
Sequential MCR via Staudinger/Aza-Wittig versus Cycloaddition Reaction to Access Diversely Functionalized 1-Amino-1H-Imidazole-2(3H)-Thiones


 , and
, and
Abstract
:
1. Introduction
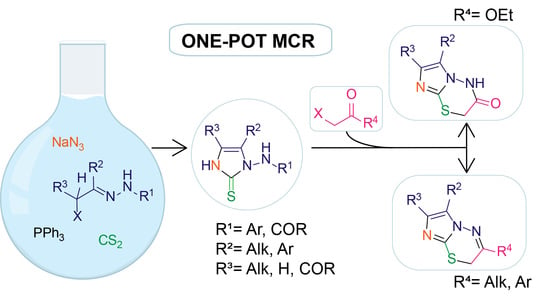
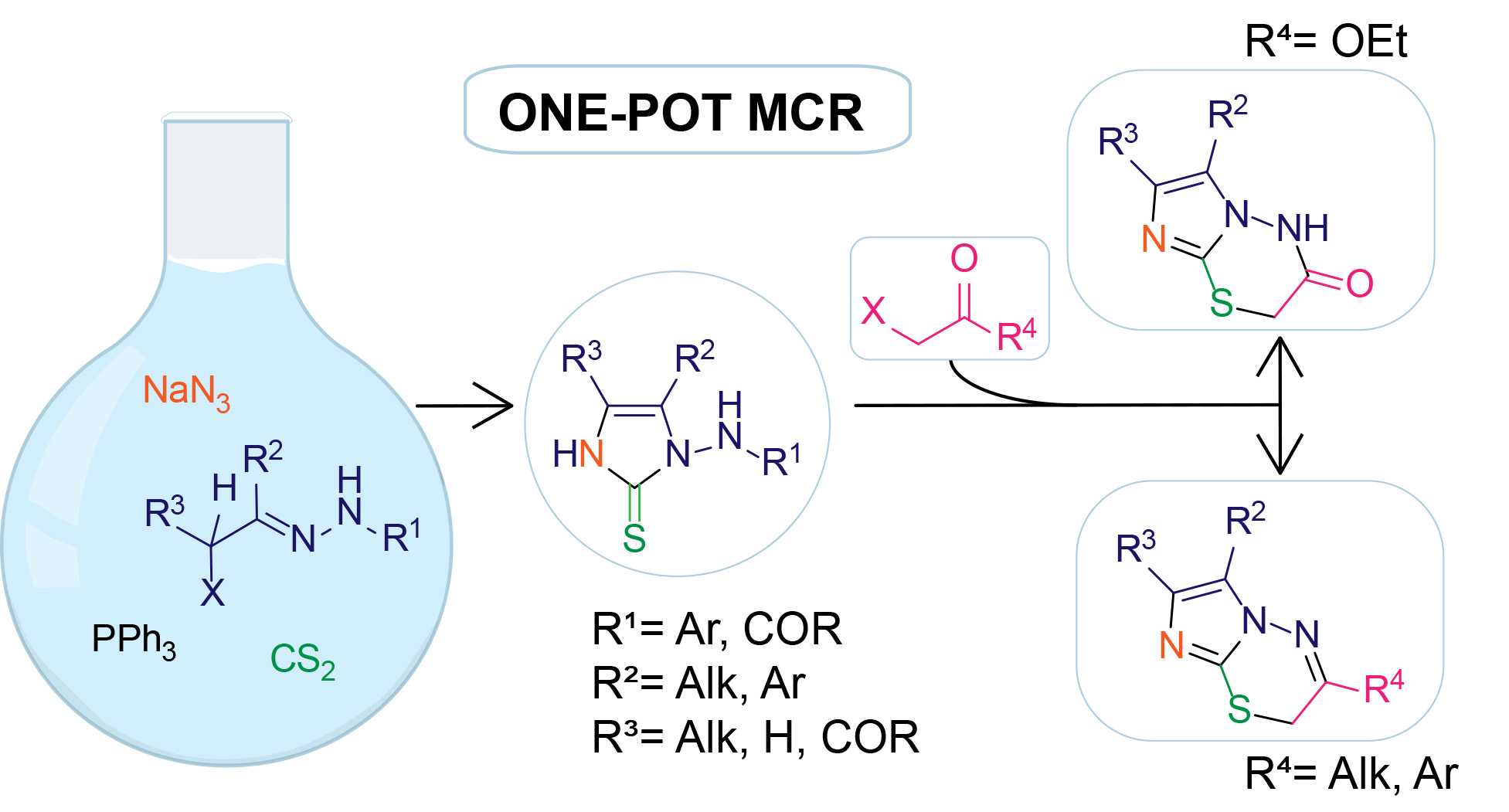
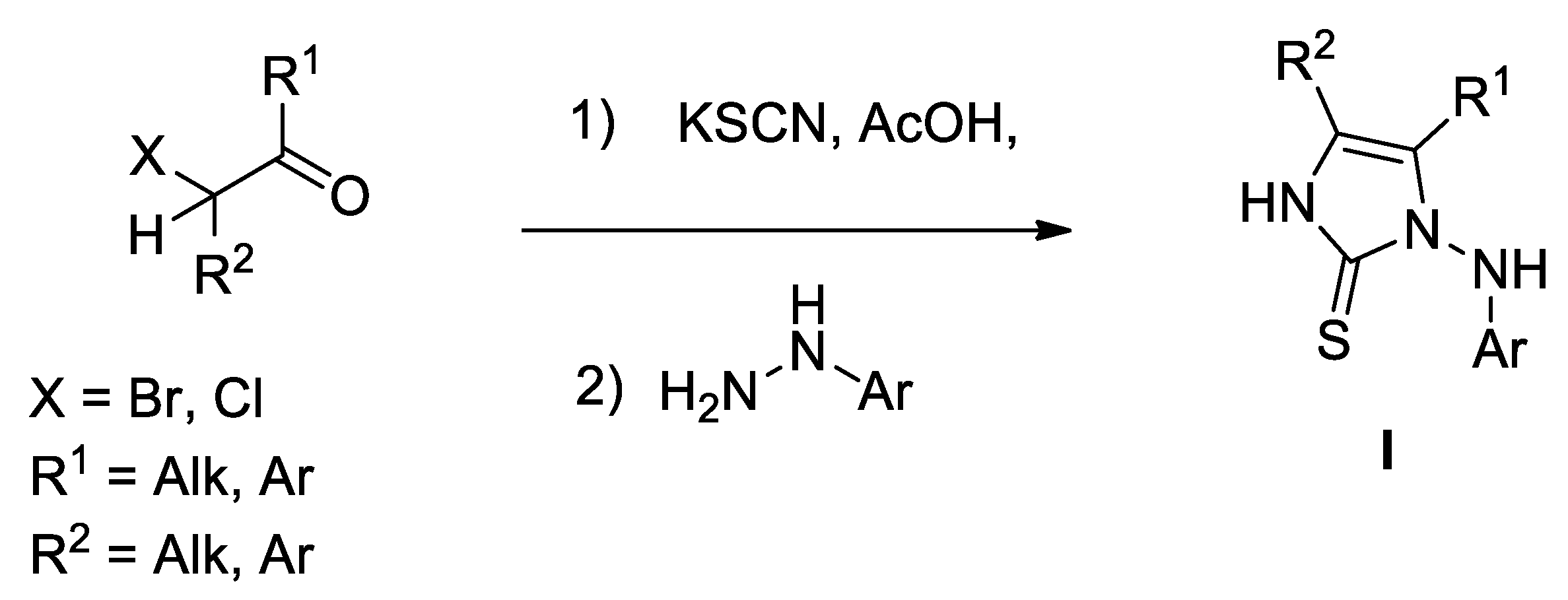
