [PubMed]
- Roscales, S.; Csákÿ, A.G. Synthesis of Di (hetero)arylamines from Nitrosoarenes and Boronic Acids: A General, Mild, and Transition-Metal-Free Coupling. Org. Lett. 2018, 20, 1667–1671. [Google Scholar] [CrossRef] [PubMed]






















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
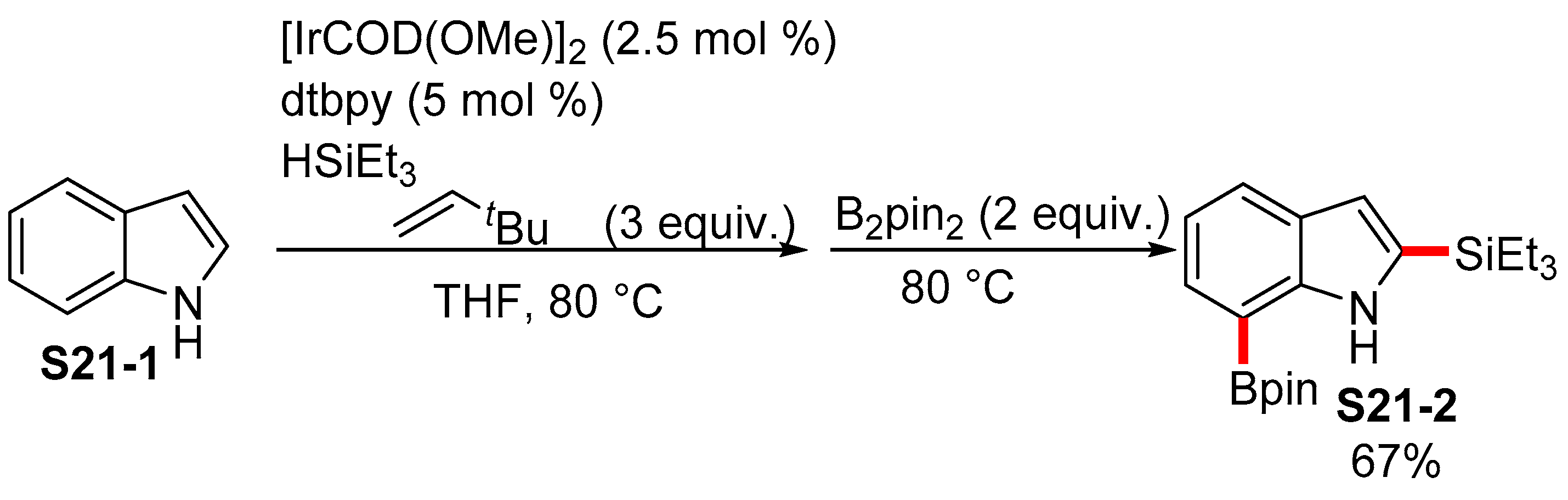{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
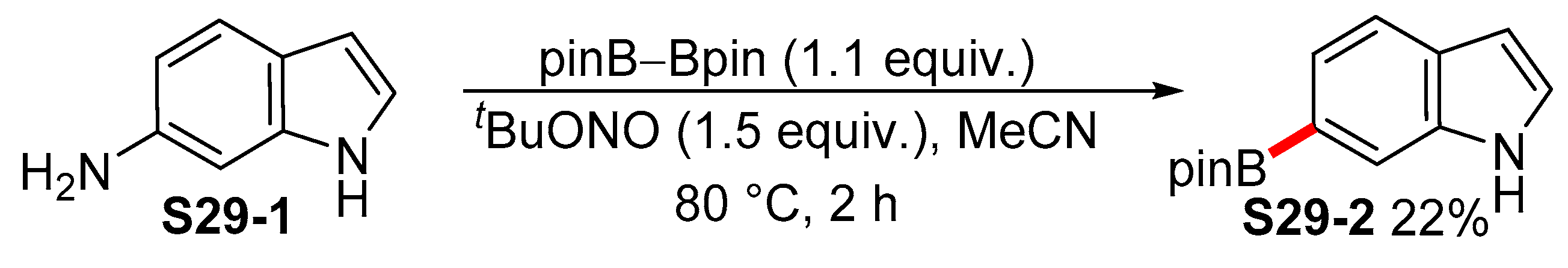{kind=link}
{kind=link}
{kind=link}
{kind=link}
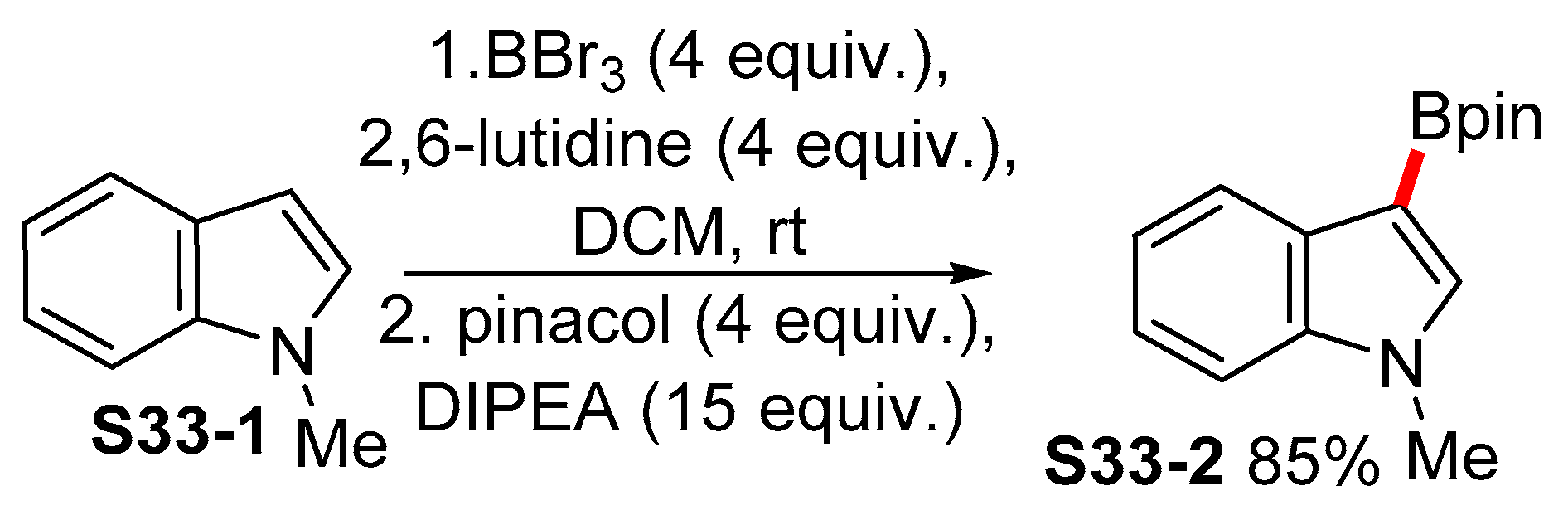{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
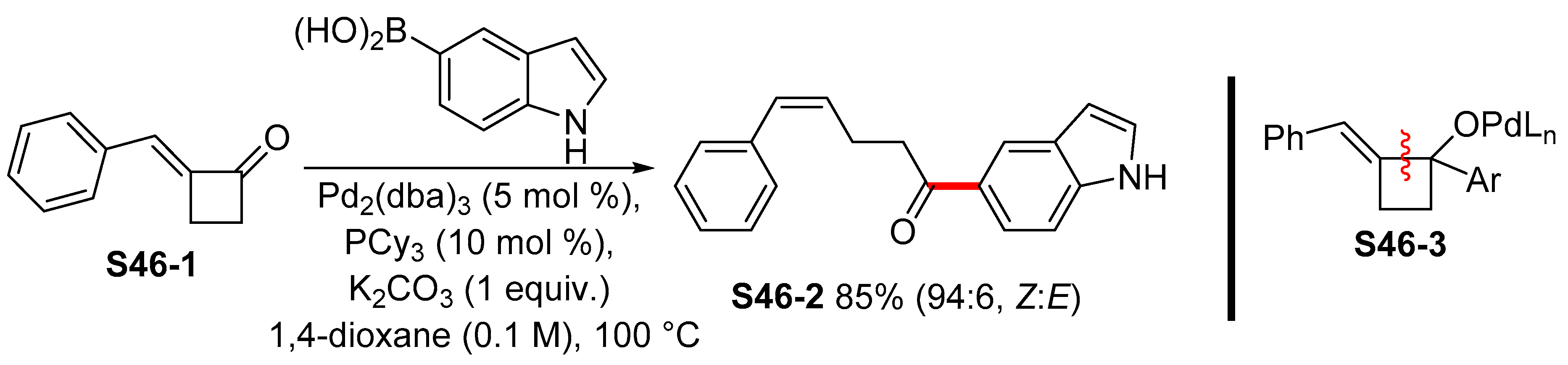{kind=link}
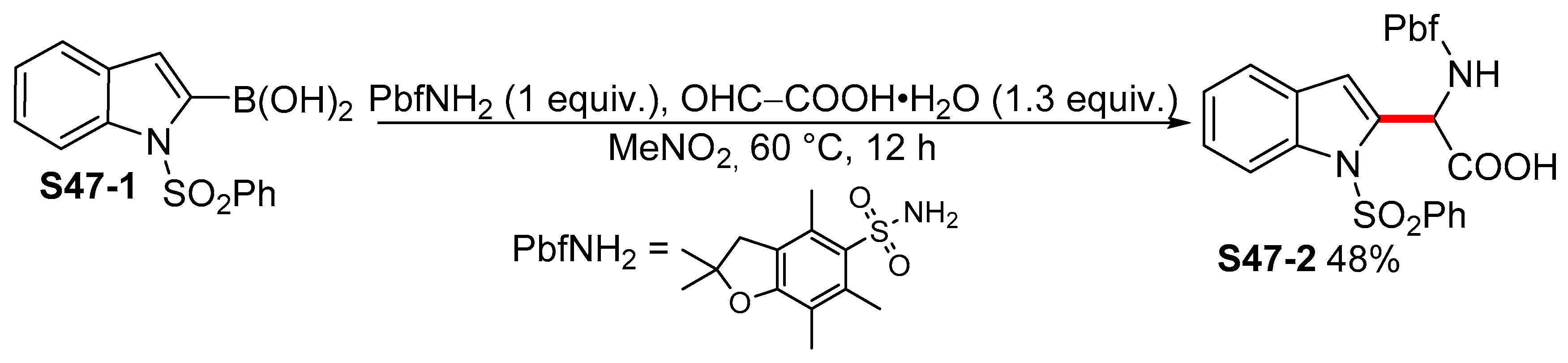{kind=link}
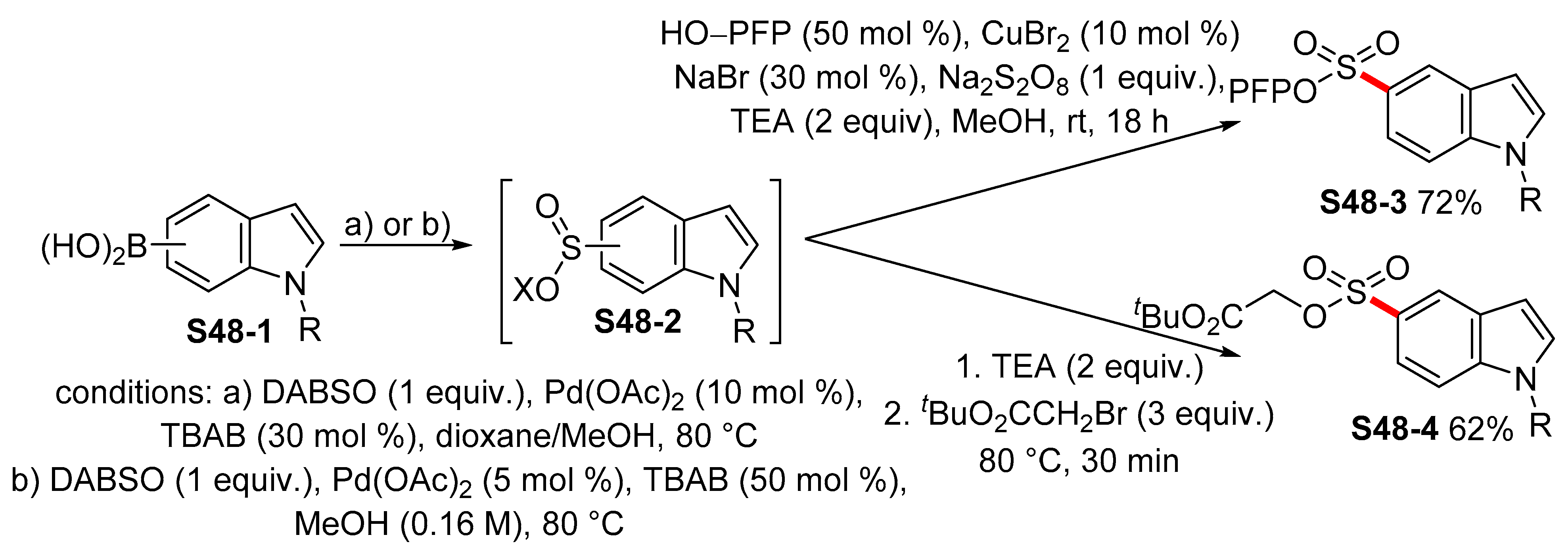{kind=link}
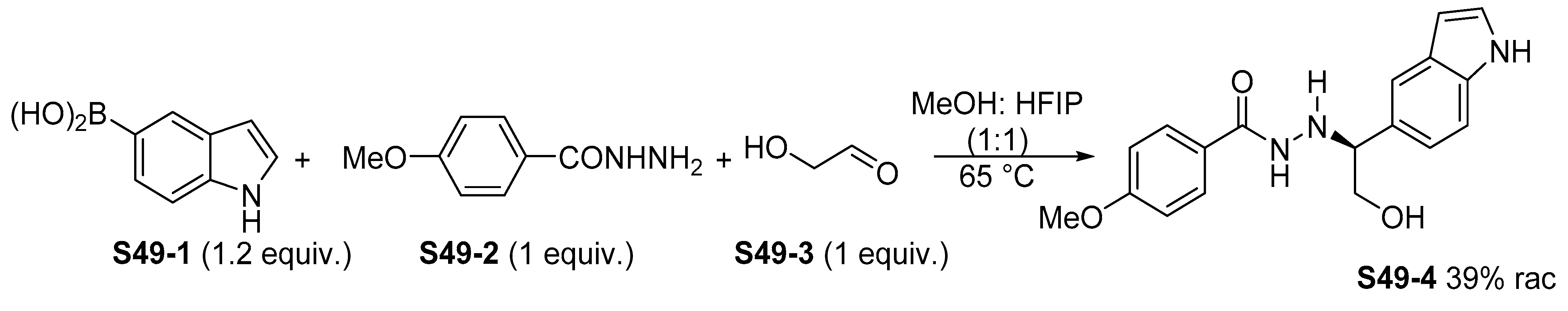{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Entry | LG | B-reagent | Conditions | T1-2 |
| 1 | I | pinB-Bdan (2 equiv.) | Pd2(dba)3 (5 mol %), Cs2CO3, MeOH, reflux |  |
| 2 | Cl | H2B-NiPr2 | Pd(OAc)2 (2 mol %), XPhos (6 mol %), KI (2 mol %), TEA, EtOAc, 50 °C, 16 h |  |
