
Halogenating Enzymes for Active Agent Synthesis: First Steps Are Done and Many Have to Follow


Abstract
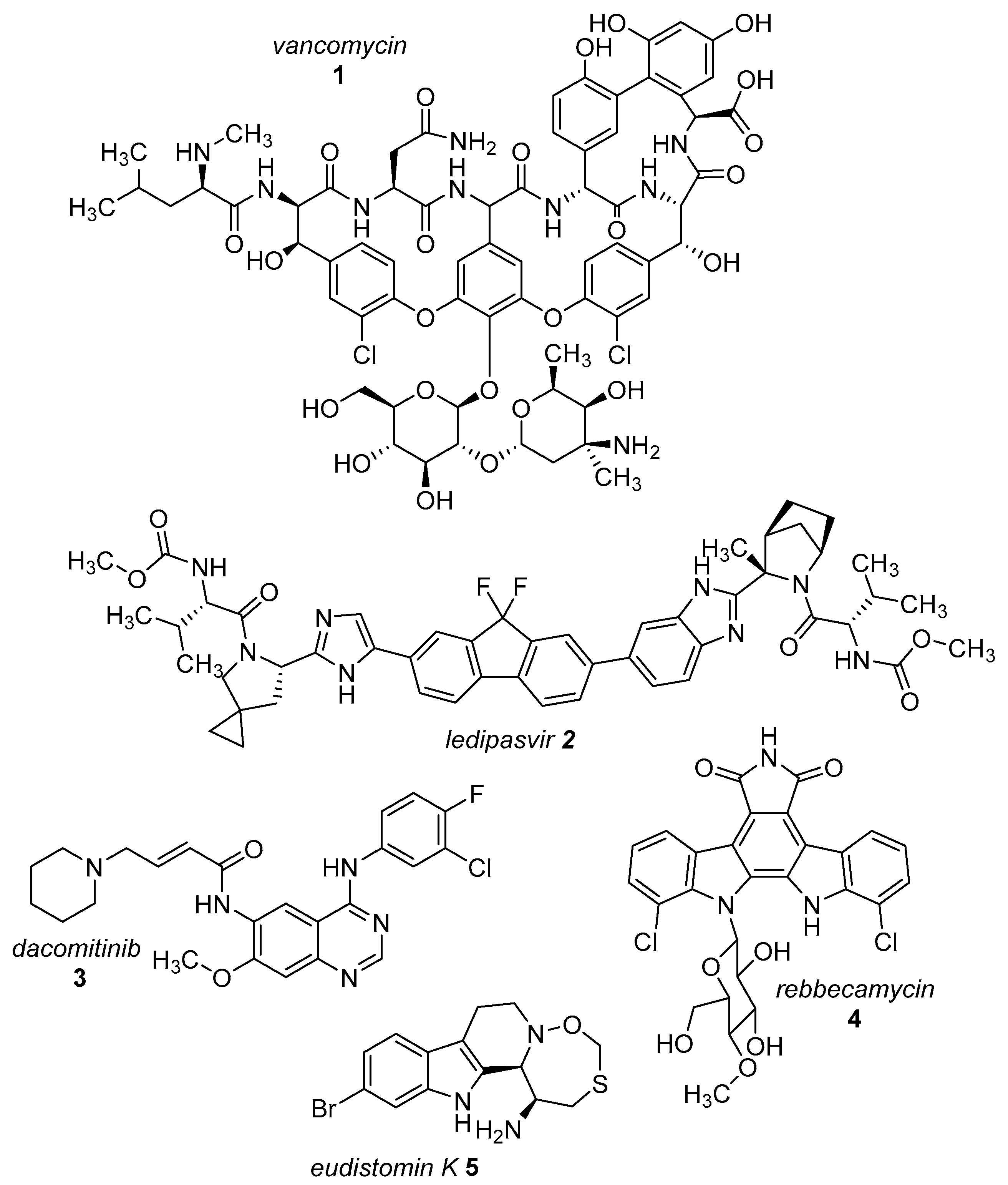
:1. Still Up-to-Date—Halogens in Active Agents
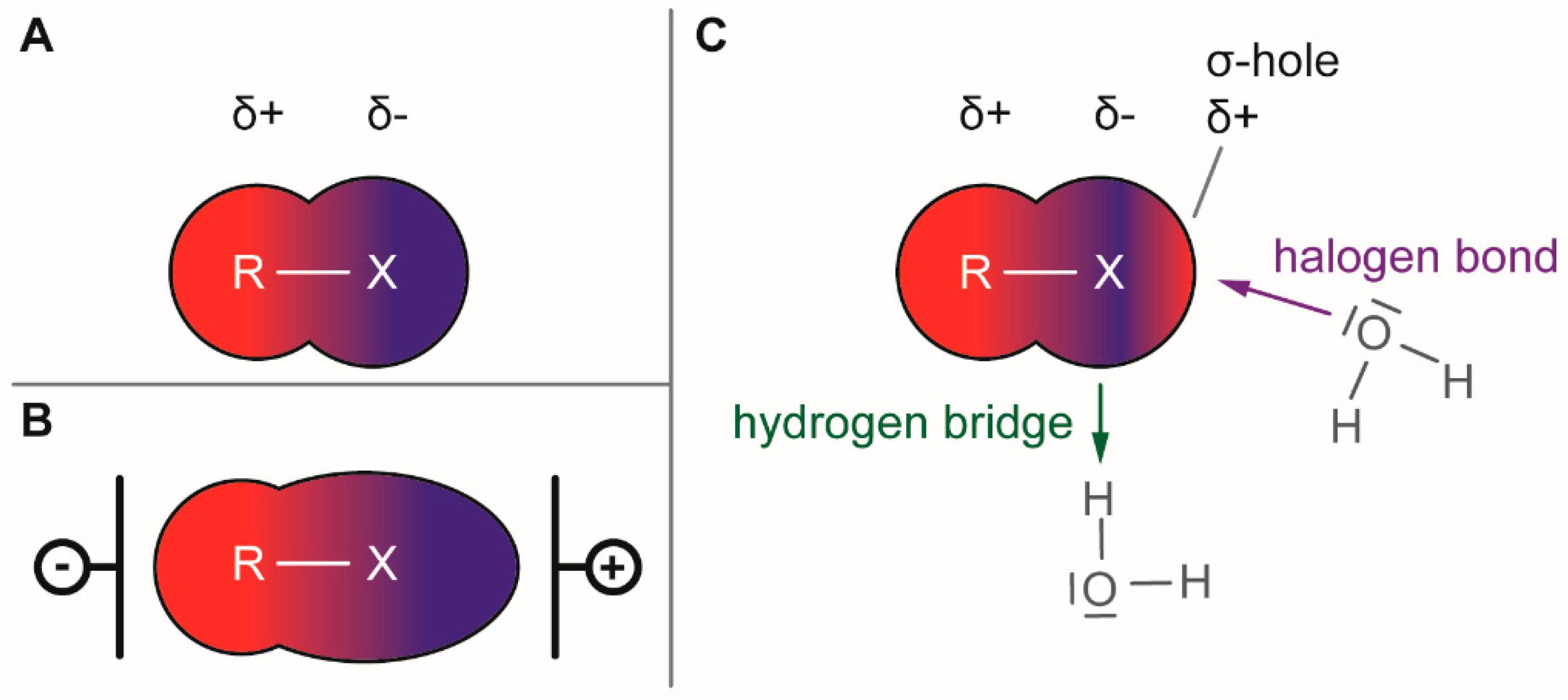
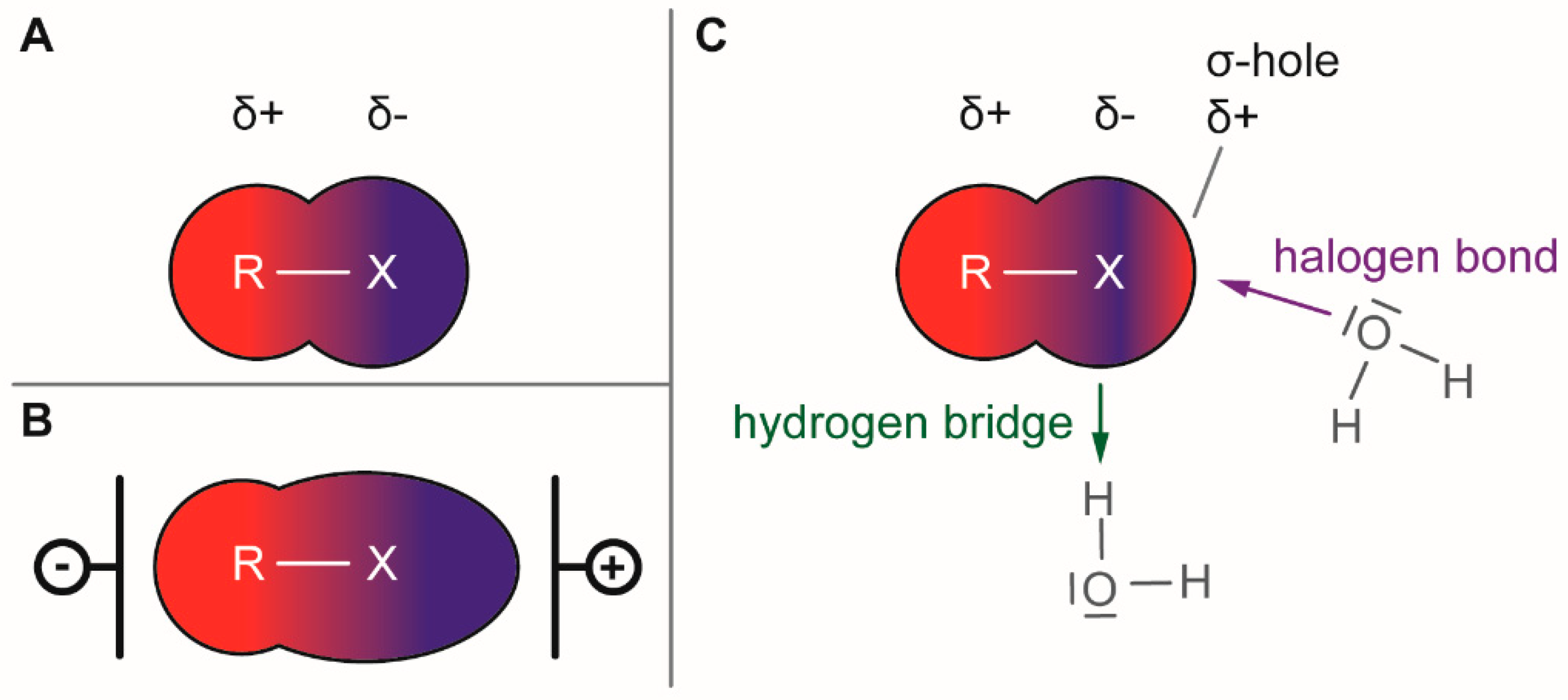
1.1. Electronical Properties of Halogen Moieties
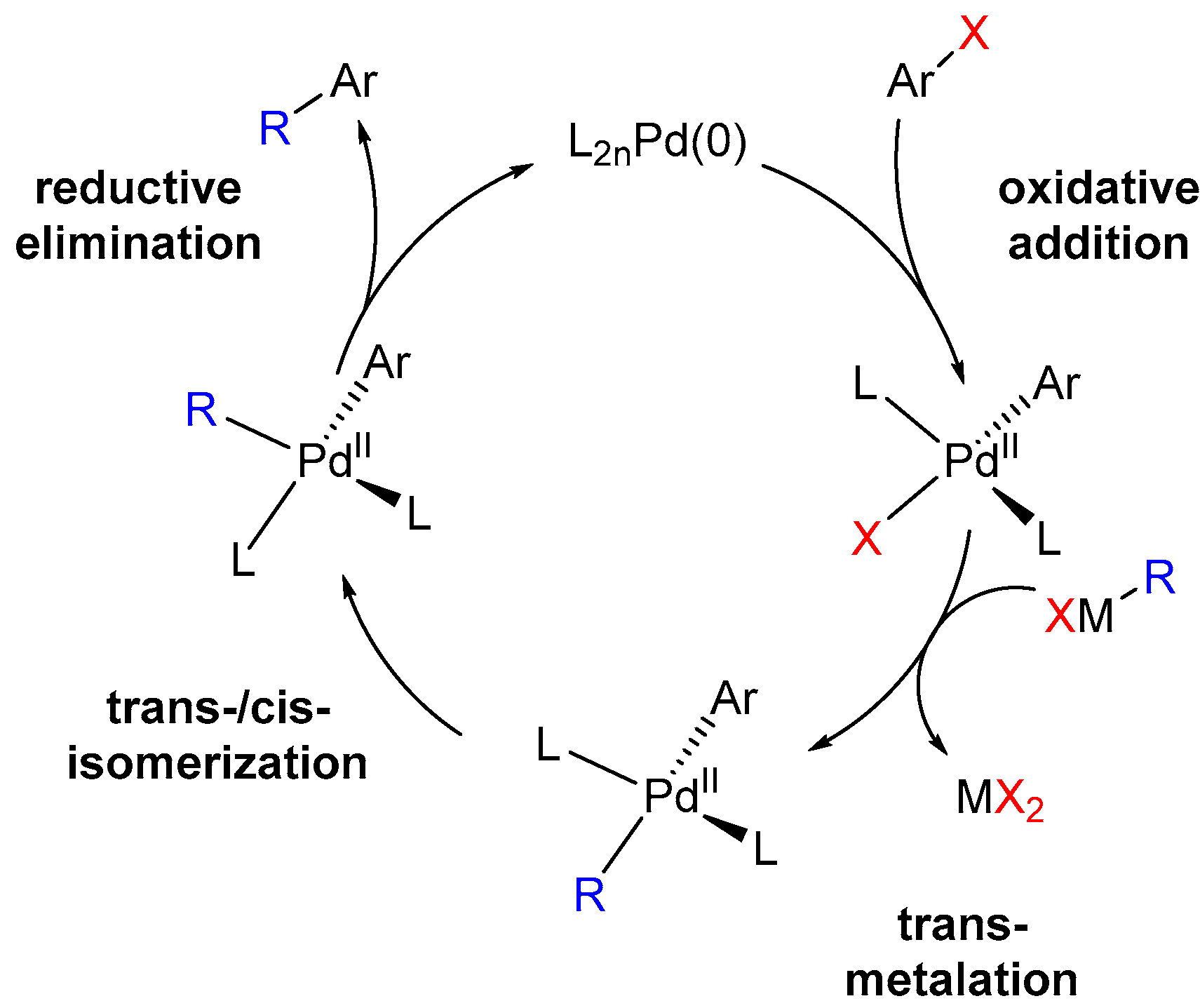
1.2. Halogens as Synthetic Tools
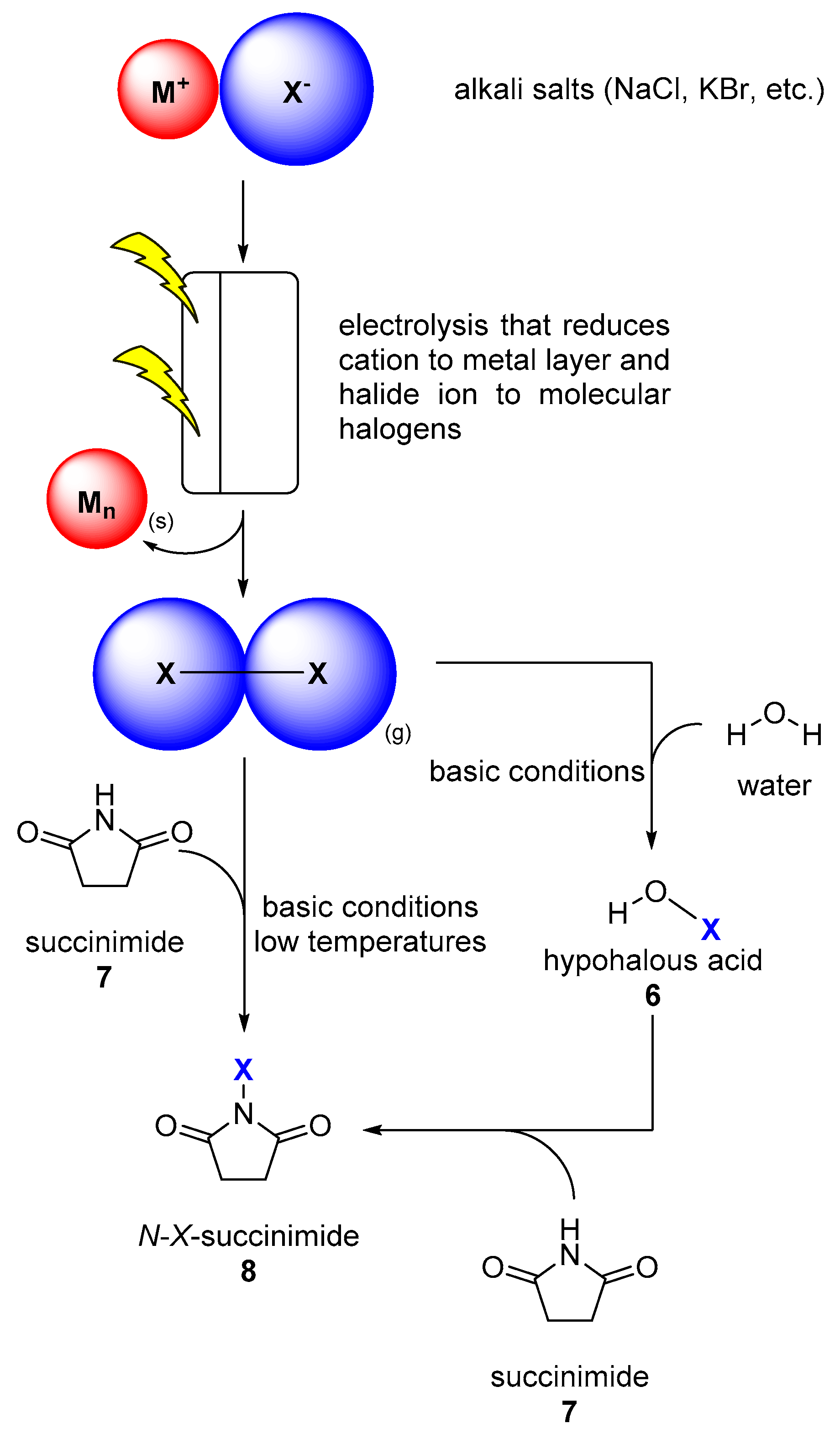
1.3. Halogen Chemistry is Energy-Demanding
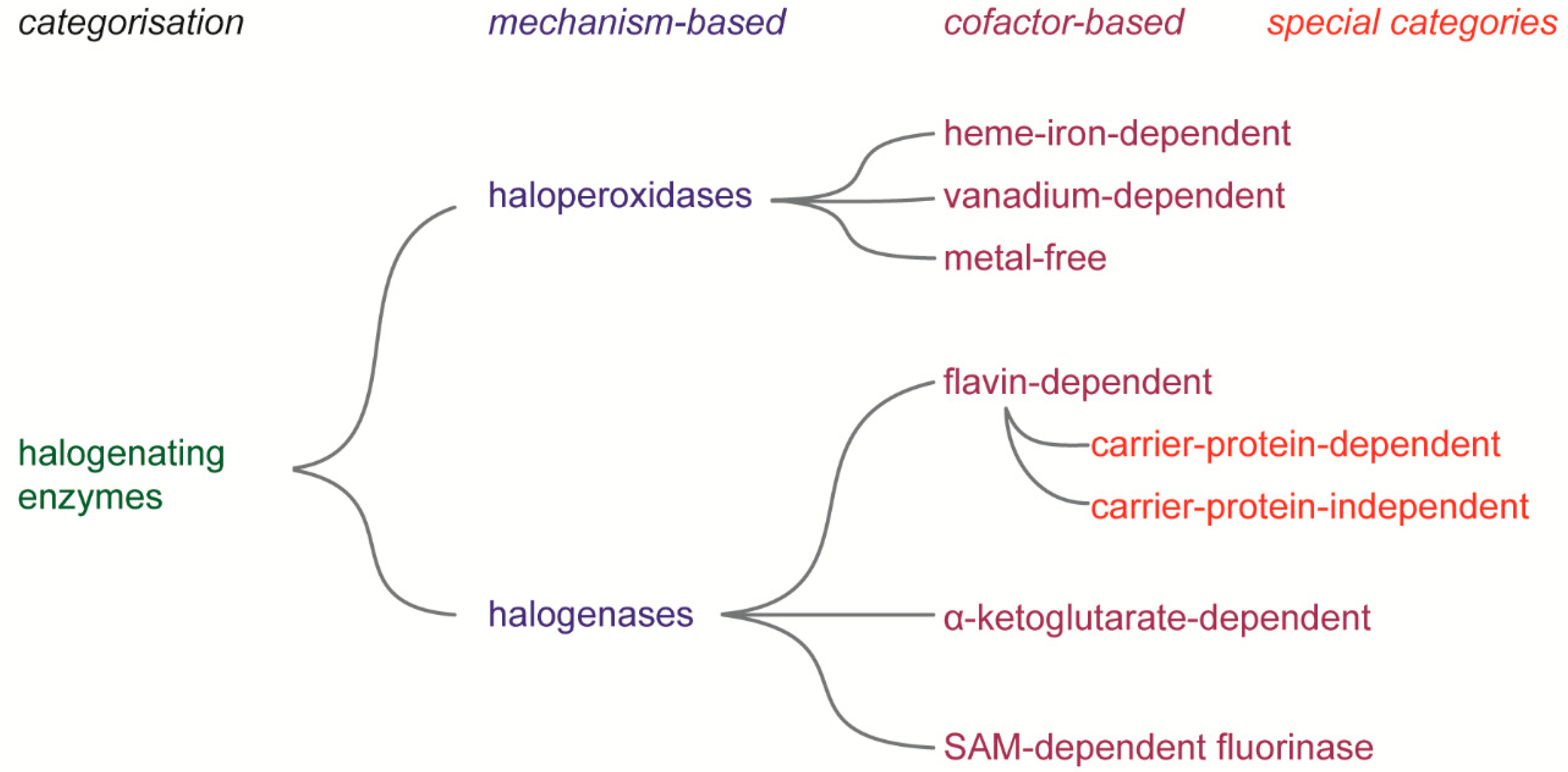
2. Halogenating Enzymes
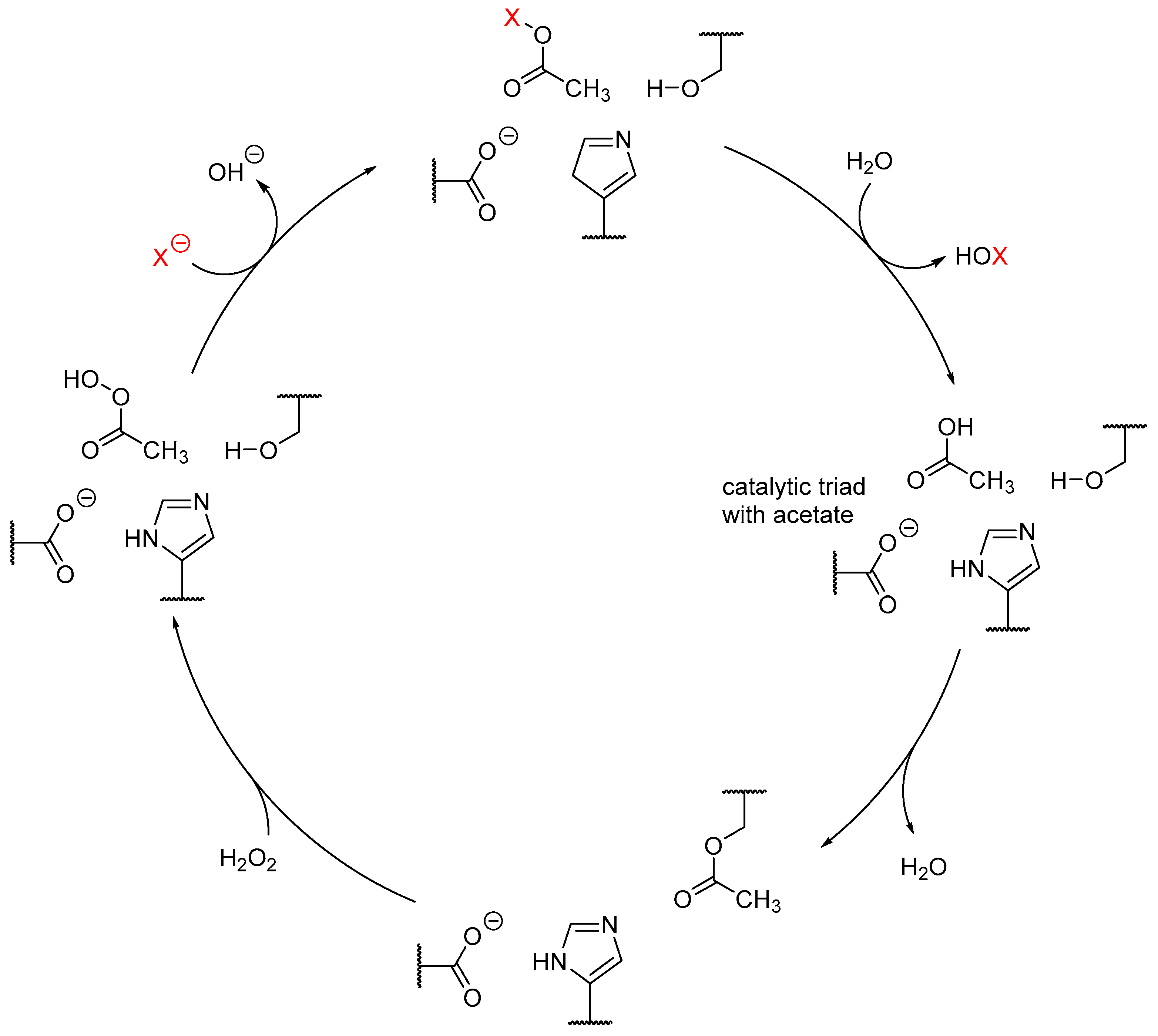
Haloperoxidases
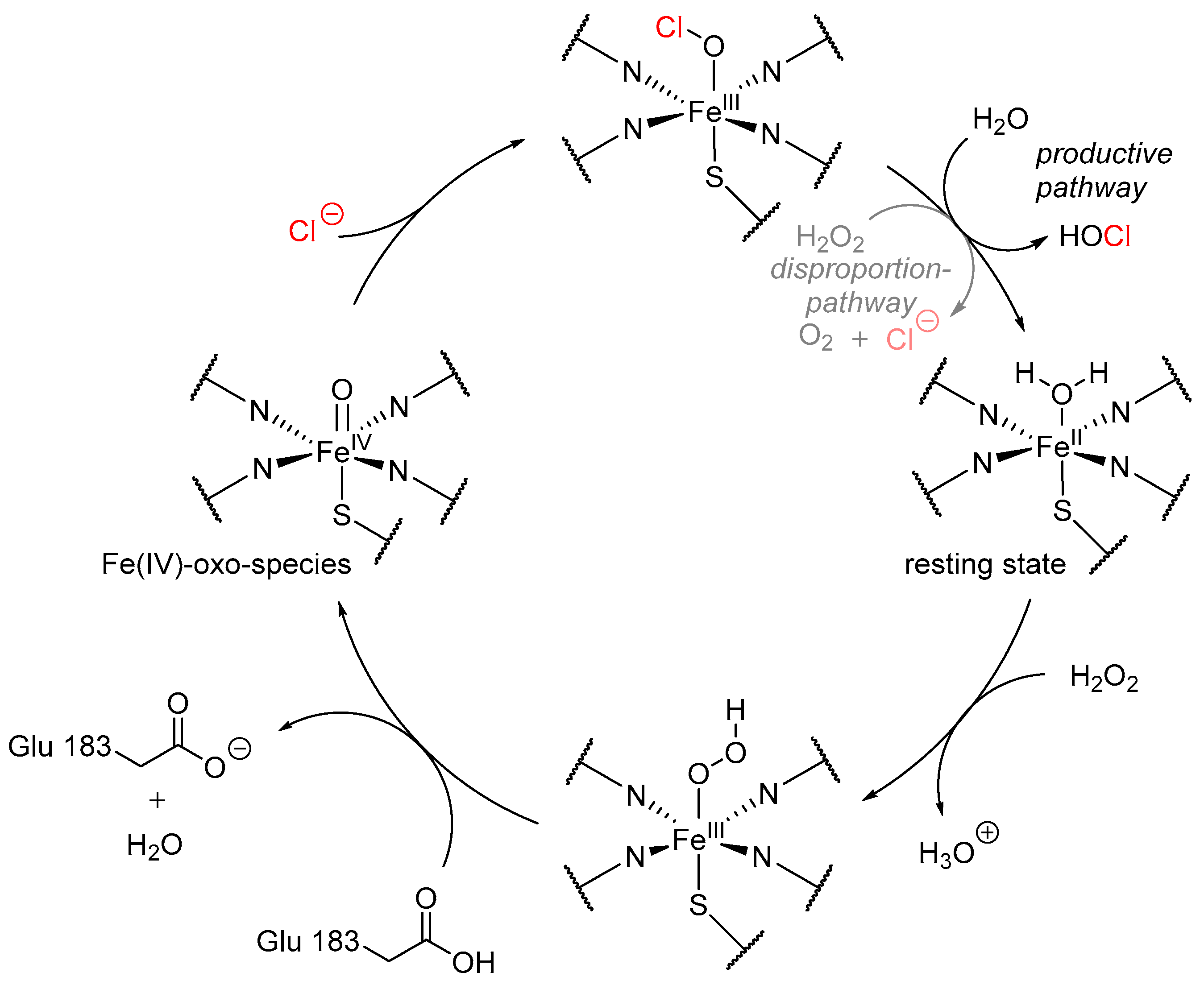
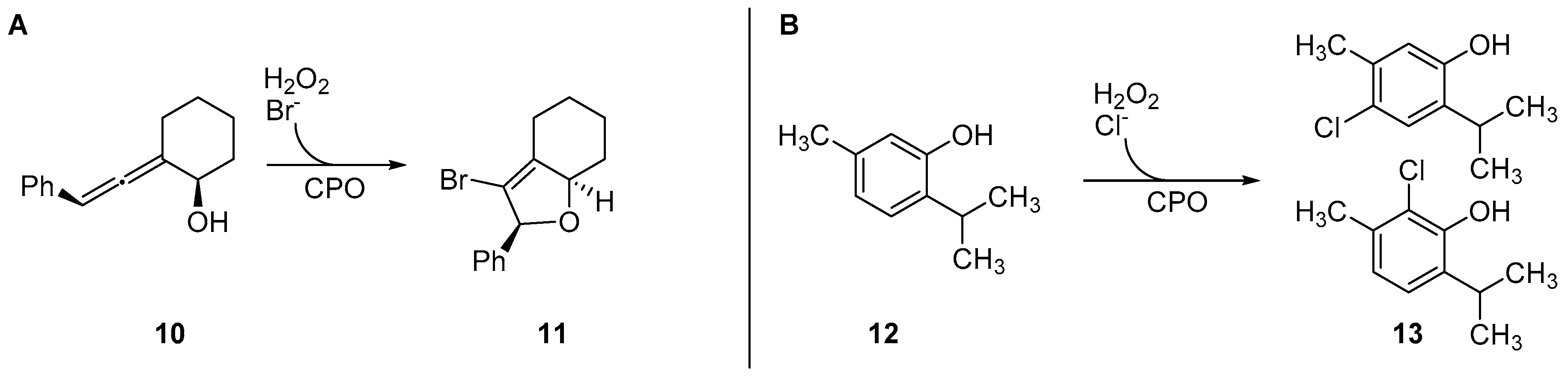
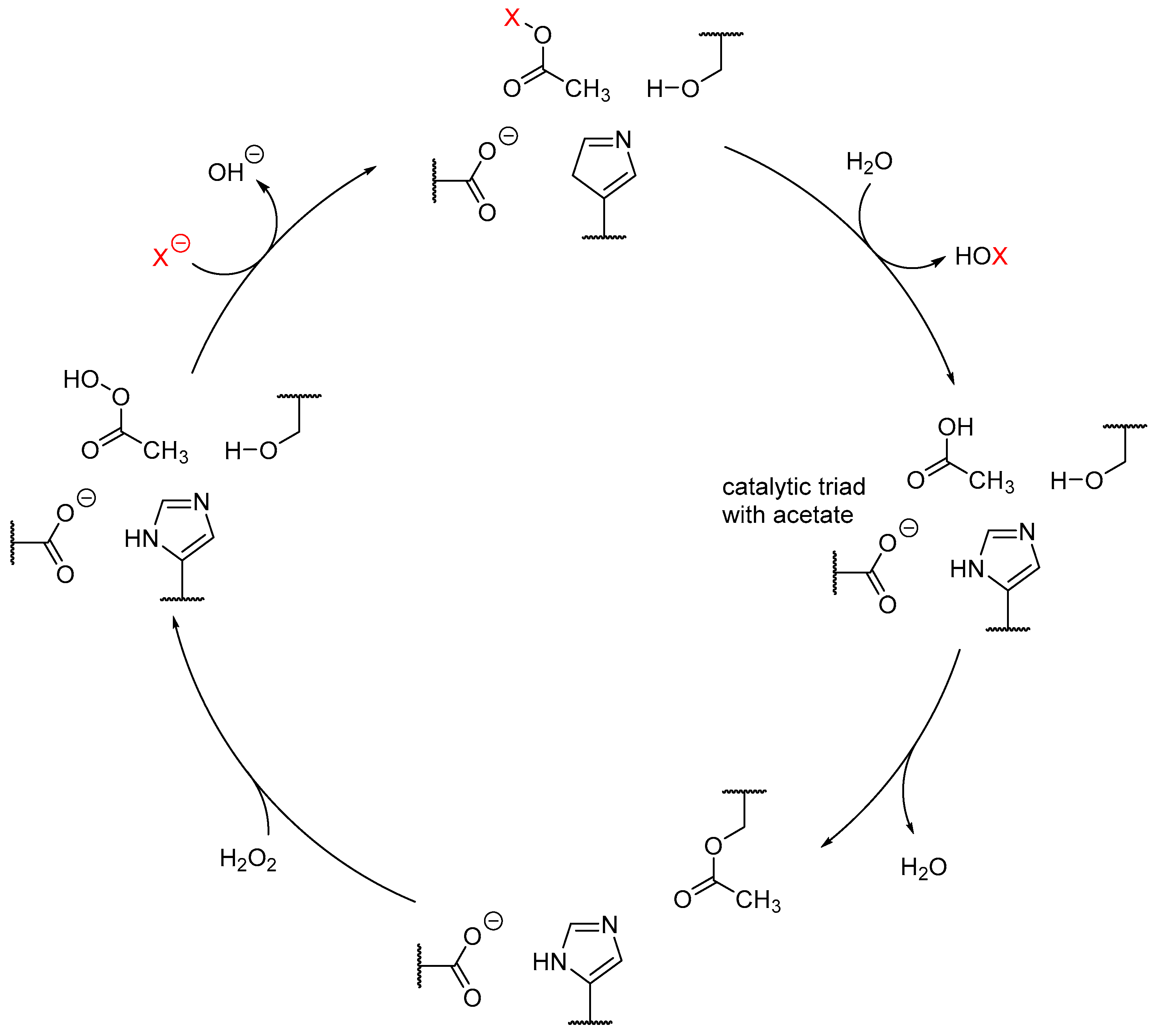
3. Heme-Iron-Dependent Haloperoxidases
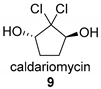
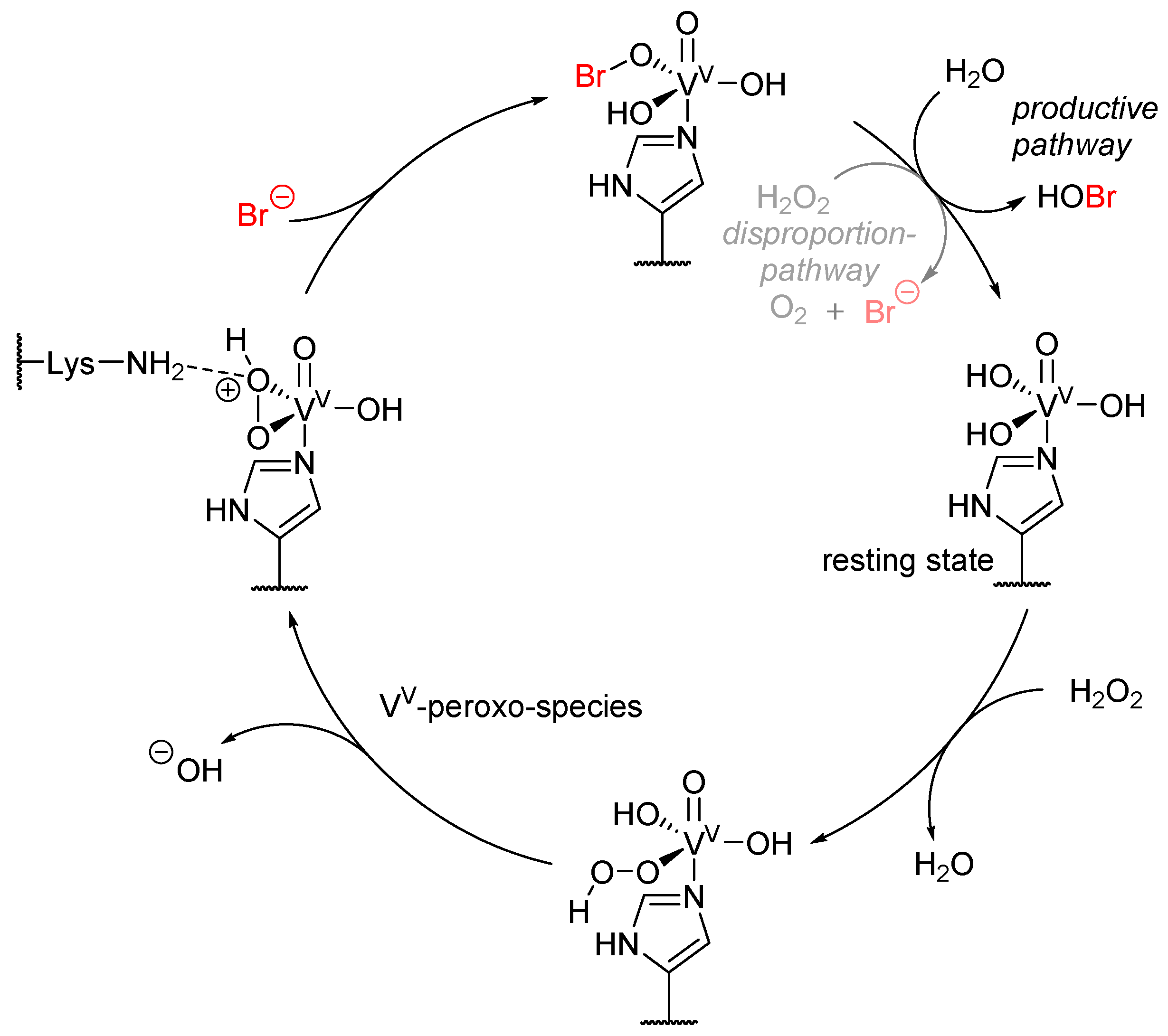
4. Vanadium-Dependent Haloperoxidases
5. Metal-Free Haloperoxidases/Perhydrolases
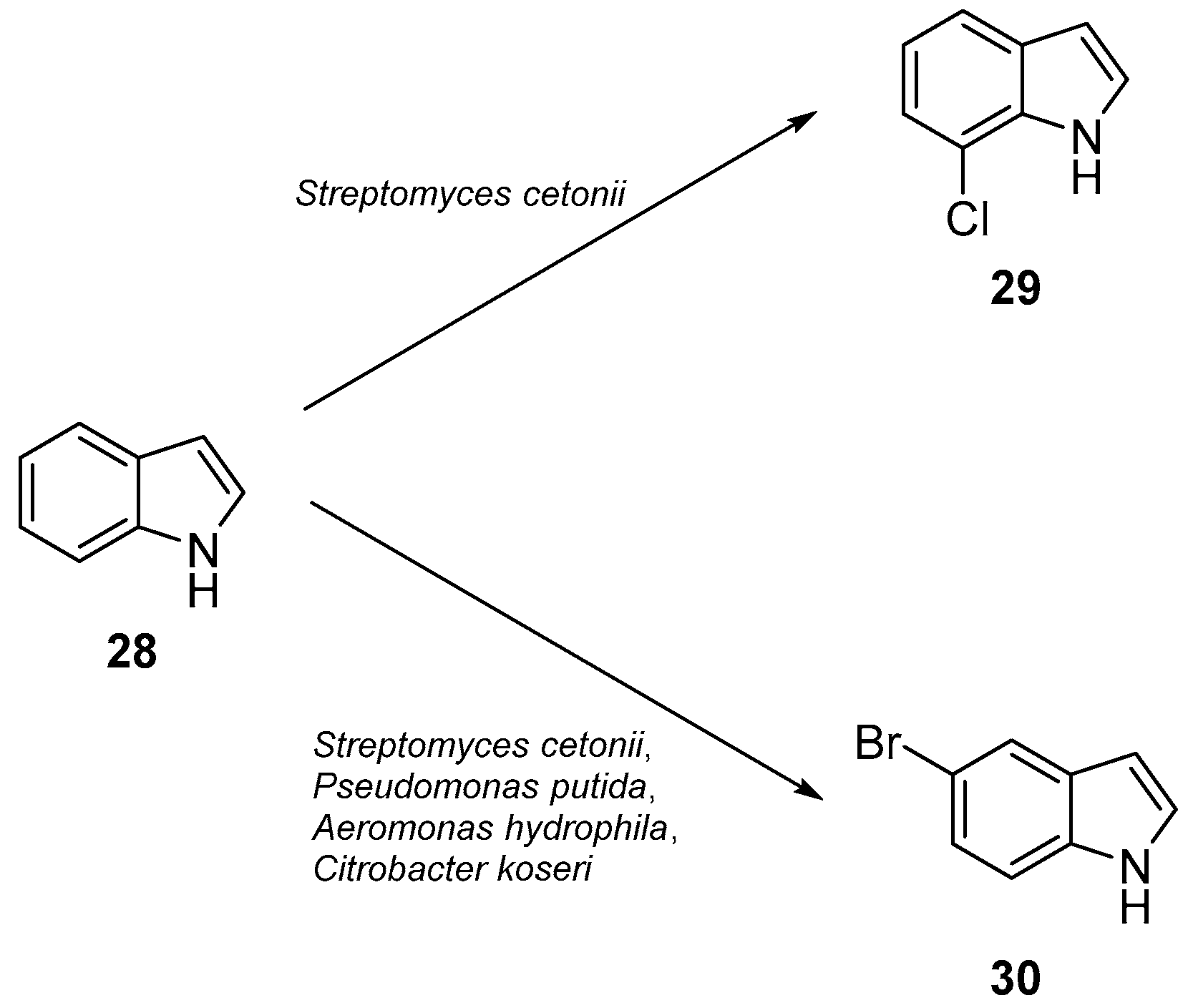
5.1. Flavin-Dependent Halogenases
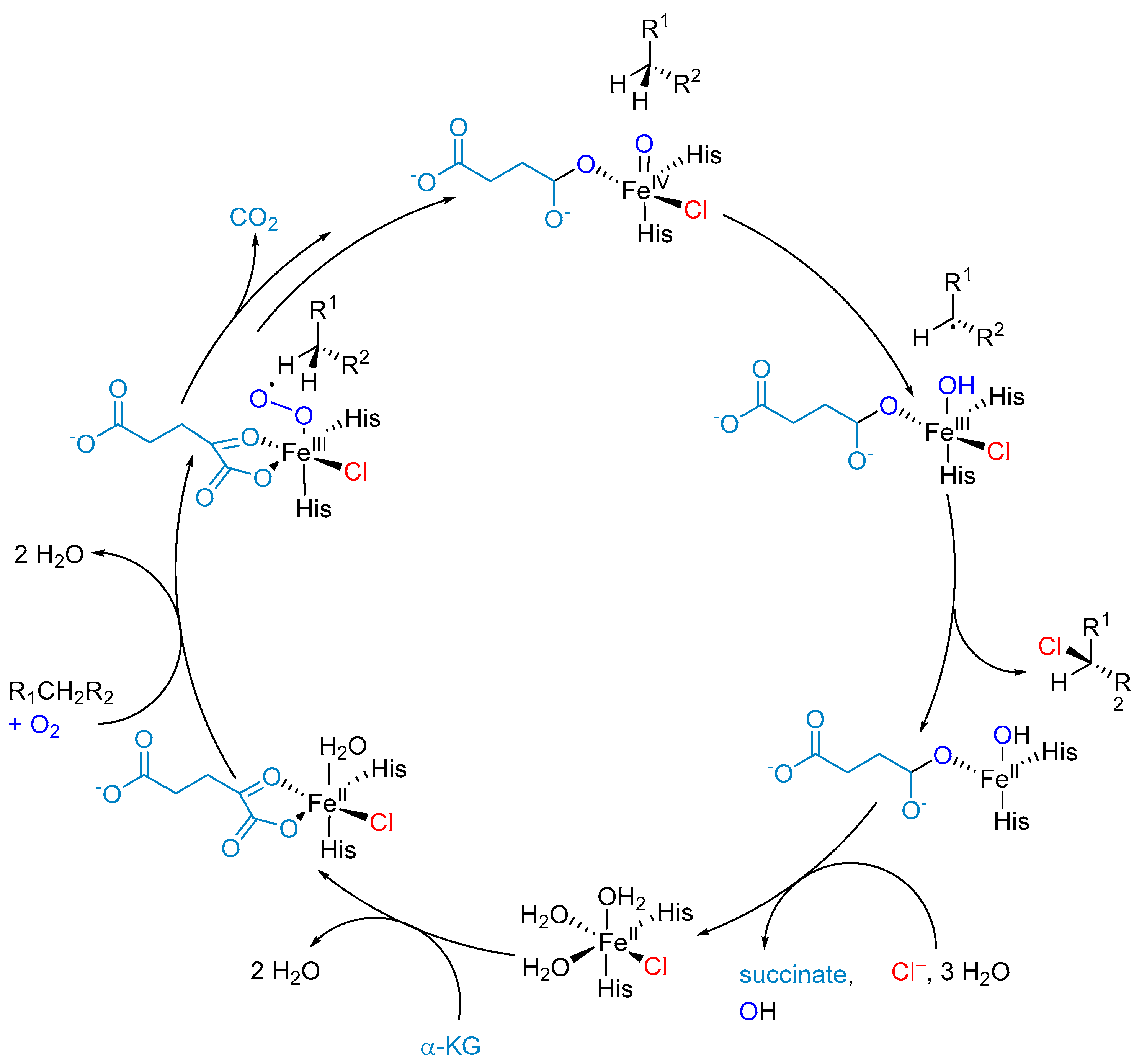
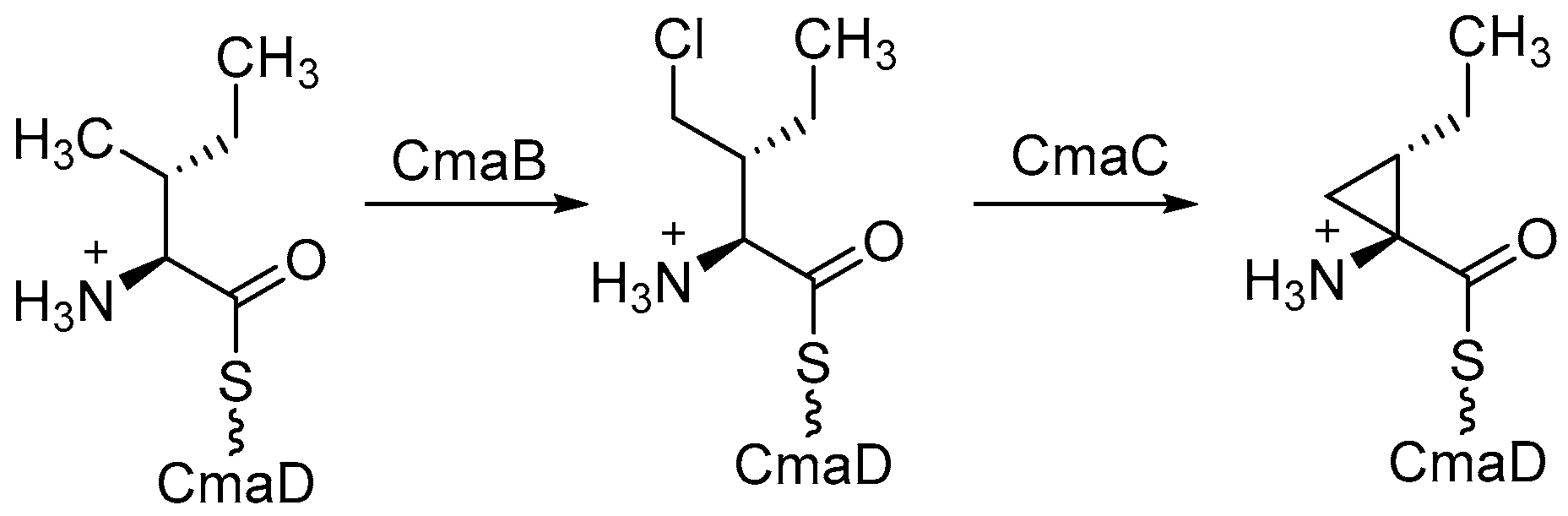
5.2. α-Ketoglutarate-Dependent Halogenases
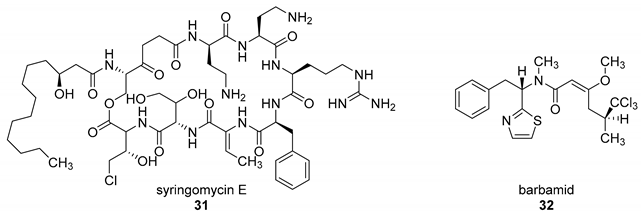
5.3. Fluorinase
6. Conclusion on Halogens in Active Agent (Syntheses)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| αKG | α-ketoglutarate |
| BPO | bromoperoxidase |
| CLEA | cross-linked enzyme aggregate |
| CPO | chloroperoxidase |
| FDA | 5′-fluoro-5′deoxyadenosine |
| Fhal, FDH | flavin-dependent halogenase |
| FlA | fluorinase A |
| g | gasous |
| Hal | halogen |
| HOX | hypohalous acid |
| HPO | haloperoxidase |
| MCD | monochlorodimedone |
| Mn | elemental metal |
| MOH | metal cation hydroxid salts |
| NBS | N-bromo-succinimde |
| NCS | N-chloro-succinimide |
| NRPS | non-ribosomal protein synthesis |
| NXS | N-halogen-succinimide |
| PKS | polyketide |
| PVC | polyvinyl chlroide |
| s | solid |
| SAM | S-adenosyl methionine |
| X | halide ion |
| X2 | molecular halogen |
| Y | non-halogen heteroatom |
References
- Gribble, G.W. Natural Organohalogens: A New Frontier for Medicinal Agents? J. Chem. Educ. 2004, 81, 1441. [Google Scholar] [CrossRef]
- Chast, F. A history of drug discovery: From first steps of chemistry to achievements in molecular pharmacology. In The Practice of Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2008; pp. 1–62. [Google Scholar]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; de Azevedo Junior, W.F.; Leite, A.C.L. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, L.; Cui, D.; Yao, Z.; Gao, B.; Lin, J.; Wei, D. The Important Role of Halogen Bond in Substrate Selectivity of Enzymatic Catalysis. Sci. Rep. 2016, 6, 34750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Miles, Z.D.; Winter, J.M.; Eustáquio, A.S.; EL Gamal, A.A.; Moore, B.S. Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev. 2017, 117, 5619–5674. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, L.C.; Drennan, C.L. Structural perspective on enzymatic halogenation. Acc. Chem. Res. 2008, 42, 147–155. [Google Scholar] [CrossRef]