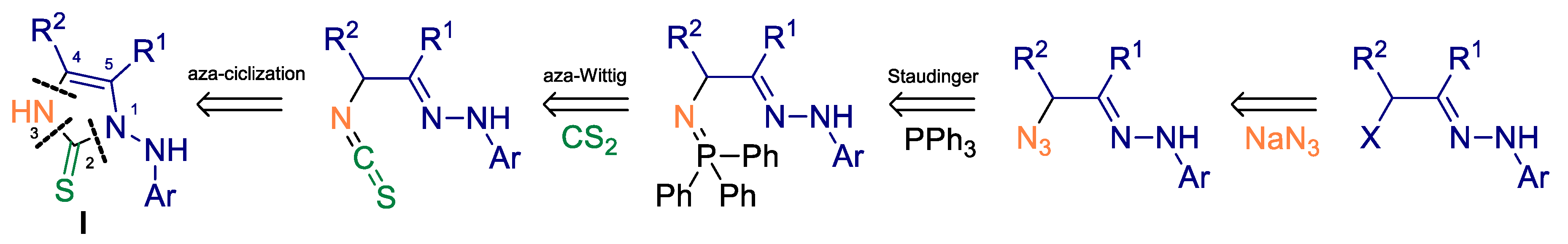
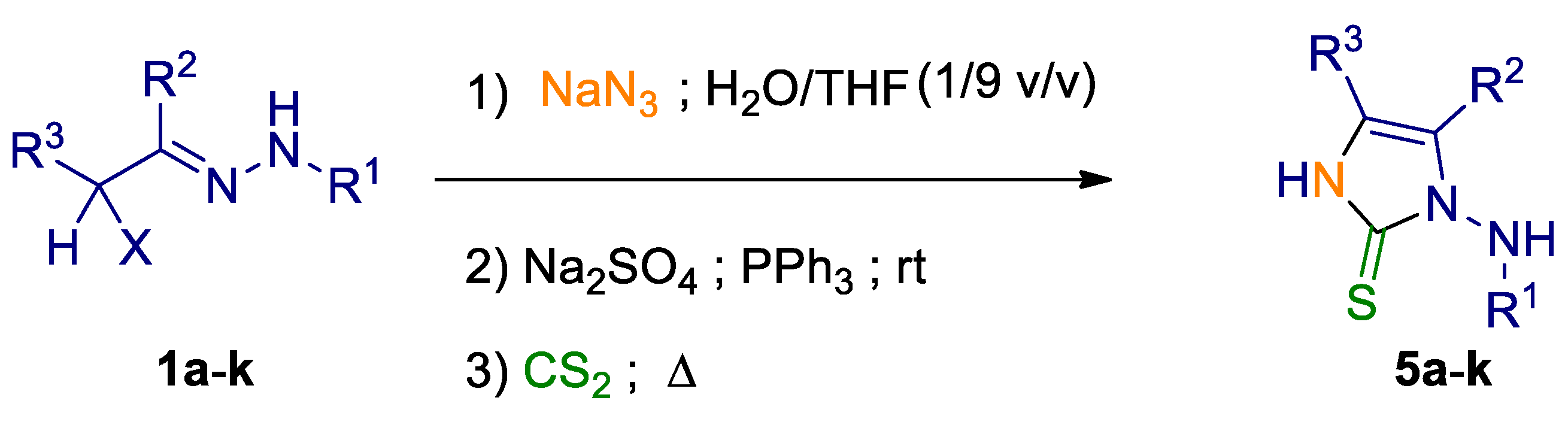
2. Results and Discussion
3. Experimental Section
3.1. General
3.2. Step-By-Step Synthetic Method for 5a
3.2.1. Synthesis of tert-butyl 2-(3-azido-4-(dimethylamino)-4-oxobutan-2-ylidene)hydrazinecarboxylate (2a)
3.2.2. Synthesis of tert-butyl 2-(4-(dimethylamino)-4-oxo-3-((triphenylphosphoranylidene)amino)butan-2-ylidene)hydrazinecarboxylate (3a)
3.2.3. Synthesis of tert-butyl (4-(dimethylcarbamoyl)-5-methyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)carbamate (5a)
