
Improved Access to Chiral Tetranaphthoazepinium-Based Organocatalysts Using Aqueous Ammonia as Nitrogen Source

Abstract
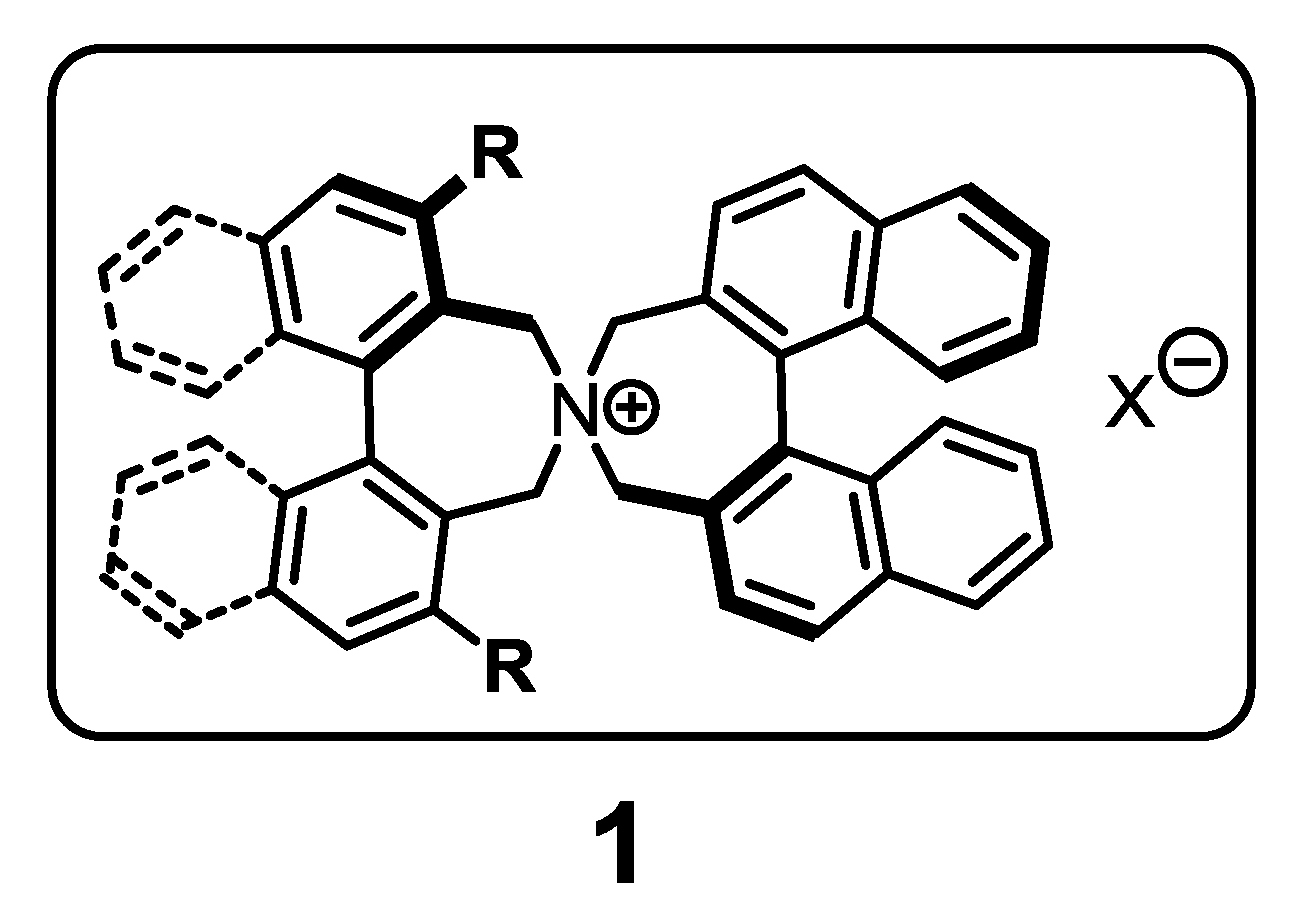
:1. Introduction
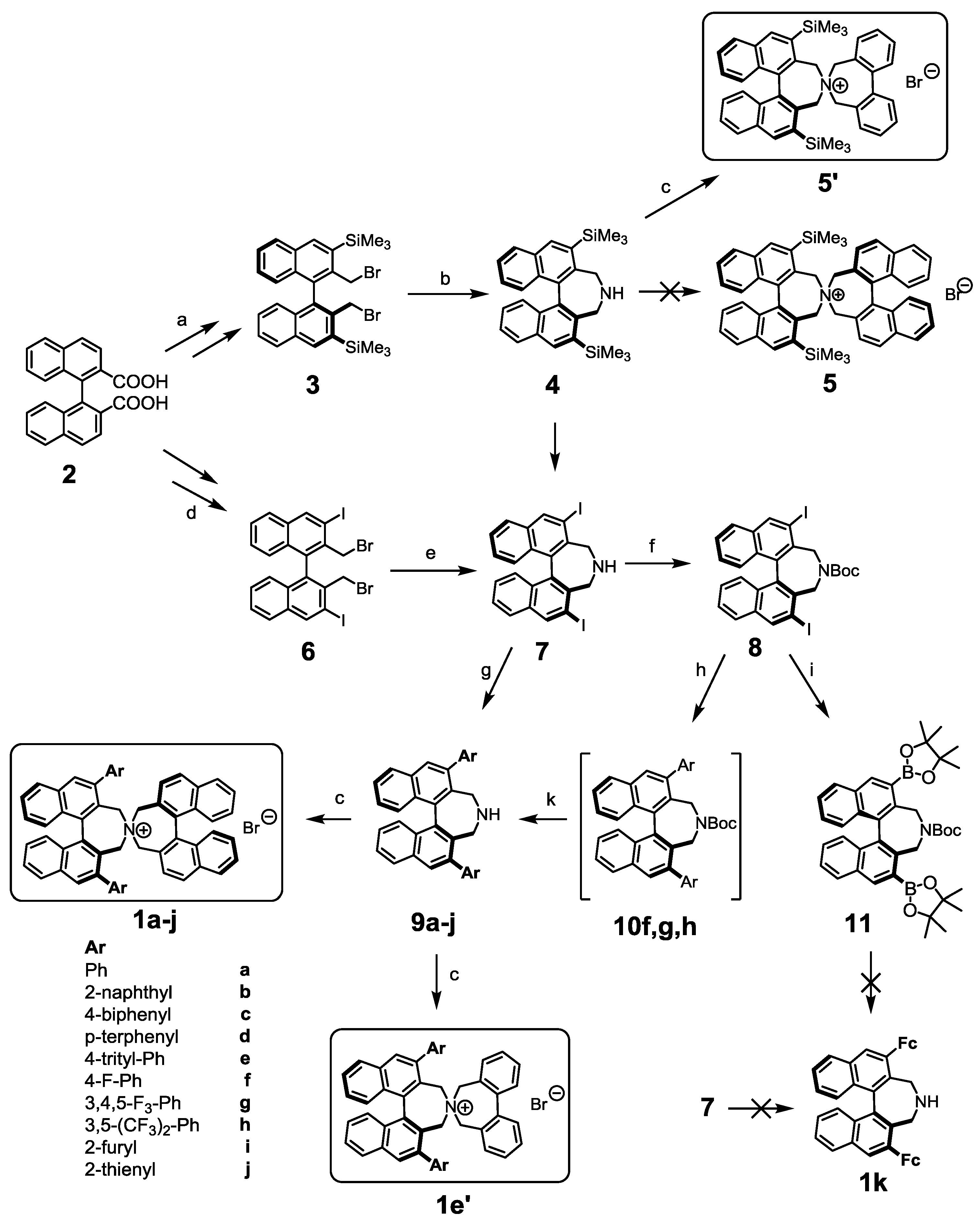
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis
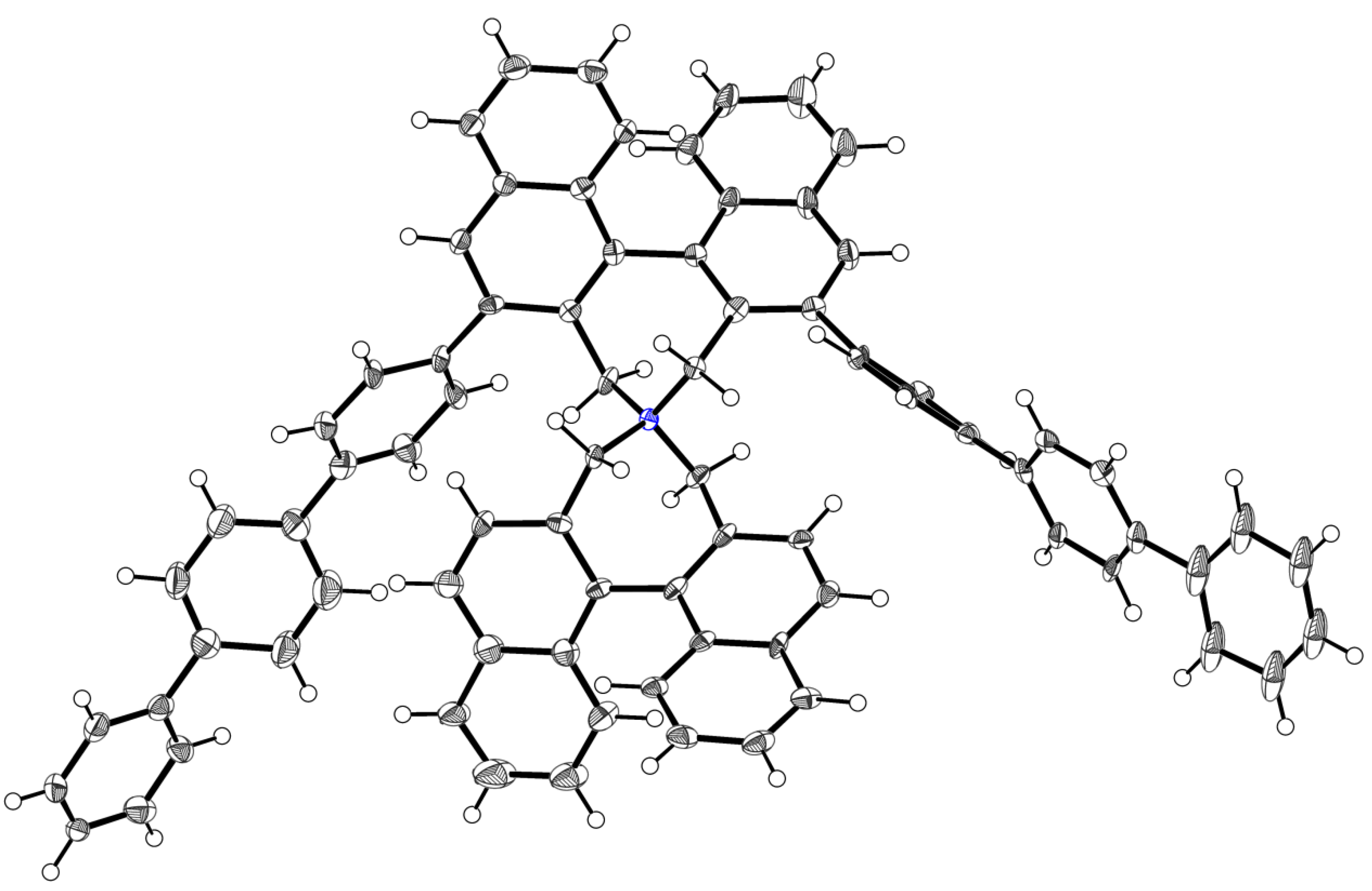
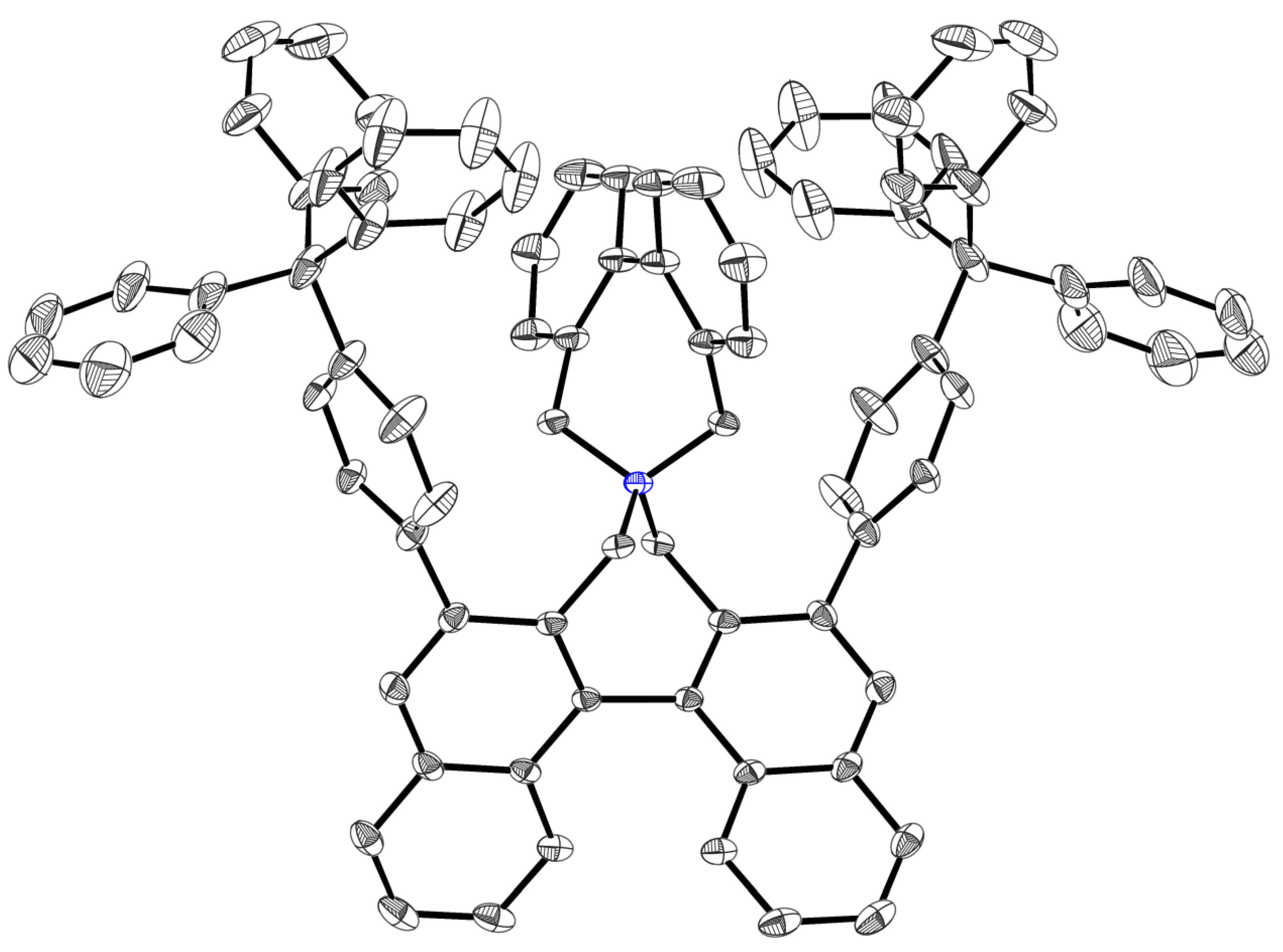
3.3. X-ray Structure Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Wabnitz, T.; Reiser, O. Binaphthyls: Universal Ligands for Catalysis, in Organic Synthesis Highlights IV; Schmalz, H.-G., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2000; Chapter 23; pp. 155–165. [Google Scholar]
- Pereira, M.M.; Calvete, M.J.F.; Carrilho, R.M.B.; Abreu, A.R. Synthesis of binaphthyl based phosphine and phosphite ligands. Chem. Soc. Rev. 2013, 42, 6990–7027. [Google Scholar] [CrossRef] [PubMed]
- Bhadury, P.S.; Yao, Y.; He, Y. Organocatalytic application of axially dissymmetric BINOLs and their conversion into binaphthyl phosphoric acids. Curr. Org. Chem. 2012, 16, 1730–1753. [Google Scholar] [CrossRef]
- Ding, K.; Li, X.; Ji, B.; Guo, H.; Kitamura, M. Ten years of research on NOBIN chemistry. Curr. Org. Synth. 2005, 2, 499–545. [Google Scholar] [CrossRef]
- Brunel, J.M. BINOL: A Versatile Chiral Reagent. Chem. Rev. 2005, 105, 857–897. [Google Scholar] [CrossRef]
- Telfer, S.G.; Kuroda, R. 1,1’-Binaphthyl-2,2’-diol and 2,2’-diamino-1,1’-binaphthyl: Versatile frameworks for chiral ligands in coordination and metallosupramolecular chemistry. Coord. Chem. Rev. 2003, 242, 33–46. [Google Scholar] [CrossRef]
