PD-1-Targeted Discovery of Peptide Inhibitors by Virtual Screening, Molecular Dynamics Simulation, and Surface Plasmon Resonance

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Synthetic Peptides and Proteins
2.3. Human PD-1 3D Structure Preparation
2.4. Virtual Screening and Molecular Docking
2.5. Molecular Dynamics (MD) Simulation
2.6. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Calculation
2.7. Surface Plasmon Resonance (SPR) Analysis
3. Results and Discussion
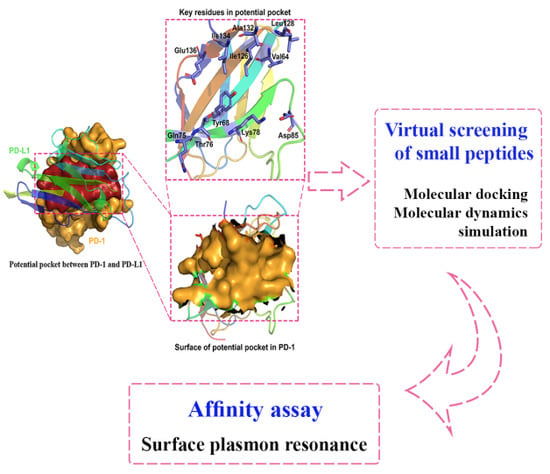
3.1. Potential Binding Site on PD-1 Protein Interface
3.2. In-House Peptides as PD-1 Inhibitors/Modulator
3.3. Detailed Interactions and MD Simulation of PD-1/Peptides
3.3.1. Interactions between PD-1 and WANG-003
3.3.2. Interactions between PD-1 and WANG-004
3.3.3. Interactions between PD-1 and WANG-005
3.3.4. SPR-Based Binding Studies on Designed Peptides and PD-1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480. [Google Scholar] [CrossRef] [PubMed]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Rmili, C.W.; Kiialainen, A.; Kienast, Y.; Mueller, H.-J.; Ooi, C.-H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef] [PubMed]
- Prieto, P.A.; Yang, J.C.; Sherry, R.M.; Hughes, M.S.; Kammula, U.S.; White, D.E.; Levy, C.L.; Rosenberg, S.A.; Phan, G.Q. CTLA-4 blockade with ipilimumab: Long-term follow-up of 177 patients with metastatic melanoma. Clin. Cancer Res. 2012, 18, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Brandsma, I.; Fleuren, E.D.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef]
- Badr, M.E.S.G.; Hata, K.; Furuhata, M.; Toyota, H.; Yokosuka, T. The Multifaceted Role of PD-1 in Health and Disease. In Chronic Inflammation; Springer: Berlin/Heidelberg, Germany, 2016; pp. 441–457. [Google Scholar]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015, 4, e1008824. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef]
- Moskowitz, C.H.; Ribrag, V.; Michot, J.-M.; Martinelli, G.; Zinzani, P.L.; Gutierrez, M.; De Maeyer, G.; Jacob, A.G.; Giallella, K.; Anderson, J.W.; et al. PD-1 blockade with the monoclonal antibody pembrolizumab (MK-3475) in patients with classical Hodgkin lymphoma after brentuximab vedotin failure: Preliminary results from a phase 1b study (KEYNOTE-013). Am. Soc. Hematol. 2014, 124, 290. [Google Scholar]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Chin, K.; Chand, V.; Nuyten, D. Avelumab: Clinical trial innovation and collaboration to advance anti-PD-L1 immunotherapy. Ann. Oncol. 2017, 28, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.-L.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558. [Google Scholar] [CrossRef]
- Segal, N.H.; Antonia, S.J.; Brahmer, J.R.; Maio, M.; Blake-Haskins, A.; Li, X.; Vasselli, J.; Ibrahim, R.A.; Lutzky, J.; Khleif, S. Preliminary data from a multi-arm expansion study of MEDI4736, an anti-PD-L1 antibody. Am. Soc. Clin. Oncol. 2014, 32, 3002. [Google Scholar] [CrossRef]
- Tykodi, S.S.; Brahmer, J.R.; Hwu, W.-J.; Chow, L.Q.; Topalian, S.L.; Hwu, P.; Odunsi, K.; Camacho, L.H.; Kauh, J.S.; Pitot, H.C.; et al. PD-1/PD-L1 pathway as a target for cancer immunotherapy: Safety and clinical activity of BMS-936559, an anti-PD-L1 antibody, in patients with solid tumors. Am. Soc. Clin. Oncol. 2012, 30, 2510. [Google Scholar]
