Insulin Hot-Spot Analogs Formed with N-Methylated Amino Acid Residues Inhibit Aggregation of Native Hormone

 and
and
Abstract
:1. Introduction
2. Results and Discussion
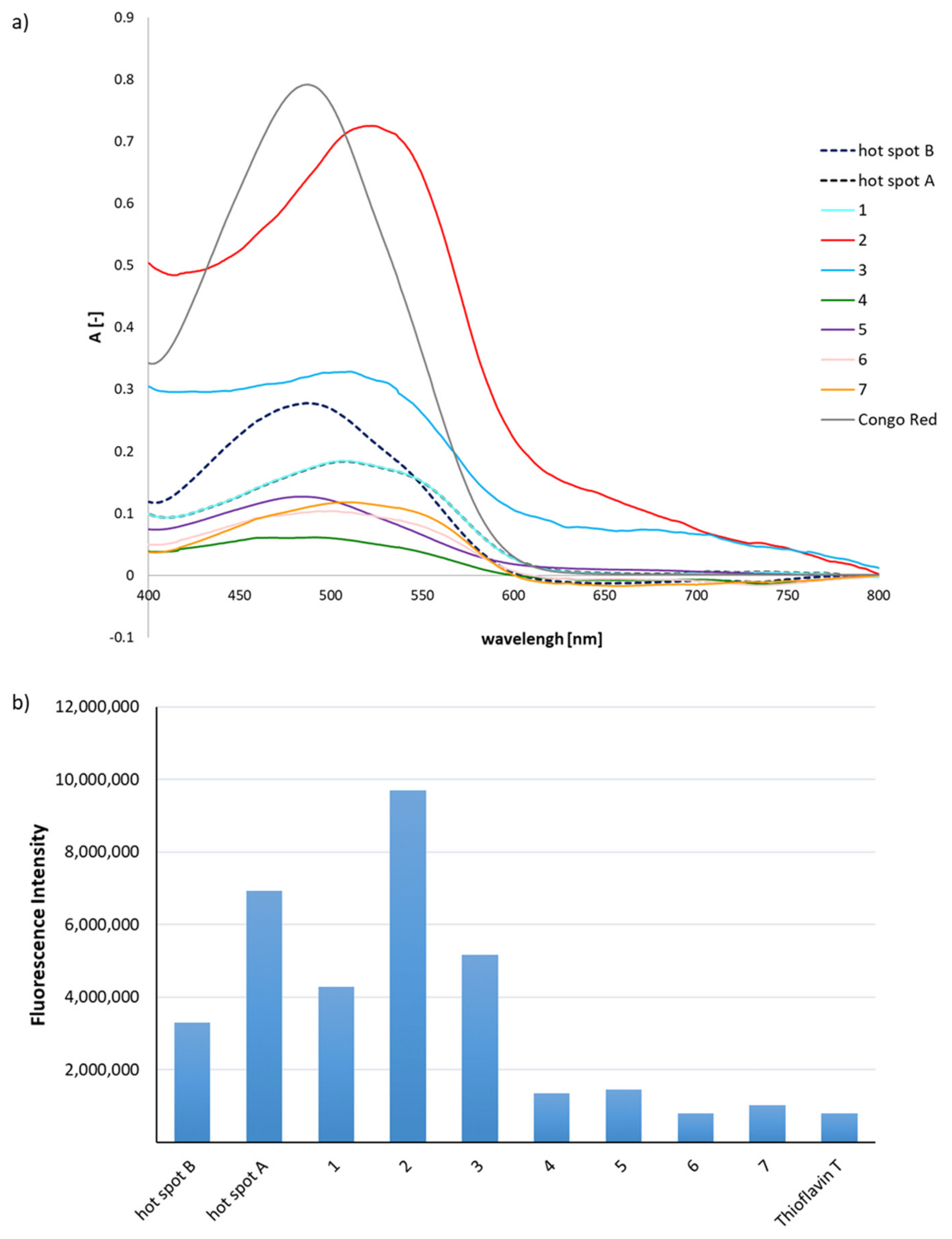
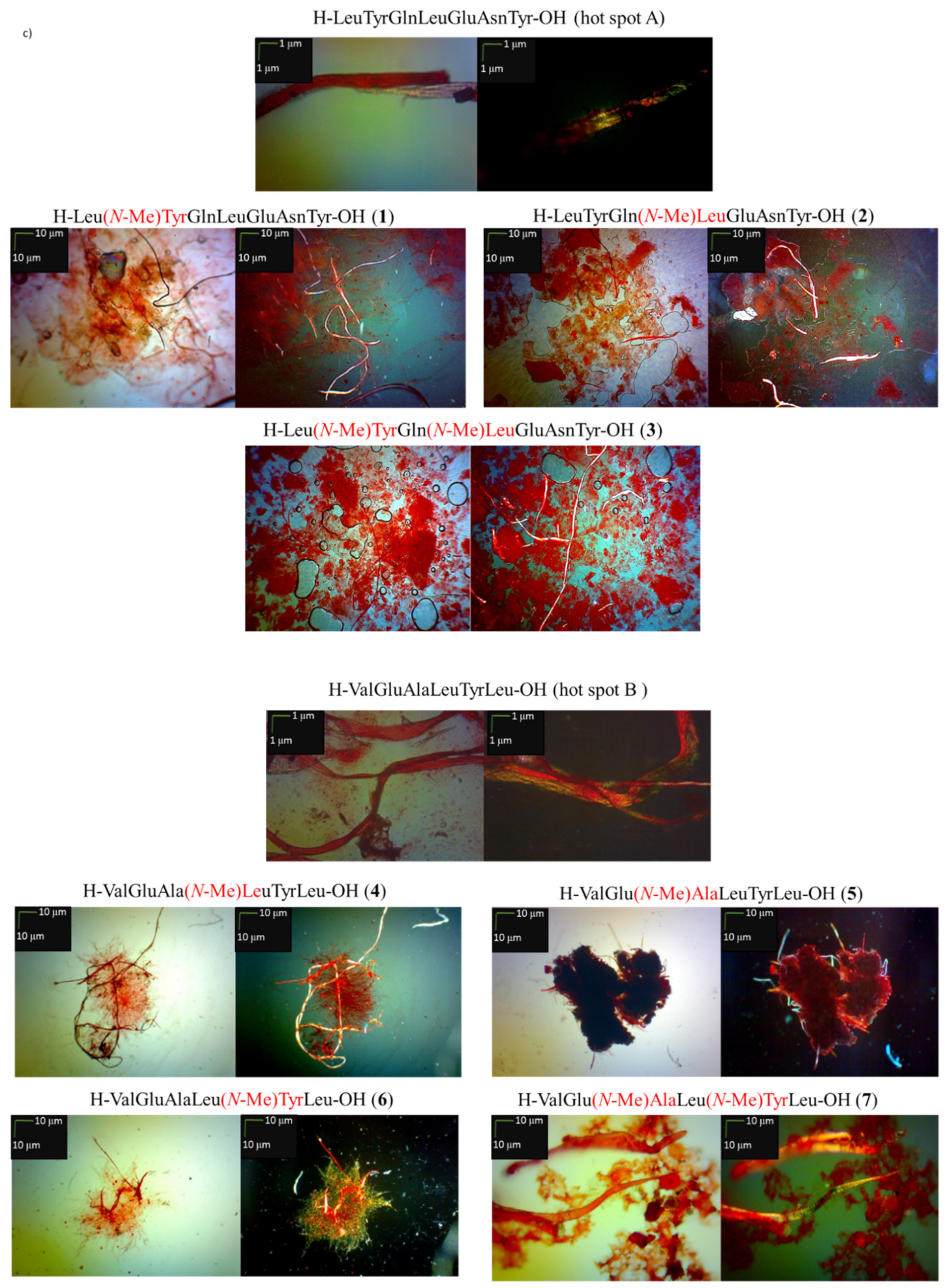
2.1. Predisposition of Peptides 1–7 to Aggregate
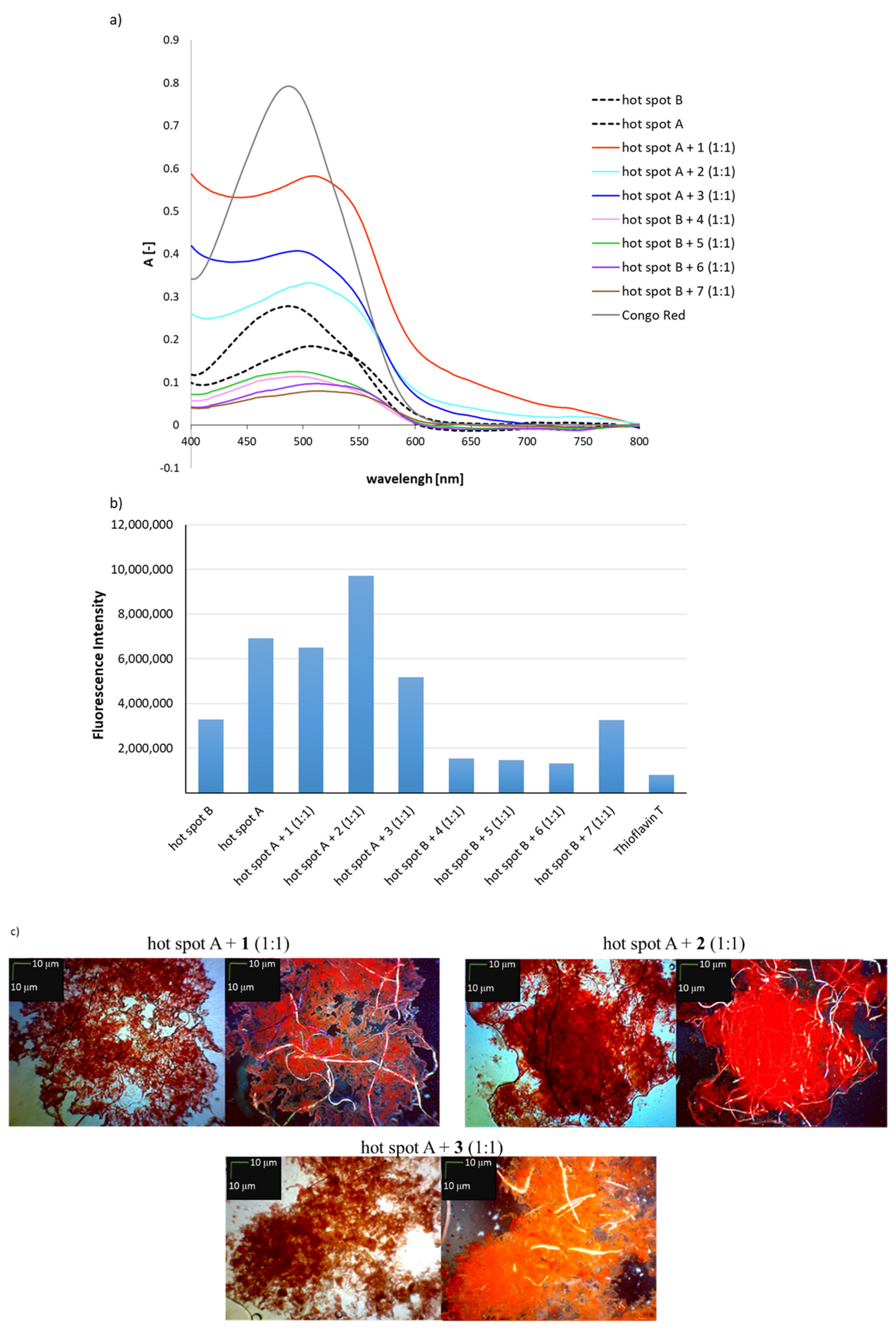
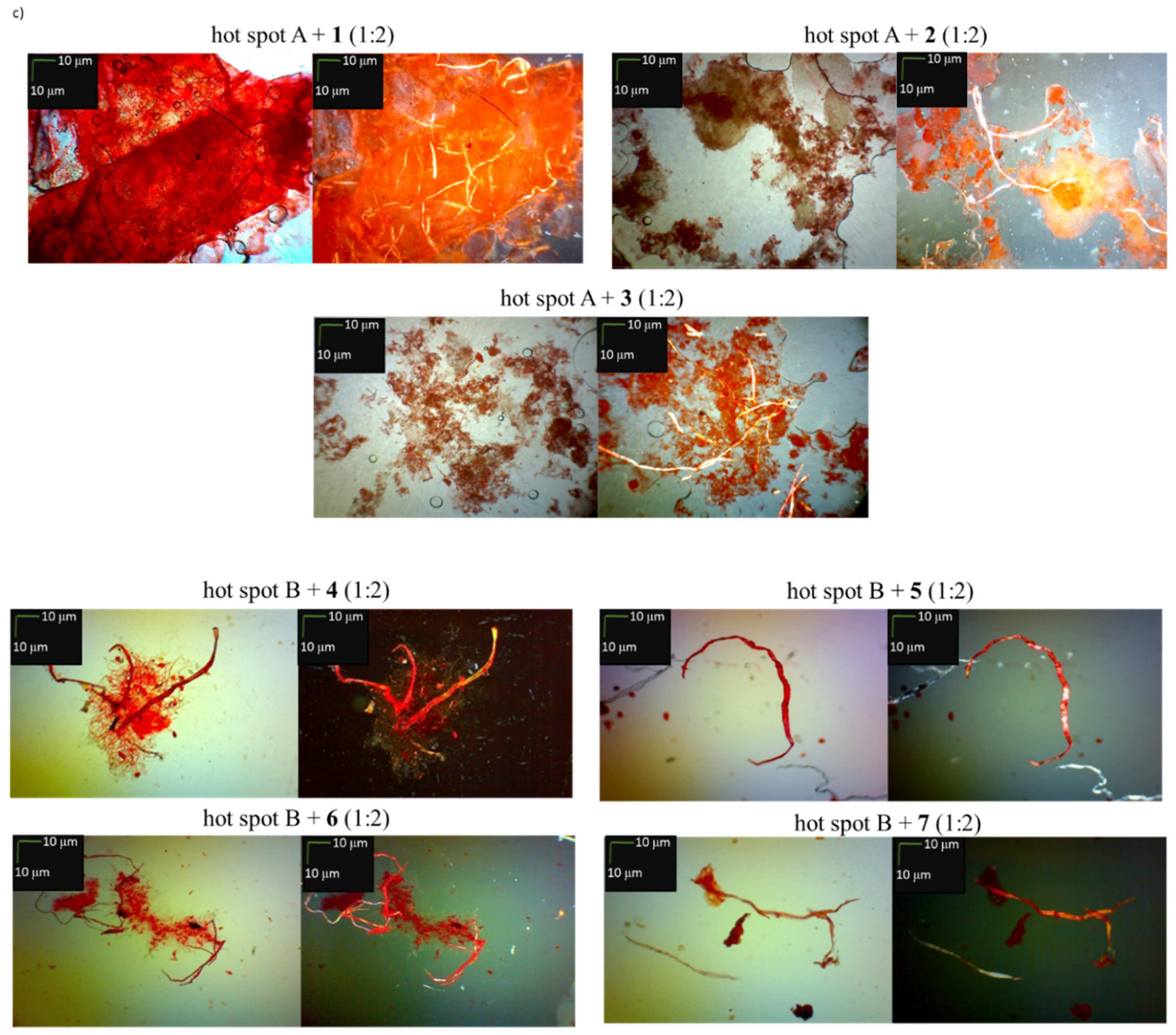
2.2. Effects of Peptides 1–7 on the Aggregation of Native Insulin Hot-Spots A and B
2.3. Effects on Aggregation of Human Insulin
3. Materials and Methods
3.1. General Methods for Solid-Phase Peptide Synthesis
3.1.1. Method 1. Resin Loading
3.1.2. Method 2. Determination of Resin Loading
3.1.3. Method 3. Amino Acid Coupling
3.1.4. Method 4. Fmoc Deprotection
3.1.5. Method 5. Cleavage from Resin
3.2. General Synthesis Procedure for Peptides 1–7
3.3. General Methods for Assays with Congo Red and Thioflavin T to Determine Aggregation Propensity
3.3.1. Spectroscopic Measurements
3.3.2. Fluorescence Measurements
3.4. Microscopic Measurements after Incubation with Congo Red
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ahmad, A.; Uversky, V.N.; Hong, D.; Fink, A.L. Early events in the fibrillation of monomeric insulin. J. Biol. Chem. 2005, 280, 42669–42675. [Google Scholar] [CrossRef] [PubMed]