3.3. Typical MCR Procedure for the Synthesis of N-Substituted 1-Amino-1H-Imidazole-2(3H)-Thione Derivatives 5a–k
3.4. General Procedure for the Synthesis of α-(Imidazol-2-Ylthio) Carbonyl Compounds 7a–c.
3.5. General Procedure for the Synthesis of N-Bridgeheaded Heterobicyclic Derivatives 8a–c.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uçucu, O.; Karaburun, N.G.; Işikdağ, I. Synthesis and analgesic activity of some 1-benzyl-2-substituted-4,5-diphenyl-1H-imidazole derivatives. Il Farmaco 2001, 56, 285–290. [Google Scholar] [CrossRef]
- Rani, N.; Sharma, A.; Gupta, G.K.; Singh, R. Imidazoles as potential antifungal agents: A review. Mini-Rev. Med. Chem. 2013, 13, 1626–1655. [Google Scholar] [CrossRef] [PubMed]
- Khalafi-Nezhad, A.; Rad, M.N.S.; Mohabatkar, H.; Asrari, Z.; Hemmateenejad, B. Design, synthesis, antibacterial and QSAR studies of benzimidazole and imidazole chloroaryloxyalkyl derivatives. Bioorg. Med. Chem. 2005, 13, 1931–1938. [Google Scholar] [CrossRef]
- Kerru, N.; Bhaskaruni, S.V.H.S.; Gummidi, L.; Maddila, S.N.; Maddila, S.; Jonnalagadda, S.B. Recent advances in heterogeneous catalysts for the synthesis of imidazole derivatives. Synthetic Commun. 2019, 49, 2437–2459. [Google Scholar] [CrossRef]
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. MedChemComm 2017, 8, 1742–1773. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, V.; Kharb, R.; Kumar, S.; Sharma, P.C.; Pathak, D.P. Imidazole derivatives as potential therapeutic agents. Curr. Pharm. Des. 2016, 22, 3265–3301. [Google Scholar] [CrossRef]
- Bellina, F.; Cauteruccio, S.; Rossi, R. Synthesis and biological activity of vicinal diaryl-substituted 1H-imidazoles. Tetrahedron 2007, 63, 4571–4624. [Google Scholar] [CrossRef]
- Savjani, J.K.; Gajjar, A.K. Pharmaceutical importance and synthetic strategies for imidazolidine-2-thione and imidazole-2-thione derivatives. Pak. J. Biol. Sci. 2011, 14, 1076–1089. [Google Scholar] [CrossRef]
- Isaia, F.; Aragoni, M.C.; Arca, M.; Demartin, F.; Devillanova, F.A.; Floris, G.; Garau, A.; Hursthouse, M.B.; Lippolis, V.; Medda, R.; et al. Interaction of methimazole with I2: X-ray crystal structure of the charge transfer complex methimazole-I2. implications for the mechanism of action of methimazole-based antithyroid drugs. Med. Chem. 2008, 51, 4050–4053. [Google Scholar] [CrossRef]
- Cesarini, S.; Spallarossa, A.; Ranise, A.; Schenone, S.; Rosano, C.; La Colla, P.; Sanna, G.; Busonera, B.; Loddo, R. N-acylated and N,N’-diacylated imidazolidine-2-thione derivatives and N,N’-diacylated tetrahydropyrimidine-2(1H)-thione analogues: Synthesis and antiproliferative activity. Eur. J. Med. Chem. 2009, 44, 1106–1118. [Google Scholar] [CrossRef]
- Sheppeck, J.; Gilmore, J.L. Substituted 1.3-dihydro-imidazol-2-one and 1.3-dihydro-imidazol-2-thione derivatives as inhibitors of matrix metallo proteinases and/or TNF-α converting enzyme (TACE). U.S. Patent 20050075384, 7 April 2005. [Google Scholar]