- Gribble, G.W. The diversity of naturally produced organohalogens. Chemosphere 2003, 52, 289–297. [Google Scholar] [CrossRef]
- Latham, J.; Brandenburger, E.; Shepherd, S.A.; Menon, B.R.K.; Micklefield, J. Development of Halogenase Enzymes for Use in Synthesis. Chem. Rev. 2018, 118, 232–269. [Google Scholar] [CrossRef]
- Schnepel, C.; Sewald, N. Enzymatic Halogenation: A Timely Strategy for Regioselective C−H Activation. Chem. Eur. J. 2017, 23, 12064–12086. [Google Scholar] [CrossRef]
- Senn, H.M. Insights into enzymatic halogenation from computational studies. Front. Chem. 2014, 2, 98. [Google Scholar] [CrossRef]
- Smith, D.R.; Grüschow, S.; Goss, R.J. Scope and potential of halogenases in biosynthetic applications. Curr. Opin. Chem. Biol. 2013, 17, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.H.; Yeh, E.; Vosburg, D.A.; Garneau-Tsodikova, S.; Walsh, C.T. Nature’s inventory of halogenation catalysts: Oxidative strategies predominate. Chem. Rev. 2006, 106, 3364–3378. [Google Scholar] [CrossRef] [PubMed]
- Weichold, V.; Milbredt, D.; van Pée, K.-H. Specific Enzymatic Halogenation—From the Discovery of Halogenated Enzymes to Their Applications In Vitro and In Vivo. Angew. Chem. Int. Ed. 2016, 55, 6374–6389. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhan, J. Chlorinated Natural Products and Related Halogenases. Isr. J. Chem. 2019, 59, 1–17. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen bond: Its role beyond drug–target binding affinity for drug discovery and development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen interactions in protein–ligand complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discov. 2012, 7, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, Z.; Chen, T.; Chen, T.; Wang, Z.; Tian, G.; Shi, J.; Wang, X.; Lu, Y.; Yan, X.; et al. Utilization of halogen bond in lead optimization: A case study of rational design of potent phosphodiesterase type 5 (PDE5) inhibitors. J. Med. Chem. 2011, 54, 5607–5611. [Google Scholar] [CrossRef]
- Harris, C.M.; Kannan, R.; Kopecka, H.; Harris, T.M. The role of the chlorine substituents in the antibiotic vancomycin: Preparation and characterization of mono-and didechlorovancomycin. J. Am. Chem. Soc. 1985, 107, 6652–6658. [Google Scholar] [CrossRef]