- Dirix, L.; Takacs, I.; Nikolinakos, P.; Jerusalem, G.; Arkenau, H.-T.; Hamilton, E.P.; Von Heydebreck, A.; Grote, H.-J.; Chin, K.; Lippman, M.E. Abstract S1-04: Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase Ib JAVELIN solid tumor trial. Breast Cancer Res Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-molecule inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Han, Y.; Gao, Y.; He, T.; Wang, D.; Guo, N.; Zhang, X.; Chen, S.; Wang, H. PD-1/PD-L1 inhibitor screening of caffeoylquinic acid compounds using surface plasmon resonance spectroscopy. Anal. Biochem. 2018, 547, 52–56. [Google Scholar] [CrossRef]
- Donnelly, D.J.; Smith, R.A.; Morin, P.; Lipovsek, D.; Gokemeijer, J.; Cohen, D.; Lafont, V.; Tran, T.; Cole, E.L.; Wright, M.; et al. Synthesis and biologic evaluation of a novel (18)F-labeled adnectin as a PET radioligand for imaging PD-L1 expression. J. Nucl. Med. 2018, 59, 529–535. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Alsina, J.; Albericio, F. Solid-phase synthesis of C-terminal modified peptides. Pept. Sci. 2003, 71, 454–477. [Google Scholar] [CrossRef] [PubMed]
- Shenmar, K.; Sharma, K.K.; Wangoo, N.; Maurya, I.K.; Kumar, V.; Khan, S.I.; Jacob, M.R.; Tikoo, K.; Jain, R. Synthesis, stability and mechanistic studies of potent anticryptococcal hexapeptides. Eur. J. Med. Chem. 2017, 132, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Domling, A.; Dubin, G.; Holak, T.A. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring noncovalent protein-ligand interactions: A continuous differentiable function tuned to compute binding affinities. J. Comput. Aided Mol. Des. 1996, 10, 427–440. [Google Scholar] [CrossRef]
- Feng, Z.; Pearce, L.V.; Xu, X.; Yang, X.; Yang, P.; Blumberg, P.M.; Xie, X.-Q. Structural insight into tetrameric hTRPV1 from homology modeling, molecular docking, molecular dynamics simulation, virtual screening and bioassay validations. J. Chem. Inf. Model. 2015, 55, 572–588. [Google Scholar] [CrossRef]
- Chen, J.-Z.; Wang, J.; Xie, X.-Q. GPCR structure-based virtual screening approach for CB2 antagonist search. J. Chem. Inf. Model. 2007, 47, 1626–1637. [Google Scholar] [CrossRef]
- Sheng, S.; Wang, J.; Wang, L.; Liu, H.; Li, P.; Liu, M.; Long, C.; Xie, C.; Xie, X.; Su, W. Network pharmacology analyses of the antithrombotic pharmacological mechanism of Fufang Xueshuantong Capsule with experimental support using disseminated intravascular coagulation rats. J. Ethnopharmacol. 2014, 154, 735–744. [Google Scholar] [CrossRef]
- Feng, Z.; Kochanek, S.; Close, D.; Wang, L.; Srinivasan, A.; Almehizia, A.A.; Iyer, P.; Xie, X.-Q.; Johnston, P.A.; Gold, B. Design and activity of AP endonuclease-1 inhibitors. J. Chem. Biol. 2015, 8, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Pearce, L.V.; Zhang, Y.; Xing, C.; Herold, B.K.; Ma, S.; Hu, Z.; Turcios, N.A.; Yang, P.; Tong, Q.; et al. Multi-functional diarylurea small molecule inhibitors of TRPV1 with therapeutic potential for neuroinflammation. AAPS J. 2016, 18, 898–913. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Case, D.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; Greene, D.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Chem. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf. Model. 2012, 52, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Case, D.A. Theory and applications of the generalized born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Bashford, D.; Case, D.A. Generalized born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from Linear Combinations of Pairwise Overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Hu, J.; Feng, Z.; Ma, S.; Zhang, Y.; Tong, Q.; Alqarni, M.H.; Gou, X.; Xie, X.-Q. Difference and influence of inactive and active states of cannabinoid receptor subtype CB2: From conformation to drug discovery. J. Chem. Inf. Model. 2016, 56, 1152–1163. [Google Scholar] [CrossRef]