- Lagoja, I.M.; Pannecouque, C.; Van Aerschot, A.; Witvrouw, M.; Debyser, Z.; Balzarini, J.; Herdewijn, P.; De Clercq, E.J. N-Aminoimidazole derivatives inhibiting retroviral replication via a yet unidentified mode of action. Med. Chem. 2003, 46, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Schantl, J.G.; Prean, M. Addition products of hydrazine derivatives to azo-alkenes, part V: The reaction of α-(1-phenylhydrazino)alkanone phyenylhydrazones with acids and acid derivatives. Monatsh. Chem. 1993, 124, 299–308. [Google Scholar] [CrossRef]
- Schantl, J.G.; Kahlig, H.; Preans, M. 1-Arylamino-2,3-dihydro-1H-imidazole-2-thiones from the reaction of 1-[2-(2-arylhydrazono)alkyl]pyridinium iodides with potassium thiocyanate. Heterocycles 1994, 37, 1873–1878. [Google Scholar] [CrossRef]
- Schantl, J.G.; Lagoia, I. Direct synthetic approach to N-Substituted 1-amino-2,3-dihydro-1H-imidazole-2-thiones. Heterocycles 1997, 45, 691–700. [Google Scholar] [CrossRef]
- Schantl, J.; Nádenik, P. Tandem [3+2] cycloaddition reaction of azo-alkenes and thiocyanic acid: Extending the scope of the classical “criss-cross” cycloaddition reaction. Synlett 1998, 786–788. [Google Scholar] [CrossRef]
- Yurttaş, L.; Ertas, M.; Gulsen, A.C.; Temel, H.E.; Demirayak, Ş. Novel benzothiazole based imidazole derivatives as new cytotoxic agents against glioma (C6) and liver (HepG2) cancer cell lines. Acta Pharm. Sci. 2017, 55, 39–47. [Google Scholar] [CrossRef]
- Neochoritis, C.; Tsoleridis, C.A.; Stephanidou-Stephanatou, J. 1-Arylaminoimidazole-2-thiones as intermediates in the synthesis of imidazo[2,1-b][1,3,4]thiadiazines. Tetrahedron 2008, 64, 3527–3533. [Google Scholar] [CrossRef]
- Grimmett, M.R. Imidazoles. In Science of Synthesis; Neier, D., Bellus, D., Eds.; G. Thieme Verlag: Stuttgart, Germany, 2002; Volume 12, pp. 325–328. [Google Scholar] [CrossRef]
- Schantl, J.G. Azomethine imines. In Science of Synthesis; Padwa, A., Ed.; G. Thieme Verlag: Stuttgart, Germany, 2004; Volume 27, pp. 731–738. [Google Scholar] [CrossRef]
- Schantl, J.G. Cyclic azomethine imines from diazenes (azo compounds). In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: Oxford, UK; Volume 99, pp. 185–207. [CrossRef]
- Attanasi, O.A.; Favi, G.; Filippone, P.; Perrulli, F.R.; Santeusanio, S. Direct access to variously substituted 2-imino-4-thiazolines. Synlett 2010, 1859–1861. [Google Scholar] [CrossRef]
- Attanasi, O.A.; Bartoccini, S.; Favi, G.; Filippone, P.; Perrulli, F.R.; Santeusanio, S. Tandem aza-Wittig/carbodiimide-mediated annulation applicable to 1,2-diaza-1,3-dienes for the one-pot synthesis of fully substituted 1,2-diaminoimidazoles. J. Org. Chem. 2012, 77, 9338–9343. [Google Scholar] [CrossRef]
- Xie, H.; Liu, J.-C.; Wu, L.; Ding, M.-W. Unexpected synthesis of 2,4,5-trisubstituted oxazoles via a tandem aza-Wittig/Michael/isomerization reaction of vinyliminophosphorane. Tetrahedron 2012, 68, 7984–7990. [Google Scholar] [CrossRef]
- Santhosh, L.; Durgamma, S.; Sureshbabu, V.V. Staudinger/aza-Wittig reaction to access Nβ-protected amino alkyl isothiocyanates. Org. Biomol. Chem. 2018, 16, 4874–4880. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.M.M.; Lemos, A.; Pinho e Melo, T.M.V.D. Reactivity of dipyrromethanes towards azoalkenes: Synthesis of functionalized dipyrromethanes, calix[4]pyrroles, and bilanes. Eur. J. Org. Chem. 2014, 7039–7048. [Google Scholar] [CrossRef]