- Schlosser, M.; Michel, D. About the “physiological size” of fluorine substituents: Comparison of sensorially active compounds with fluorine and methyl substituted analogues. Tetrahedron 1996, 52, 99–108. [Google Scholar] [CrossRef]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Thiehoff, C.; Rey, Y.P.; Gilmour, R. The Fluorine Gauche Effect: A Brief History. Isr. J. Chem. 2017, 57, 92–100. [Google Scholar] [CrossRef]
- Berkowitz, D.B.; Karukurichi, K.R.; de la Salud-Bea, R.; Nelson, D.L.; McCune, C.D. Use of fluorinated functionality in enzyme inhibitor development: Mechanistic and analytical advantages. J. Fluor. Chem. 2008, 129, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Blackhall, F.; Krzakowski, M.; Barrios, C.H.; Park, K.; Bover, I.; Heo, D.S.; Rosell, R.; Talbot, D.C.; Frank, R. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan–human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3337. [Google Scholar] [CrossRef]
- Bush, J.A.; Long, B.H.; Catino, J.J.; Bradner, W.T.; Tomita, K. Production and biological activity of rebeccamycin, a novel antitumor agent. J. Antibiot. 1987, 40, 668–678. [Google Scholar] [CrossRef]
- Lake, R.J.; Brennan, M.M.; Blunt, J.W.; Munro, M.H.; Pannell, L.K. Eudistomin K sulfoxide-an antiviral sulfoxide from the New Zealand ascidian Ritterella sigillinoides. Tetrahedron Lett. 1988, 29, 2255–2256. [Google Scholar] [CrossRef]
- Braghirolli, A.M.S.; Waissmann, W.; da Silva, J.B.; dos Santos, G.R. Production of iodine-124 and its applications in nuclear medicine. Appl. Radiat. Isot. 2014, 90, 138–148. [Google Scholar] [CrossRef]
- Crawford, C.C.; Evans, T.W. Production of Hypohalous Acid Solutions. U.S. Patent 2347151, 18 April 1944. [Google Scholar]
- Duncan, B.L.; Ness, R.C. Process for the Production of Highly Pure Concentrated Slurries of Sodium Hypochlorite. U.S. Patent 5194238A, 16 March 1993. [Google Scholar]
- Howarth, J.N.; Dadgar, A.; Sergent, R.H. Recovery of Bromine and Preparation of Hypobromous acid from Bromide Solution. U.S. Patent 5385650A, 31 January 1995. [Google Scholar]
- Kesner, M. Bromine and Bromine Compounds from the Dead Sea, Israel Products in the Service of People; The Weizmann Institute of Science, Israel: Rehovot, Israel, 1999. [Google Scholar]
- Groweiss, A. Use of Sodium Bromate for Aromatic Bromination: Research and Development. Org. Process Res. Dev. 2000, 4, 30–33. [Google Scholar] [CrossRef]
- Nishi, Z.; Kudoh, K.; Okamoto, N. Method of Forming Hypobromous Acid in Aqueous System. U.S. PATENT 7785559B2, 31 August 2010. [Google Scholar]
- Ziegler, K.; Späth, A.; Schaaf, E.; Schumann, W.; Winkelmann, E. Die halogenierung ungesättigter Substanzen in der Allylstellung. Justus Liebigs Ann. Chem. 1942, 551, 80–119. [Google Scholar] [CrossRef]