- Frankær, C.G.; Sønderby, P.; Bang, M.B.; Mateiu, R.V.; Groenning, M.; Bukrinski, J.; Harris, P. Insulin fibrillation: The influence and coordination of Zn2+. J. Struct. Biol. 2017, 199, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Koltun, W.L.; Waugh, D.F.; Bear, R.S. An X-Ray Diffraction investigation of selected types of insulin fibrils. J. Am. Chem. Soc. 1954, 76, 413–417. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Sawaya, M.R.; Wall, J.S.; Eisenberg, D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA 2009, 106, 18990–18995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surin, A.K.; Grishin, S.Y.; Galzitskaya, O.V. Identification of Amyloidogenic Regions in the Spine of Insulin Fibrils. Biochem. (Mosc) 2019, 84, 47–55. [Google Scholar] [CrossRef]
- Banerjee, V.; Rajiv, K.; Kali, P. Use of a Small Peptide Fragment as an Inhibitor of Insulin Fibrillation Process: A Study by High and Low Resolution Spectroscopy. PLoS ONE 2013, 8, e72318. [Google Scholar] [CrossRef]
- Buchner, J.; Rudolph, R. Renaturation, purification and characterization of recombinant Fab-fragments produced in Escherichia coli. Nat. Biotechnol. 1991, 9, 157–162. [Google Scholar]
- Tanaja, S.; Ahmad, F. Increased thermal stability of proteins in the presence of amino acids. Biochem. J. 1994, 303, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Z.S.; Guo, Z.Y.; Feng, Y.M. Putative disulfide-forming pathway of porcine insulin precursor during its refolding in vitro. Biochemistry 2001, 40, 2662–2668. [Google Scholar] [CrossRef]