- Kocovsky, P.; Vyskocil, S.; Smrcina, M. Non-Symmetrically Substituted 1,1’-Binaphthyls in Enantioselective Catalysis. Chem. Rev. 2003, 103, 3213–3245. [Google Scholar] [CrossRef]
- Pu, L. Synthesis and study of binaphthyl-based chiral dendrimers. J. Photochem. Photobiol. A: Chem. 2003, 155, 47–55. [Google Scholar] [CrossRef]
- Kaneko, S.; Kumatabara, Y.; Shirakawa, S. A new generation of chiral phase-transfer catalysts. Org. Biomol. Chem. 2016, 14, 5367–5376. [Google Scholar] [CrossRef] [Green Version]
- Maruoka, K. Practical Aspects of Recent Asymmetric Phase-Transfer Catalysis. Organic Process Research & Development 2008, 12, 679–697. [Google Scholar]
- Ooi, T.; Maruoka, K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef]
- Shirakawa, S.; Wang, L.; He, R.; Arimitsu, S.; Maruoka, K. A Base-Free Neutral Phase-Transfer Reaction System. Chem. Asian J. 2014, 9, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Hayashi, H.; Kazuaki, I. High-turnover hypoiodite catalysis for asymmetric synthesis of tocopherols. Science 2014, 345, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Hayashi, Y.; Maruoka, M. Construction of a Chiral Quaternary Carbon Center by CatalyticAsymmetric Alkylation of 2-Arylcyclohexanones under Phase-Transfer Conditions. J. Am. Chem. Soc. 2013, 135, 7134–7137. [Google Scholar] [CrossRef] [PubMed]
- Hua, M.-Q.; Cui, H.-F.; Wang, L.; Nie, J.; Ma, J.-A. Reversal of Enantioselectivity by Tuning the Conformational Flexibility of Phase-Transfer Catalysts. Angew. Chem. Int. Ed. 2010, 49, 2772–2776. [Google Scholar] [CrossRef]