| 3 | Br | H-Bpin (2.5 equiv.) | CuI (10 mol %), Fe(acac)3 (10 mol %), TMEDA (1.5 equiv.), NaH (1.5 equiv.), -10 °C, 18 h |  |
| 4 | I, Br | pinB-Bpin | [Cu(DPEphos)(DMEGqu)]PF6 (2.5 mol %) DIPEA (1.2 equiv.), pyridine (20 mol %), MeCN/H2O, 12 h blue LED |  |
| 5 | Br | pinB-Bpin (1.3 equiv.) | Co(IMes)2Cl2 (2.5 mol %), KOMe (1.3 equiv.), MTBE, 50 °C, 8 h |  |
| 6 | F | H-Bpin (2 equiv.) | CoCl2 (10 mol %), L1 (15 mol %), Mg (1.5 equiv.), THF, 60 °C, 16 h |  |
| 7 | F | nepB-Bnep (3 equiv.) | Ni(cod)2 (5 mol %), PCy3 (20 mol %), NaOPh (3 equiv.), THF, 110 °C |  |
| 8 | CN | nepB-Bnep (2 equiv.) | [RhCl(cod)]2 (5 mol %), Xantphos (20 mol %), DABCO (1.0 equiv.), toluene, 100 °C, 15 h |  |
| 9 | Cl | pinB-Bpin | hν, TMEDA, TBAF, MeCN/H2O, acetone |  |
| 10 | Br | PhMe2Si-Bpin (1.5 equiv.) | KOMe (1.2 equiv.), DME, 30 °C, 1 h |  |
 | |||
| Entry | R | Conditions | T2-2 |
| 1 | OH | pinB−Bpin (1.5 equiv.), Pd(OAc)2, dppb (10 mol %), TEA, piv2O, dioxane 160 °C, 15 h |  |
| 2 | SEt | pinB−Bpin (2 equiv.), [Rh(OH)(cod)]2 (5 mol %), P(nBu)3 (50 mol %), KOAc (20 mol %), CPME, 80 °C, 24 h |  |
| 3 | ONPhth |  |  |