- Shiraki, K.; Kudon, M.; Fujiwara, S.; Imanaka, T.; Takagi, M. Biophysical effect of amino acids on the prevention of protein aggregation. J. Biochem. 2001, 132, 591–595. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K. The effects of arginine on refolding of aggregated proteins: Not facilitate refolding but suppress aggregation. Biochem. Biophys. Res. Commun. 2003, 304, 148–152. [Google Scholar] [CrossRef]
- Tsumoto, K.; Umetsu, M.; Kumagai, J.; Eijma, D.; Philo, J.S.; Arakawa, T. The role of arginine in protein refolding, solubilization and purification. Biotechnol. Prog. 2004, 20, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Guo, T.; Lu, J.; Zhou, H.M. The guanidine like effects of arginine on aminoacylase and salt-induced molten globule state. Int. J. Biochem. Cell Biol. 2004, 36, 296–306. [Google Scholar]
- Gibson, T.J.; Murphy, R.M. Inhibition of insulin fibriliogenesis with targeted peptides. Protein Sci. 2006, 15, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Varughese, M.; Newman, J. Inhibitory Effects of Arginine on the Aggregation of Bovine Insulin. J. Biophys. 2012, 2012, 434289. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Sciaretta, K.L.; Meredith, S.C. Inhibition of β-amyloid(40) fibrillogenesis and disassembly of β-amyloid(40) fibrils by short β-amyloid cogeners containing N-methyl amino acids at alternate residues. Biochemistry 2001, 40, 8237–8245. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Tappe, R.; Meredith, S.C. Design and characterization of membrane permeable N-methyl amino acid-containing peptide that inhibits Aβ(1-40) fibrillogenesis. J. Pept. Res. 2002, 60, 37–55. [Google Scholar] [CrossRef]