- Torborg, C.; Beller, M. Recent applications of palladium-catalyzed coupling reactions in the pharmaceutical, agrochemical, and fine chemical industries. Adv. Synth. Catal. 2009, 351, 3027–3043. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. Copper in cross-coupling reactions: The post-Ullmann chemistry. Coord. Chem. Rev. 2004, 248, 2337–2364. [Google Scholar] [CrossRef]
- Xi, Z.; Liu, B.; Chen, W. Room-temperature Kumada cross-coupling of unactivated aryl chlorides catalyzed by N-heterocylic carbene-based nickel (II) complexes. J. Org. Chem. 2008, 73, 3954–3957. [Google Scholar] [CrossRef]
- Gomes, F.; Echeverria, P.G.; Fürstner, A. Iron-or Palladium-Catalyzed Reaction Cascades Merging Cycloisomerization and Cross-Coupling Chemistry. Chem. Eur. J. 2018, 24, 16814–16822. [Google Scholar] [CrossRef]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-Catalyzed Cross-Couplings in the Synthesis of Pharmaceuticals: In Pursuit of Sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116–11128. [Google Scholar] [CrossRef]
- Cahiez, G.R.; Lefèvre, G.; Moyeux, A.; Guerret, O.; Gayon, E.; Guillonneau, L.; Lefèvre, N.; Gu, Q.; Zhou, E. Gram-Scale, Cheap, and Eco-Friendly Iron-Catalyzed Cross-Coupling between Alkyl Grignard Reagents and Alkenyl or Aryl Halides. Org. Lett. 2019, 21, 2679–2683. [Google Scholar] [CrossRef]
- Cooke, J.W.; Bright, R.; Coleman, M.J.; Jenkins, K.P. Process research and development of a dihydropyrimidine dehydrogenase inactivator: Large-scale preparation of eniluracil using a Sonogashira coupling. Org. Process Res. Dev. 2001, 5, 383–386. [Google Scholar] [CrossRef]
- Wallace, M.D.; McGuire, M.A.; Yu, M.S.; Goldfinger, L.; Liu, L.; Dai, W.; Shilcrat, S. Multi-kiloscale enantioselective synthesis of a vitronectin receptor antagonist. Org. Process Res. Dev. 2004, 8, 738–743. [Google Scholar] [CrossRef]
- Benkeser, R.A.; Siklosi, M.P.; Mozdzen, E.C. Reversible Grignard and organolithium reactions. J. Am. Chem. Soc. 1978, 100, 2134–2139. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J.K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E.-I. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Corriu, R.; Masse, J. Activation of Grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyls. Chem. Commun. 1972, 144. [Google Scholar] [CrossRef]
- Global Chlorine Market Set for Rapid Growth to Reach around USD 38.4 Billion in 2021. Available online: https://www.zionmarketresearch.com/news/global-chlorine-market (accessed on 23 August 2019).
- Ausfelder, F. Flexibilitätsoptionen in der Grundstoffindustrie: Methodik, Potenziale, Hemmnisse: Bericht des AP V. 6 “Flexibilitätsoptionen und Perspektiven in der Grundstoffindustrie ”im Kopernikus-Projekt “SynErgie-Synchronisierte und Energieadaptive Produktionstechnik zur Flexiblen Ausrichtung von Industrieprozessen Auf Eine Fluktuierende Energieversorgung”; DECHEMA Gesellschaft für Chemische Technik und Biotechnologie eV: Frankfurt, Germany, 2018. [Google Scholar]