| 4 | ONPhth | pinB−Bpin (3.5 equiv.), pyridine (1.5 equiv.) Cs2CO3 (0.5 equiv.), EtOAc, 35 °C, 400 nm, 24 h |  |
 | |||
| Entry | R1, R2 | Conditions | T3-2 |
| 1 | H, Ts | BCl3 (2 equiv.), DCM rt, then pinacol, TEA, rt 1 h | 36−83%, R3 = Ph, 4-MePh, 4-MeOPh, 3-thienyl, 2-naphthyl, etc. |
| 2 | R1 = cat, dan R2 = Me or Bn | B(C6F5)3 (5 mol %) | 42−60%, R3 = Ph, 4-BrPh, 4-tBuPh, nBu, ferrocenyl |
| 3 | R1 = cat R2 = Mbs, Ts | IPrAuTFA (5 mol %), NaTFA (20 mol %), toluene, 80 °C, 20 h then pinacol (3 equiv.), THF, rt, 1 h | 55−80%, R3 = Ph, 2-thienyl, nBu, cyPr, 2-BrPh, TBDPSOCH2, etc |
| 4 | R1 = Ts, Ms R2 = H | pinB-Bpin (2 equiv.), Pd2dba3 (5 mol %), AsPh3 (10 mol %), Cs2CO3, DMA, 60 °C, 30 min. | 51−79%, R3 = Ph, Bu, BnOCH2, etc |
| 5 |  | ||
 | ||
| Entry | Conditions | T4-2 |
| 1 | H-Bcat (0.55 equiv.) B(C6F5)3 (5 mol %), C6D6, rt, 2 h |  |
| 2 | H-Bcat (4 equiv.) B(C6F5)3 (10 mol %), toluene, 120 °C, 72 h 2. pinacol (3 equiv.), TEA, rt, 1 h |  |
| 3 |  |  |
 | |||
| Entry | T5-1 | Conditions | R, yield (%) |
| 1 |  | Ph3PAuNTf2 (5 mol %), dimethyl carbonate, μW, H2O (10 equiv.), 90 °C, 1 h | −H, 73 |