- Hughes, E.; Burke, R.M.; Doig, A.J. Inhibition of toxicity in β-amyloid peptide fragment β(25-35) using N-methylated drivatives-general strategy to prevent amyloid formation. J. Biol. Chem. 2000, 275, 25109–25115. [Google Scholar] [CrossRef]
- Adessi, C.; Frossard, M.J.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13905–13911. [Google Scholar] [CrossRef]
- Kapurniotu, A.; Schmauder, A.; Tenidis, K. Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxity. J. Mol. Biol. 2002, 315, 339–350. [Google Scholar] [CrossRef]
- Tanaka, M. Design and synthesis of chiral α,α-Disubstituted Amino Acids and Conformational Study of Their Oligipeptides. Chem. Pharm. Bull. 2007, 55, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Kolesinska, B.; Rozniakowski, K.K.; Fraczyk, J.; Relich, I.; Papini, A.M.; Kaminski, Z.J. The effect of counterion and tertiary amine on the efficiency of N-triazinylammonium sulfonates in solution and solid-phase peptide synthesis. Eur. J. Org. Chem. 2015, 401–408. [Google Scholar] [CrossRef]
- Swiontek, M.; Fraczyk, J.; Wasko, J.; Chaberska, A.; Pietrzak, L.; Kaminski, Z.J.; Szymanski, L.; Wiak, S.; Kolesinska, B. Search for New Aggregable Fragments of Human Insulin. Molecules 2019, 24, 1600. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Fisher, A.D.; Amori, A.R.; Welchons, D.P.; McKnight, R.E. Spectroscopic and calorimetric studies of congo red dye-amyloid β peptide complexes. JBPC 2010, 1, 153–163. [Google Scholar] [CrossRef]