- Chlor-Alkali Industry Review 2017/2018. Available online: https://www.eurochlor.org/wp-content/uploads/2019/05/euro_chlor_industry_review_FINAL.pdf (accessed on 30 October 2019).
- The Electrolysis Process and the Real Costs of Production. Available online: https://www.eurochlor.org/wp-content/uploads/2019/04/12-electrolysis_production_costs.pdf (accessed on 23 August 2019).
- Morris, D.R.; Hager, L.P.; Chloroperoxidase, I. Isolation and properties of the crystalline glycoprotein. J. Biol. Chem. 1966, 241, 1763–1768. [Google Scholar] [PubMed]
- Kuhnel, K.; Blankenfeldt, W.; Terner, J.; Schlichting, I. Crystal structures of chloroperoxidase with its bound substrates and complexed with formate, acetate, and nitrate. J. Biol. Chem. 2006, 281, 23990–23998. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Sandy, M. Mechanistic considerations of halogenating enzymes. Nature 2009, 460, 848. [Google Scholar] [CrossRef] [PubMed]
- Buchhaupt, M.; Huttmann, S.; Sachs, C.C.; Bormann, S.; Hannappel, A.; Schrader, J. Caldariomyces fumago DSM1256 Contains Two Chloroperoxidase Genes, Both Encoding Secreted and Active Enzymes. J. Mol. Microbiol. Biotechnol. 2015, 25, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.M.; Xu, L.; Drenzek, N.J.; Sturchio, N.C.; Heraty, L.J.; Kimblin, C.; Butler, A. A chlorine isotope effect for enzyme-catalyzed chlorination. J. Am. Chem. Soc. 2002, 124, 14526–14527. [Google Scholar] [CrossRef]
- Pickard, M.A. A defined growth medium for the production of chloroperoxidase by Caldariomyces fumago. Can. J. Microbiol. 1981, 27, 1298–1305. [Google Scholar] [CrossRef]
- Getrey, L.; Krieg, T.; Hollmann, F.; Schrader, J.; Holtmann, D. Enzymatic halogenation of the phenolic monoterpenes thymol and carvacrol with chloroperoxidase. Green Chem. 2014, 16, 1104–1108. [Google Scholar] [CrossRef]
- Naapuri, J.; Rolfes, J.D.; Keil, J.; Manzuna Sapu, C.; Deska, J. Enzymatic halocyclization of allenic alcohols and carboxylates: A biocatalytic entry to functionalized O-heterocycles. Green Chem. 2017, 19, 447–452. [Google Scholar] [CrossRef]
- Blanke, S.R.; Yi, S.; Hager, L.P. Development of Semi-Continuous and Continuous-Flow Bioreactors for the High-Level Production of Chloroperoxidase. Biotechnol. Lett. 1989, 11, 769–774. [Google Scholar] [CrossRef]
- Yamada, H.; Itoh, N.; Izumi, Y. Chloroperoxidase-catalyzed halogenation of trans-cinnamic acid and its derivatives. J. Biol. Chem. 1985, 260, 11962–11969. [Google Scholar] [PubMed]
- Allain, E.J.; Hager, L.P.; Deng, L.; Jacobsen, E.N. Highly Enantioselective Epoxidation of Disubstituted Alkenes with Hydrogen-Peroxide Catalyzed by Chloroperoxidase. J. Am. Chem. Soc. 1993, 115, 4415–4416. [Google Scholar] [CrossRef]