| 2 |  | NaN3 (1.5 equiv.), Cu(OAc)2 (0.1 equiv.), MeOH (0.2 M), 55 °C, 24 h | −N3, 51 |
| 3 |  | [11C]NH4CN/NH3, PdCl2(PPh3)2, K2CO3, DMF, 100 °C, 5 min | −11CN, 90 ± 15 (rcy) |
 | |||
| Entry | T6-1 | Halogen source, conditions | T6-2 |
| 1 |  | I2 (1 equiv.), KF (3.3 equiv.) 1,4-dioxane (0.1 M), 80 °C, 1 h |  |
| 2 |  | (tBuCN)2CuOTf (2 equiv.), [Me3py]PF6 (3 equiv.), AgF (2 equiv.), THF, 50 °C, 18 h |  |
| 3 |  | [18F]F-, trifluoromethanesulfonate, Cu(OTf)2Py4, DMA, air, 110 °C, 20 min |  |
| 4 |  | (i) elution of [18F]F- with Et4NHCO3 in nBuOH (ii) [Cu(OTf)2(py)4], DMA, 110 °C, 20 min, air |  |
 | |||
| Entry | BX2 | [CF3], conditions | T7-2 |
| 1 | B(OH)2 | TMSCF3 (2 equiv.), Cu(OAc)2 (1 equiv.), 1,10-phenanthroline (1.1 equiv.), CsF (2 equiv.), O2, iPrCN, DCE, 4 Å MS, rt |  |
| 2 | Bpin | K[CF3B(OMe)3] (2 equiv.), Cu(OAc)2 (1 equiv.), O2,DMSO, 60 °C |  |
| 3 | B(OH)2 | (trifluoromethyl)dibenzothiophenium triflate, CuOAc (20 mol %), 2,4,6-trimethylpyridine (2 equiv.), DMAC, 0 °C |  |
| 4 | B(OH)2 | Togni’s reagent, CuI (2 mol %), phenanthroline, K2CO3 (2 equiv.), 35 °C, DME |  |