- Chang, H.N.; Liu, B.Y.; Qi, Y.K.; Zhou, Y.; Chen, Y.P.; Pan, K.M.; Li, W.W.; Zhou, X.M.; Ma, W.W.; Fu, C.Y.; et al. Blocking of the PD-1/PD-L1 Interaction by a D-Peptide Antagonist for Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2015, 127, 11926–11930. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed Atlas of Surface Topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Shulman-Peleg, A.; Shatsky, M.; Nussinov, R.; Wolfson, H.J. MultiBind and MAPPIS: Webservers for multiple alignment of protein 3D-binding sites and their interactions. Nucleic Acids Res. 2008, 36, W260–W264. [Google Scholar] [CrossRef] [PubMed]
- Gabdoulline, R.R.; Wade, R.C.; Walther, D. MolSurfer: A macromolecular interface navigator. Nucleic Acids Res. 2003, 31, 3349–3351. [Google Scholar] [CrossRef]
- Saha, R.P.; Bahadur, R.P.; Pal, A.; Mandal, S.; Chakrabarti, P. ProFace: A server for the analysis of the physicochemical features of protein-protein interfaces. BMC Struct. Biol. 2006, 6, 11. [Google Scholar]
Sample Availability: Samples of the compounds including WANG-003, WANG-004, WANG-005, WANG-006, and WANG-007 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Name | Peptide/Protein | MW (g/mol) | HPLC | |
|---|---|---|---|---|---|
| Calculated | Observed | Purity | |||
| 1 | - | PD-L1 | 26000 | - | - |
| 2 | WANG-003 | KRWWR-NH2 | 831.00 | 830.00 | 95.96% |
| 3 | WANG-004 | FRWWR-NH2 | 849.00 | 848.40 | 98.79% |
| 4 | WANG-005 | RRWQWR-NH2 | 1045.23 | 1045.20 | 98.50% |
| 5 | WANG-006 | YVAM-NH2 | 481.60 | 481.20 | 98.93% |
| 6 | WANG-007 | YVAE-NH2 | 479.52 | 479.20 | 98.34% |
| No. | Name | Peptide | KD (μM) | Docking Score | Figure |
|---|---|---|---|---|---|
| 1 | PD-L1 | - | 0.8825 ± 0.0050 | - | S4a |
| 2 | WANG-003 | KRWWR-NH2 | 3.3527 ± 1.0276 | 9.36 | S4b |
| 3 | WANG-004 | FRWWR-NH2 | 1.6333 ± 0.3088 | 9.93 | S4c |
| 4 | WANG-005 | RRWQWR-NH2 | 5.1537 ± 2.9329 | 10.01 | S4d |
| 5 | WANG-006 | YVAM-NH2 | NA | 5.34 | - |
| 6 | WANG-007 | YVAE-NH2 | NA | 5.91 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Guo, H.; Feng, Z.; Wang, S.; Wang, Y.; He, Q.; Li, G.; Lin, W.; Xie, X.-Q.; Lin, Z. PD-1-Targeted Discovery of Peptide Inhibitors by Virtual Screening, Molecular Dynamics Simulation, and Surface Plasmon Resonance. Molecules 2019, 24, 3784. https://doi.org/10.3390/molecules24203784
Wang Y, Guo H, Feng Z, Wang S, Wang Y, He Q, Li G, Lin W, Xie X-Q, Lin Z. PD-1-Targeted Discovery of Peptide Inhibitors by Virtual Screening, Molecular Dynamics Simulation, and Surface Plasmon Resonance. Molecules. 2019; 24(20):3784. https://doi.org/10.3390/molecules24203784
Chicago/Turabian StyleWang, Yuanqiang, Haiqiong Guo, Zhiwei Feng, Siyi Wang, Yuxuan Wang, Qingxiu He, Guangping Li, Weiwei Lin, Xiang-Qun Xie, and Zhihua Lin. 2019. "PD-1-Targeted Discovery of Peptide Inhibitors by Virtual Screening, Molecular Dynamics Simulation, and Surface Plasmon Resonance" Molecules 24, no. 20: 3784. https://doi.org/10.3390/molecules24203784