- Manthey, J.A.; Hager, L.P. Purification and properties of bromoperoxidase from Penicillus capitatus. J. Biol. Chem. 1981, 256, 11232–11238. [Google Scholar] [PubMed]
- van Pée, K.H.; Lingens, F. Purification of bromoperoxidase from Pseudomonas aureofaciens. J. Bacteriol. 1985, 161, 1171–1175. [Google Scholar]
- Baden, D.G.; Corbett, M.D. Bromoperoxidases from Penicillus capitatus, Penicillus lamourouxii and Rhipocephalus phoenix. Biochem. J. 1980, 187, 205–211. [Google Scholar] [CrossRef]
- Kaup, B.-A.; Ehrich, K.; Pescheck, M.; Schrader, J. Microparticle-enhanced cultivation of filamentous microorganisms: Increased chloroperoxidase formation by Caldariomyces fumago as an example. Biotechnol. Bioeng. 2008, 99, 491–498. [Google Scholar] [CrossRef]
- Conesa, A.; van de Velde, F.; van Rantwijk, F.; Sheldon, R.A.; van den Hondel, C.A.M.J.J.; Punt, P.J. Expression of the Caldariomyces fumago chloroperoxidase in Aspergillus niger and characterization of the recombinant enzyme. J. Biol. Chem. 2001, 276, 17635–17640. [Google Scholar] [CrossRef]
- Coughlin, P.; Roberts, S.; Rush, C.; Willetts, A. Biotransformation of Alkenes by Haloperoxidases - Regiospecific Bromohydrin Formation from Cinnamyl Substrates. Biotechnol. Lett. 1993, 15, 907–912. [Google Scholar] [CrossRef]
- van Schijndel, J.W.; Vollenbroek, E.G.; Wever, R. The chloroperoxidase from the fungus Curvularia inaequalis; a novel vanadium enzyme. Biochim. Biophys. Acta 1993, 1161, 249–256. [Google Scholar] [CrossRef]
- Van Schijndel, J.W.P.M.; Barnett, P.; Roelse, J.; Vollenbroek, E.G.M.; Wever, R. The stability and steady-state kinetics of vanadium chloroperoxidase from the fungus Curvularia inaequalis. Eur. J. Biochem. 1994, 225, 151–157. [Google Scholar] [CrossRef]
- Rush, C.; Willetts, A.; Davies, G.; Dauter, Z.; Watson, H.; Littlechild, J. Purification, crystallisation and preliminary X-ray analysis of the vanadium-dependent haloperoxidase from Corallina officinalis. FEBS Lett. 1995, 359, 244–246. [Google Scholar] [CrossRef]
- Wever, R. Vanadium: Biochemical and Molecular Biological Approaches; Springer: New York, NY, USA, 2012. [Google Scholar]
- Leblanc, C.; Vilter, H.; Fournier, J.B.; Delage, L.; Potin, P.; Rebuffet, E.; Michel, G.; Solari, P.L.; Feiters, M.C.; Czjzek, M. Vanadium haloperoxidases: From the discovery 30 years ago to X-ray crystallographic and V K-edge absorption spectroscopic studies. Coord. Chem. Rev. 2015, 301–302, 134–146. [Google Scholar] [CrossRef]