| 5 | BF3K | NaSO2CF3 (3 equiv.), TBHP (5 equiv.), CuCl (1 equiv.), DCM/MeOH/H2O, rt |  |
 | ||||
| Entry | LG | BX2 | Conditions | T8-3 |
| 1 |  | B(OH)2 | PhenNi(OAc)2∙xH2O (5 mol %), K3PO4 (3.4 equiv.), EtOH (5 equiv), dioxane (0.5 M), 60 °C, 5 h |  |
| 2 | OMe | Bnep |  |  |
 | ||||
| Entry | BX2 | Reagent | Conditions | T9-2 |
| 1 | Bpin | BrF2CCONEt2 (2 equiv.), CO | Pd(PPh)4 (5 mol %), Xantphos (10 mol %), K2CO3 (4 equiv.), toluene/H2O (9:1), 80 °C |  |
| 2 | B(OH)2 |  | Pd(OAc)2 (3 mol %), PCy3HBF4 (12 mol %), K2CO3, H3BO3, THF, 65 °C, 15 h |  |
| 3 | Bpin | CO (1 atm) | Pd(OAc)2/2PPh3 (5 mol%), pbq (1 equiv.), MeOH, rt, 18 h |  |
| 4 | B(OH)2 | ClCO−NMe(OMe) | PdCl2(PPh3)2, K3PO4∙H2O, EtOH, 65 °C |  |
 | ||||
| Entry | LG | BX2 | Conditions | T10-3 |
 | Bnep | [RhCl(cod)] (5 mol %), I(2Ad)∙HCl (20 mol %), NaOEt (2 equiv.), toluene, 130 °C, 20 h |  | |
| -SMe | B(OH)2 | Pd2(dba)3 (2.5 mol %), TFP (7.5 mol %), CuTC (3 equiv.), THF, 55 °C |  | |
 | |||
| Entry | T11-1 | Conditions | T11-3 |
| 1 |  |  |  |
| 2 |  | ArB(OH)2 (1.5 equiv.), [Rh(cod)Cl]2 (1 mol %), KOH (1 equiv.), dioxane/H2O (10:1), 60 °C, 1 h |  |