- Wang, X.; Lan, Q.; Shirakawa, S.; Maruoka, K. Chiral bifunctional phase transfer catalysts for asymmetric fluorination of β-keto esters. Chem. Commun. 2010, 46, 321–323. [Google Scholar] [CrossRef]
- Uyanik, M.; Okamoto, H.; Yasui, T.; Ishihara, K. Quaternary Ammonium (Hypo)iodite Catalysis for Enantioselective Oxidative Cycloetherification. Science 2010, 328, 1376–1379. [Google Scholar] [CrossRef]
- Arakawa, Y.; Haraguchi, N.; Itsuno, S. An Immobilization Method of Chiral Quaternary Ammonium Salts onto Polymer Supports. Angew. Chem. Int. Ed. 2008, 47, 8232–8235. [Google Scholar] [CrossRef]
- Wang, X.; Kitamura, M.; Maruoka, K. New, Chiral Phase Transfer Catalysts for Effecting Asymmetric Conjugate Additions of α-Alkyl-α-cyanoacetates to Acetylenic Esters. J. Am. Chem. Soc. 2007, 129, 1038–1039. [Google Scholar] [CrossRef]
- Ooi, T.; Uematsu, Y.; Kameda, M.; Maruoka, K. Asymmetric phase-transfer catalysis of homo- and heterochiral quaternary ammonium salts: Development and application of conformationally flexible chiral phase-transfer catalysts. Tetrahedron 2006, 62, 11425–11436. [Google Scholar] [CrossRef]
- Kitamura, M.; Shirakawa, S.; Maruoka, K. Powerful Chiral Phase-Transfer Catalysts for the Asymmetric Synthesis of α-Alkyl- and α,α-Dialkyl-α-amino Acids. Angew. Chem. Int. Ed. 2005, 44, 1549–1551. [Google Scholar] [CrossRef]
- Ooi, T.; Takeuchi, M.; Kato, D.; Uematsu, Y.; Tayama, E.; Sakai, D.; Maruoka, K. Highly Enantioselective Phase-Transfer-Catalyzed Alkylation of Protected α-Amino Acid Amides toward Practical Asymmetric Synthesis of Vicinal Diamines, α-Amino Ketones, and α-Amino Alcohols. J. Am. Chem. Soc. 2005, 127, 5073–5083. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Ohara, D.; Tamura, M.; Maruoka, K. Design of New Chiral Phase-Transfer Catalysts with Dual Functions for Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2004, 126, 6844–6845. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Takahashi, M.; Doda, K.; Maruoka, K. Asymmetric Induction in the Neber Rearrangement of Simple Ketoxime Sulfonates under Phase-Transfer Conditions: Experimental Evidence for the Participation of an Anionic Pathway. J. Am. Chem. Soc. 2002, 124, 7640–7641. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Kameda, M.; Maruoka, K. Design of N-Spiro C2-Symmetric Chiral Quaternary Ammonium Bromides as Novel Chiral Phase-Transfer Catalysts: Synthesis and Application to Practical Asymmetric Synthesis of α Amino Acids. J. Am. Chem. Soc. 2003, 125, 5139–5151. [Google Scholar] [CrossRef]
- Ooi, T.; Uematsu, Y.; Maruoka, K. New, Improved Procedure for the Synthesis of Structurally Diverse N-Spiro C2-Symmetric Chiral Quaternary Ammonium Bromides. J. Org. Chem. 2003, 68, 4576–4578. [Google Scholar] [CrossRef]
- Kitamura, M.; Arimura, Y.; Shirakawa, S.; Maruoka, K. Combinatorial approach for the design of new, simplified chiral phase-transfer catalysts with high catalytic performance for practical asymmetric synthesis of α-alkyl-α-amino acids. Tetrahedron Lett. 2008, 49, 2026–2030. [Google Scholar] [CrossRef]
- Kitamura, M.; Shirakawa, S.; Arimura, Y.; Wang, X.; Maruoka, K. Combinatorial Design of Simplified High-Performance Chiral Phase-Transfer Catalysts for Practical Asymmetric Synthesis of α-Alkyl- and α,α-Dialkyl-α-Amino Acids. Chem. Asian J. 2008, 3, 1702–1714. [Google Scholar] [CrossRef]