- Hemrika, W.; Renirie, R.; Macedo-Ribeiro, S.; Messerschmidt, A.; Wever, R. Heterologous Expression of the Vanadium-containing Chloroperoxidase from Curvularia inaequalis in Saccharomyces cerevisiae and Site-directed Mutagenesis of the Active Site Residues His496, Lys353, Arg360, and Arg490. J. Biol. Chem. 1999, 274, 23820–23827. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fueyo, E.; van Wingerden, M.; Renirie, R.; Wever, R.; Ni, Y.; Holtmann, D.; Hollmann, F. Chemoenzymatic Halogenation of Phenols by using the Haloperoxidase from Curvularia inaequalis. ChemCatChem 2015, 7, 4035–4038. [Google Scholar] [CrossRef]
- Dong, J.J.; Fernandez-Fueyo, E.; Li, J.; Guo, Z.; Renirie, R.; Wever, R.; Hollmann, F. Halofunctionalization of alkenes by vanadium chloroperoxidase from Curvularia inaequalis. Chem. Commun. 2017, 53, 6207–6210. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fueyo, E.; Younes, S.H.; Rootselaar, S.V.; Aben, R.W.; Renirie, R.; Wever, R.; Holtmann, D.; Rutjes, F.P.; Hollmann, F. A biocatalytic aza-Achmatowicz reaction. ACS Catal. 2016, 6, 5904–5907. [Google Scholar] [CrossRef]
- But, A.; van Noord, A.; Poletto, F.; Sanders, J.P.M.; Franssen, M.C.R.; Scott, E.L. Enzymatic halogenation and oxidation using an alcohol oxidase-vanadium chloroperoxidase cascade. Mol. Catal. 2017, 443, 92–100. [Google Scholar] [CrossRef]
- Coupe, E.E.; Smyth, M.G.; Fosberry, A.P.; Hall, R.M.; Littlechild, J.A. The dodecameric vanadium-dependent haloperoxidase from the marine algae Corallina officinalis: Cloning, expression, and refolding of the recombinant enzyme. Protein Expr. Purif. 2007, 52, 265–272. [Google Scholar] [CrossRef]
- Littlechild, J. Haloperoxidases and their role in biotransformation reactions. Curr. Opin. Chem. Biol. 1999, 3, 28–34. [Google Scholar] [CrossRef]
- Andersson, M.; Willetts, A.; Allenmark, S. Asymmetric sulfoxidation catalyzed by a vanadium-containing bromoperoxidase. J. Org. Chem. 1997, 62, 8455–8458. [Google Scholar] [CrossRef]
- Carter, J.N.; Beatty, K.E.; Simpson, M.T.; Butler, A. Reactivity of recombinant and mutant vanadium bromoperoxidase from the red alga Corallina officinalis. J. Inorg. Biochem. 2002, 91, 59–69. [Google Scholar] [CrossRef]
- Van Peè, K.-H. Microbial biosynthesis of halometabolites. Arch. Microbiol. 2001, 175, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Tolzer, S.; Pelletier, I.; Altenbuchner, J.; van Pee, K.H.; Hecht, H.J. Structural investigation of the cofactor-free chloroperoxidases. J. Mol. Biol. 1998, 279, 889–900. [Google Scholar] [CrossRef]
- van Peé, K.H.; Dong, C.; Flecks, S.; Naismith, J.; Patallo, E.P.; Wage, T. Biological halogenation has moved far beyond haloperoxidases. Adv. Appl. Microbiol. 2006, 59, 127–157. [Google Scholar]
- Medici, R.; Garaycoechea, J.I.; Dettorre, L.A.; Iribarren, A.M.; Lewkowicz, E.S. Biocatalysed halogenation of nucleobase analogues. Biotechnol. Lett. 2011, 33, 1999–2003. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Ayub, M.J.; Furnham, N.; Thornton, J.M.; Laskowski, R.A. Chopping and changing: The evolution of the flavin-dependent monooxygenases. J. Mol. Biol. 2016, 428, 3131–3146. [Google Scholar] [CrossRef]