- Wolfe, L.S.; Calabrese, M.F.; Nath, A.; Blaho, D.V.; Miranker, A.D.; Xiong, Y. Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc. Natl. Acad. Sci. USA 2010, 107, 16863–16868. [Google Scholar] [CrossRef] [Green Version]
- Klunk, W.E.; Pettegrew, J.W.; Abraham, D.J. Quantitative evaluation of congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. J. Histochem. Cytochem. 1989, 37, 1273–1281. [Google Scholar] [CrossRef]
- Klunk, W.E.; Jacob, R.F.; Mason, R.P. Quantifying amyloid by congo red spectral shift assay. Methods Enzym. 1999, 309, 285–305. [Google Scholar]
- Klunk, W.E.; Jacob, R.F.; Mason, R.P. Quantifying amyloid beta-peptide (Abeta) aggregation using the Congo red-Abeta (CR-abeta) spectrophotometric assay. Anal. Biochem. J. 1999, 266, 66–76. [Google Scholar]
- O’Nuallain, B.; Shivaprasad, S.; Kheterpal, I.; Wetzel, R. Thermodynamics of A beta(1-40) amyloid fibril elongation. Biochemistry 2005, 44, 12709–12718. [Google Scholar] [CrossRef]
- Shivaprasad, S.; Wetzel, R. Scanning cysteine mutagenesis analysis of Abeta-(1-40) amyloid fibrils. J. Biol. Chem. 2006, 281, 993–1000. [Google Scholar] [CrossRef]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J. Chem. Biol. 2010, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.A.; Gestwicki, J.E. Insight into Amyloid Structure Using Chemical