- Tharamak, S.; Knittl-Frank, C.; Manaprasertsak, A.; Pengsook, A.; Suchy, L.; Schuller, P.; Happl, B.; Roller, A.; Widhalm, M. Economy of Catalyst Synthesis—Convenient Access to Libraries of Di- and Tetranaphtho Azepinium Compounds. Molecules 2018, 23, 750. [Google Scholar] [CrossRef]
- Hayashi, T.; Hayashizaki, K.; Kiyoi, T.; Ito, Y. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition-metal complexes. 6. Practical asymmetric synthesis of 1,1’-binaphthyls via asymmetric cross-coupling with a chiral [(alkoxyalkyl)ferrocenyl]monophosphine/nickel catalyst. J. Am. Chem. Soc. 1988, 110, 8153–8156. [Google Scholar] [CrossRef]
- Egami, H.; Sato, K.; Asada, J.; Kawato, Y.; Hamashima, Y. Concise synthesis of binaphthol-derived chiral dicarboxylic acids. Tetrahedron 2015, 71, 6384–6388. [Google Scholar] [CrossRef]
- Konishi, H.; Hoshino, F.; Manabe, K. Practical Synthesis of Axially Chiral Dicarboxylates via Pd-Catalyzed External-CO-Free Carbonylation. Chem. Pharm. Bull. 2016, 64, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Ito, M.; Inagaki, K.; Takaya, H. A convenient synthesis of optically pure dimethyl 1,1′-binaphthalene-2,2′-dicarboxylate from 1,1′-binaphthalene-2,2′-diol. Tetrahedron Lett. 1993, 10, 1615–1616. [Google Scholar] [CrossRef]
- For example see:Oi, S.; Matsunaga, K.; Hattori, T.; Miyano, S. Convenient Synthesis of 1,1′-Binaphthyl-2.2′-dicarboxylic Acid. Synthesis 1993, 895–898. [Google Scholar] [CrossRef]
- Berger, M. Novel Binaphthyl Auxiliaries and Their Application in Asymmetric Intermolecular Bromofunctionalisation of Olefins. Master Thesis, University of Vienna, Vienna, 2014. [Google Scholar]