| 3 |  | ArB(OH)2 (2 equiv.), Pd(OAc)2 (10 mol %), NaF (2 equiv.), TFT, 100 °C, 12 h |  |
| 4 |  | ArB(OH)2 (1.5 equiv.), Cu(OTf)2 (10 mol %), toluene, 70 °C, 18 h |  |
| 5 |  | ArB(OH)2 (2.5 equiv.), Ni(cod)2 (10 mol %), TrixiePhos (10 mol %), THF (0.05 M), rt, 24 h |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čubiňák, M.; Edlová, T.; Polák, P.; Tobrman, T. Indolylboronic Acids: Preparation and Applications. Molecules 2019, 24, 3523. https://doi.org/10.3390/molecules24193523
Čubiňák M, Edlová T, Polák P, Tobrman T. Indolylboronic Acids: Preparation and Applications. Molecules. 2019; 24(19):3523. https://doi.org/10.3390/molecules24193523
Chicago/Turabian StyleČubiňák, Marek, Tereza Edlová, Peter Polák, and Tomáš Tobrman. 2019. "Indolylboronic Acids: Preparation and Applications" Molecules 24, no. 19: 3523. https://doi.org/10.3390/molecules24193523
APA StyleČubiňák, M., Edlová, T., Polák, P., & Tobrman, T. (2019). Indolylboronic Acids: Preparation and Applications. Molecules, 24(19), 3523. https://doi.org/10.3390/molecules24193523