Probes. Chem. Biol. Drug Des. 2011, 77, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiontek, M.; Rozniakowski, K.; Fraczyk, J.; Lipinski, W.; Galecki, K.; Wysocki, S.; Dupont, B.G.R.; Kaminski, Z.J.; Kolesinska, B. The quest for the shortest fragments of A (13-19) and B (12-17) responsible for the aggregation of human insulin. Nanomedicine 2016, 11, 2083–2101. [Google Scholar] [CrossRef]
- Clemens, A.; Allsop, D.; Walsh, D.M.; Williams, C.H. Aggregation and metal-binding properties of mutant forms of the amyloid A beta peptide of Alzheimer’s disease. J. Neurochem. 1996, 19, 740–747. [Google Scholar]
- Soto, C.; Castano, E.M. The conformation of Alzheimer’s beta peptide determines the rate of amyloid formation and its resistance to proteolysis. Biochem. J. 1996, 314, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [PubMed]
- Dunstan, D.E.; Hamilton-Brown, P.; Asimakis, P.; Ducker, W.; Bertolini, J. Shear flow promotes amyloid-β fibrilization. Protein Eng. Des. Sel. 2009, 22, 741–746. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 3095–3103. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insulin Complex with Peptides 1–7 | CR Assay | ThT Assay | Microscopic Examination |
|---|---|---|---|
| Complex (1:1) H-Leu(N-Me)TyrGlnLeuGluAsnTyr-OH (1) with Insulin | - | - | - |
| Complex (1:1) H-LeuTyrGln(N-Me)LeuGluAsnTyr-OH (2) with Insulin | +/− | +/− | - |
| Complex (1:1) H-Leu(N-Me)TyrGln(N-Me)LeuGluAsnTyr-OH (3) with Insulin | - | +/− | - |