- Vial, L.; Lacour, J. Conformational Preference and Configurational Control of Highly Symmetric Spirobi[dibenzazepinium] Cation. Org. Lett. 2002, 4, 3939–3942. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Lan, Q.; Wang, X.; Maruoka, K. Effects of Aromatic Substituents on Binaphthyl-Based Chiral Spiro-Type Ammonium Salts in Asymmetric Phase Transfer Reactions. Adv. Synth. Catal. 2007, 349, 556–560. [Google Scholar] [CrossRef]
- Shirakawa, S.; Ueda, M.; Tanaka, Y.; Hashimoto, T.; Maruoka, K. Design of Binaphthyl-Modified Symmetrical Chiral Phase-Transfer Catalysts: Substituent Effect of 4,4′,6,6′-Positions of Binaphthyl Rings in the Asymmetric Alkylation of a Glycine Derivative. Chem. Asian J. 2007, 2, 1276–1281. [Google Scholar] [CrossRef]
- Widhalm, M.; Abraham, M.; Arion, V.B.; Saarsalu, S.; Maeorg, U. A modular approach to a new class of phosphinohydrazones and their use in asymmetric allylic alkylation reactions. Tetrahedron: Asymmetry 2010, 21, 1971–1982. [Google Scholar] [CrossRef]
- Hawkins, J.M.; Fu, G.C. Asymmetric Michael Reactions of 3,5-Dihydro-4H-dinaphth[2,1-c:1’,2’-e]azepine with Methyl Crotonate. J. Org. Chem. 1986, 51, 2820–2822. [Google Scholar] [CrossRef]
- Applying similar conditions to 4,5-dihydro-3H-dinaphtho[2,1-c:1’,2’-e]azepine afforded enantiomerically enriched fractions of (R)-enantiomer from the less soluble salt (40%, 78% ee) and (S)-enantiomer from the mother liquor (40%, 78%ee). It is worth to note that two types of crystals form simultaneously with the (R)-enantiomer containing one or two molecules of dinaphthoazepine.