- Attanasi, O.A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F.R.; Santeusanio, S. Cultivating the passion to build heterocycles from 1,2-diaza-1,3-dienes: The force of imagination. Eur. J. Org. Chem. 2009, 3109–3127. [Google Scholar] [CrossRef]
- Attanasi, O.A.; De Crescentini, L.; Giorgi, R.; Perrone, A.; Santeusanio, S. Synthesis of 3-unsubstituted-1-aminopyrroles. Heterocycles 1996, 43, 1447–1457. [Google Scholar] [CrossRef]
- Attanasi, O.A.; Favi, G.; Mantellini, F.; Mantenuto, S.; Moscatelli, G.; Nicolini, S. Regioselective formation of 5-methylene-6-methoxy-1,4,5,6-tetrahydropyridazines from the [4+2]-cycloaddition reaction of in situ generated 1,2-diaza-1,3-dienes with methoxyallene. Synlett 2015, 193–196. [Google Scholar] [CrossRef]
- Attanasi, O.A.; De Crescentini, L.; Favi, G.; Mantellini, F.; Mantenuto, S.; Nicolini, S. Interceptive [4+1] annulation of in situ generated 1,2-diaza-1,3-dienes with diazo esters: Direct access to substituted mono-, bi-, and tricyclic 4,5-dihydropyrazoles. J. Org. Chem. 2014, 79, 8331–8338. [Google Scholar] [CrossRef]
- Attanasi, O.A.; Serra-Zanetti, F.; Zhiyuan, L. Easy one-pot conversion of 2-chlorohydrazone into 2-oxohydrazone derivatives via 2-azidohydrazone intermediates. Tetrahedron 1992, 48, 2785–2792. [Google Scholar] [CrossRef]
- Herrera, R.P.; Marqués-Lopez, E. Multicomponent Reactions: Concepts and Applications for Design and Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Preeti; Singh, K.N. Multicomponent reactions: A sustainable tool to 1,2- and 1,3-azoles. Org. Biomol. Chem. 2018, 16, 9084–9116. [Google Scholar] [CrossRef] [PubMed]
- Yurttaş, L.; Duran, M.; Demirayak, S.; Gençer, H.K.; Tunali, Y. Synthesis and initial biological evaluation of substituted 1-phenylamino-2-thio-4,5-dimethyl-1H-imidazole derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 6764–6768. [Google Scholar] [CrossRef]
- Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; de los Santos, J.M. The aza-Wittig reaction: An efficient tool for the construction of carbon–nitrogen double bonds. Tetrahedron 2007, 63, 523–575. [Google Scholar] [CrossRef]
- Eguchi, S. Recent progress in the synthesis of heterocyclic natural products by the Staudinger/intramolecular aza-Wittig reaction. ARKIVOC 2005, (ii), 98–119. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P. Application of iminophosphorane-based methodologies for the synthesis of natural products. Synlett 2004, 1–17. [Google Scholar] [CrossRef]
- Pavlova, A.S.; Ianova, O.A.; Chagarovskij, A.O.; Stebunov, N.S.; Orlov, N.V.; Shumsky, A.N.; Budynina, E.M.; RybaKov, V.B.; Trushkov, I.V. Domino Staudinger/aza-Wittig/Mannich reaction: An approach to diversity of di- and tetrahydropyrrole scaffolds. Chem. Eur. J. 2016, 22, 17967–17971. [Google Scholar] [CrossRef]
- Xiong, J.; Wei, X.; Ding, M.-W. New Facile synthesis of 2-alkylthiopyrimidin-4(3H)-ones by tandem aza-Wittig reaction starting from the Baylis–Hillman adducts. Synlett 2017, 1075–1078. [Google Scholar] [CrossRef]