| Complex (1:1) H-ValGluAla(N-Me)LeuTyrLeu-OH (4) with Insulin | - | - | - |
| Complex (1:1) H-ValGlu(N-Me)AlaLeuTyrLeu-OH (5) with Insulin | - | - | - |
| Complex (1:1) H-ValGluAlaLeu(N-Me)TyrLeu-OH (6) with Insulin | - | - | - |
| Complex (1:1) H-ValGlu(N-Me)AlaLeu(N-Me)TyrLeu-OH (7) with Insulin | +/− | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swiontek, M.; Wasko, J.; Fraczyk, J.; Galecki, K.; Kaminski, Z.J.; Kolesinska, B. Insulin Hot-Spot Analogs Formed with N-Methylated Amino Acid Residues Inhibit Aggregation of Native Hormone. Molecules 2019, 24, 3706. https://doi.org/10.3390/molecules24203706
Swiontek M, Wasko J, Fraczyk J, Galecki K, Kaminski ZJ, Kolesinska B. Insulin Hot-Spot Analogs Formed with N-Methylated Amino Acid Residues Inhibit Aggregation of Native Hormone. Molecules. 2019; 24(20):3706. https://doi.org/10.3390/molecules24203706
Chicago/Turabian StyleSwiontek, Monika, Joanna Wasko, Justyna Fraczyk, Krystian Galecki, Zbigniew J. Kaminski, and Beata Kolesinska. 2019. "Insulin Hot-Spot Analogs Formed with N-Methylated Amino Acid Residues Inhibit Aggregation of Native Hormone" Molecules 24, no. 20: 3706. https://doi.org/10.3390/molecules24203706
APA StyleSwiontek, M., Wasko, J., Fraczyk, J., Galecki, K., Kaminski, Z. J., & Kolesinska, B. (2019). Insulin Hot-Spot Analogs Formed with N-Methylated Amino Acid Residues Inhibit Aggregation of Native Hormone. Molecules, 24(20), 3706. https://doi.org/10.3390/molecules24203706