- The reversed sequence with 3,3′-diiodo-bisazepinium bromide (analog to structure 1 with iodo substituents instead of Ar) as a precursor for the Suzuki reactions was originally considered, too. But after several attempts it turned out that arylation afforded various products mainly triarylated species with cleavage of the C-N bond even when stoichiometric ammounts of arylboronic acids (2 equiv.) were applied [26].
- Shirakawa, S.; Maruoka, K. Chiral Onium Salts (Phase Transfer Reactions). In Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Dalko, P.I., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2013; Chapter 14; pp. 365–379. [Google Scholar]
- Tan, J.; Yasuda, N. Contemporary Asymmetric Phase Transfer Catalysis: Large-Scale Industrial Applications. Org. Process Res. Dev. 2015, 19, 1731–1746. [Google Scholar] [CrossRef]
- In the 1H-NMR spectra of crude mixtures and also during chromatography varying amounts of benzophenone have been often observed. For instance, when the product was kept in contact with SiO2 over 14 h at r.t. this resulted in formation of 40% of benzophenone. Also when of a solution of the substrate in toluene was kept for 4 weeks at 4 °C 4% of benzophenone was detected (NMR).
- Ooi, T.; Kameda, M.; Maruoka, K. Molecular Design of a C2-Symmetric Chiral Phase-Transfer Catalyst for Practical Asymmetric Synthesis of α-Amino Acids. J. Am. Chem. Soc. 1999, 121, 6519–6520. [Google Scholar] [CrossRef]
- Ooi, T.; Kameda, M.; Taniguchi, M.; Maruoka, K. Development of Highly Diastereo- and Enantioselective Direct Asymmetric Aldol Reaction of a Glycinate Schiff Base with Aldehydes Catalyzed by Chiral Quaternary Ammonium Salts. J. Am. Chem. Soc. 2004, 126, 9685–9694. [Google Scholar] [CrossRef] [PubMed]
- For a potential application of 9a–j in organocatalysis see Kano, T.; Sugimoto, H.; Tokuda, O.; Maruoka, K. Unusual anti-selective asymmetric conjugate addition of aldehydes to nitroalkenes catalyzed by a biphenylbased chiral secondary amine. Chem. Comm. 2013, 49, 7028–7030. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Yamada, S.; Kuroda, T.; Imashiro, R.; Shimizu, T. A Practical Synthesis of C2-Symmetric Chiral Binaphthyl Ketone Catalyst. Synthesis 2000, 1677–1680. [Google Scholar] [CrossRef]
- Mazaleyrat, J.-P. Synthesis and resolution of axially chiral C2-symmetric 1,1′-binaphthyl-substituted tetramethylethylenediamines. Tetrahedron: Asymmetry 1997, 8, 2709–2721. [Google Scholar] [CrossRef]
- Kanoh, S.; Hongoh, Y.; Motoi, M.; Suda, H. Convenient Optical Resolution of Axially Chiral 1,1′-Binaphthyl-2,2′-dicarboxylic Acid. Bull. Chem. Soc. Jpn. 1988, 61, 1032–1034. [Google Scholar] [CrossRef]
- Vadehra, G.S.; Jiang, X.; Dotson, J.J.; Chu, G.M.; Garcia-Garibay, M.A. High-Yielding and Divergent Paradigm for the Synthesis of D2h-Symmetric Octakis-Substituted Pentiptycenequinones. Org. Lett. 2017, 19, 1838–1841. [Google Scholar] [CrossRef]
- Akhatou, A.; Rahimi, M.; Chebou, K.; Ghosez, L.; Hanquet, G. Acid promoted enantioselective oxygen-atom transfer from N-alkyl binaphthyl-derived oxaziridines onto sulfides. Tetrahedron 2007, 63, 6232–6240. [Google Scholar] [CrossRef]