- Agami, C.; Couty, F. The reactivity of the N-Boc protecting group: An underrated feature. Tetrahedron 2002, 58, 2701–2724. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd ed.; Wiley: New York, NY, USA, 1999. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Amberlyst 15, a superior, mild, and selective catalyst for carbonyl regeneration from nitrogeneous derivatives. J. Chem. Soc. Perkin Trans. 1 1988, 2563–2565. [Google Scholar] [CrossRef]
- Sasaki, T.; Ito, E.; Shimizu, I. Ring transformations of oxazoles and their benzo analogues. New synthetic route for 2H-imidazo[2,1-b][1,3,4]thiadiazine and N-heteroaryl-o-aminophenol. Heterocycles 1982, 19, 2119–2129. [Google Scholar] [CrossRef]
- Qi, L.-W.; Mao, J.-H.; Zhang, J.; Tan, B. Organocatalytic asymmetric arylation of indoles enabled by azo groups. Nat. Chem. 2018, 10, 58–64. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 5a–k are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | α-Halohydrazone 1 |  | One-Pot MCR Yield (%) a, b | ||||
|---|---|---|---|---|---|---|---|
| 5 | |||||||
| R1 | R2 | R3 | X | ||||
| 1 | 1a | CO2But | Me | CON(Me)2 | Cl | 5a | 25 a; 79 b |
| 2 | 1b | CONHPh | Me | CON(Me)2 | Cl | 5b | 53 b |
| 3 | 1c | CO2But | Me | CON(Et)2 | Br | 5c | 72 b |
| 4 | 1d | CO2But | Me | H | Cl | 5d | 69 b |
| 5 | 1e | CO2But | Me | CONH2 | Br | 5e | 58 b |
| 6 | 1f | CO2But | Me | CONHPh | Br | 5f | 67 b |
| 7 | 1g | CONHPh | Me | H | Cl | 5g | 82 b |
| 8 | 1h | COPh | Me | H | Cl | 5h | 59 b |
| 9 | 1i | CONHPh | Me | Me | Cl | 5i | 85 b |
| 10 | 1j | 4-NO2-Ph | Me | Me | Cl | 5j | 84 b |
| 11 | 1k | CO2But | Ph | H | Br | 5k | 66 b |
| 5 | 6 | 7 | Yield (%) a | 8 | Yield (%) b | ||||
|---|---|---|---|---|---|---|---|---|---|
| R2 | R3 | X | R4 | ||||||
| 5c | Me | CON(Et)2 | 6a | Br | Ph | 7a | 84 | 8a | 82 |
| 5d | Me | H | 6b | Cl | Me | 7b | 93 | 8b | 65 |
| 5f | Me | CONHPh | 6c | Br | OEt | 7c | 92 | 8c | 74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccolini, C.; Mari, G.; Favi, G.; Mantellini, F.; De Crescentini, L.; Santeusanio, S. Sequential MCR via Staudinger/Aza-Wittig versus Cycloaddition Reaction to Access Diversely Functionalized 1-Amino-1H-Imidazole-2(3H)-Thiones. Molecules 2019, 24, 3785. https://doi.org/10.3390/molecules24203785
Ciccolini C, Mari G, Favi G, Mantellini F, De Crescentini L, Santeusanio S. Sequential MCR via Staudinger/Aza-Wittig versus Cycloaddition Reaction to Access Diversely Functionalized 1-Amino-1H-Imidazole-2(3H)-Thiones. Molecules. 2019; 24(20):3785. https://doi.org/10.3390/molecules24203785
Chicago/Turabian StyleCiccolini, Cecilia, Giacomo Mari, Gianfranco Favi, Fabio Mantellini, Lucia De Crescentini, and Stefania Santeusanio. 2019. "Sequential MCR via Staudinger/Aza-Wittig versus Cycloaddition Reaction to Access Diversely Functionalized 1-Amino-1H-Imidazole-2(3H)-Thiones" Molecules 24, no. 20: 3785. https://doi.org/10.3390/molecules24203785
APA StyleCiccolini, C., Mari, G., Favi, G., Mantellini, F., De Crescentini, L., & Santeusanio, S. (2019). Sequential MCR via Staudinger/Aza-Wittig versus Cycloaddition Reaction to Access Diversely Functionalized 1-Amino-1H-Imidazole-2(3H)-Thiones. Molecules, 24(20), 3785. https://doi.org/10.3390/molecules24203785