- Aquino, M.; Guerrero, M.D.; Bruno, I.; Terencio, M.C.; Paya, M.; Riccio, R. Development of a second generation of inhibitors of microsomal prostaglandin E synthase 1 expression bearing the γ-hydroxybutenolide scaffold. Bioorg. Med. Chem. 2008, 16, 9056–9064. [Google Scholar] [CrossRef]
- Guthrie, D.B.; Curran, D.P. Asymmetric Radical and Anionic Cyclizations of Axially Chiral Carbamates. Org. Lett. 2009, 11, 249–251. [Google Scholar] [CrossRef]
- Gagnon, H.; Beauchemin, S.; Kwiatkowska, A.; Couture, F.; D’Anjou, F.; Levesque, C.; Dufour, F.; Desbiens, A.R.; Vaillancourt, R.; Bernard, S.; et al. Optimization of Furin Inhibitors To Protect against the Activation of Influenza Hemagglutinin H5 and Shiga Toxin. J. Med. Chem. 2014, 57, 29–41. [Google Scholar] [CrossRef]
- Manning, P.T.; Misko, T.P. Preparation of Amino Acid Derivatives as Inhibitors of Inducible Nitric Oxide Synthase for Use in Combination Therapy with Alkylating Agents. PCT Int. Appl. 2005, 25620, A2. [Google Scholar]
Sample Availability: Not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| entry | cat. | yield/% b | yield/% c | ee/% d |
|---|---|---|---|---|
| 1 | 1ae | 78 | 74 | 86 |
| 2 | 1bf | 91 | 89 | 94 |
| 3 | 1c | 86 | 86 | 92 |
| 4 | 1d | 92 | 88 | 91 |
| 5 | 1e’ | 72 | 69 | rac. |
| 6 | 1fg | 82 | 79 | 94 |
| 7 | 1gh | 90 | 85 | 99 |
| 8 | 1h | 33 | 28 | 92 |
| 9 | 1i | 67 | 67 | 88 |
| 10 | 1j | 87 | 83 | 89 |
| 11 | 5′ | 92 | 91 | 61 |
| 14B | 14C | 14D | 14E | 14F | 14G | 14Hb | 14Ic | |
| cat. | y/ee d | y/ee d | y/ee d | y/ee d | y/ee d | y/ee d | y/ee d | y/ee d |
| 1c | 89/88 | 80/91 | 90/75 | 51/83 | 92/89 | 88/72 | 62/51 e | 33/57 f |
| 1d | 90/88 | 86/91 | 90/74 | 55/88 | 91/88 | 91/71 | 56/41 g | 26/51 f |
| 1g | 89/98 h | 71/95 i | 87/93 | 72/68 | 90/90 j | 81/83 | 61/79k | 23/83 f (33/88) |
| 1h | 70/98 | 45/52 | 61/84 | 66/14 | 30/90 | 83/73 | 60/80 | 20/83 i |
| (R)(R)-1d | (R)bina(S)biphe-1e | (R)-7 (S,S)-dibenzoyltartrate | |
|---|---|---|---|
| M [g/mol] | 1323.48 | 791.92 | 811.22 |
| Space group | P212121 | C2 | C2 |
| a [Å] | 9.1716(8) | 21.7990(14) | 28.7403(11) |
| b [Å] | 26.018(2) | 23.2291(13) | 12.0555(4) |
| c [Å] | 30.298(3) | 8.8684(5) | 8.9062(3) |
| α [°] | 90 | 90 | 90 |
| ß [°] | 90 | 94.251(2) | 101.095(2) |
| γ [°] | 90 | 90 | 90 |
| V [Å3] | 7229.8(11) | 4478.3(5) | 3028.14(18) |
| Z | 4 | 4 | 4 |
| Dcalc [g/cm3] | 1.216 | 1.175 | 1.779 |
| Rint | 0.1022 | 0.0294 | 0.0359 |
| Rsigma | 0.0399 | 0.0300 | 0.0175 |
| R1 (I > 2σ(I)) | 0.0812 | 0.0802 | 0.0362 |
| wR2 (all data) | 0.2433 | 0.2295 | 0.0897 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manaprasertsak, A.; Tharamak, S.; Schedl, C.; Roller, A.; Widhalm, M. Improved Access to Chiral Tetranaphthoazepinium-Based Organocatalysts Using Aqueous Ammonia as Nitrogen Source. Molecules 2019, 24, 3844. https://doi.org/10.3390/molecules24213844
Manaprasertsak A, Tharamak S, Schedl C, Roller A, Widhalm M. Improved Access to Chiral Tetranaphthoazepinium-Based Organocatalysts Using Aqueous Ammonia as Nitrogen Source. Molecules. 2019; 24(21):3844. https://doi.org/10.3390/molecules24213844
Chicago/Turabian StyleManaprasertsak, Auraya, Sorachat Tharamak, Christina Schedl, Alexander Roller, and Michael Widhalm. 2019. "Improved Access to Chiral Tetranaphthoazepinium-Based Organocatalysts Using Aqueous Ammonia as Nitrogen Source" Molecules 24, no. 21: 3844. https://doi.org/10.3390/molecules24213844