- Dong, C.; Flecks, S.; Unversucht, S.; Haupt, C.; Van Pee, K.-H.; Naismith, J.H. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. [Google Scholar] [CrossRef]
- Hohaus, K.; Altmann, A.; Burd, W.; Fischer, I.; Hammer, P.E.; Hill, D.S.; Ligon, J.M.; van Pée, K.H. NADH-Dependent Halogenases Are More Likely To Be Involved in Halometaolite Biosynthesis Than Haloperoxidases. Angew. Chem. Int. Ed. 1997, 36, 2012–2013. [Google Scholar] [CrossRef]
- Keller, S.; Wage, T.; Hohaus, K.; Hölzer, M.; Eichhorn, E.; van Pée, K.H. Purification and Partial Characterization of Tryptophan 7-Halogenase (PrnA) from Pseudomonas fluorescens. Angew. Chem. Int. Ed. 2000, 112, 2380–2382. [Google Scholar] [CrossRef]
- Yeh, E.; Garneau, S.; Walsh, C.T. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 3960–3965. [Google Scholar] [CrossRef]
- Lee, J.K.; Zhao, H. Identification and characterization of the flavin:NADH reductase (PrnF) involved in a novel two-component arylamine oxygenase. J. Bacteriol. 2007, 189, 8556–8563. [Google Scholar] [CrossRef] [PubMed]
- Eschenbrenner, M.; Covès, J.; Fontecave, M. The flavin reductase activity of the flavoprotein component of sulfite reductase from Escherichia coli a new model for the protein structure. J. Biol. Chem. 1995, 270, 20550–20555. [Google Scholar] [CrossRef] [PubMed]
- Heemstra, J.R., Jr.; Walsh, C.T. Tandem action of the O2-and FADH2-dependent halogenases KtzQ and KtzR produce 6,7-dichlorotryptophan for kutzneride assembly. J. Am. Chem. Soc. 2008, 130, 14024–14025. [Google Scholar] [CrossRef]
- Shepherd, S.A.; Karthikeyan, C.; Latham, J.; Struck, A.-W.; Thompson, M.L.; Menon, B.R.; Styles, M.Q.; Levy, C.; Leys, D.; Micklefield, J. Extending the biocatalytic scope of regiocomplementary flavin-dependent halogenase enzymes. Chem. Sci. 2015, 6, 3454–3460. [Google Scholar] [CrossRef] [Green Version]
- Unversucht, S.; Hollmann, F.; Schmid, A.; van Pée, K.H. FADH2-dependence of tryptophan 7-halogenase. Adv. Synth. Catal. 2005, 347, 1163–1167. [Google Scholar] [CrossRef]
- Schroeder, L.; Frese, M.; Müller, C.; Sewald, N.; Kottke, T. Photochemically Driven Biocatalysis of Halogenases for the Green Production of Chlorinated Compounds. ChemCatChem 2018, 10, 3336–3341. [Google Scholar] [CrossRef]
- Ortega, M.A.; Cogan, D.P.; Mukherjee, S.; Garg, N.; Li, B.; Thibodeaux, G.N.; Maffioli, S.I.; Donadio, S.; Sosio, M.; Escano, J.; et al. Two flavoenzymes catalyze the post-translational generation of 5-chlorotryptophan and 2-aminovinyl-cysteine during NAI-107 biosynthesis. ACS Chem. Biol. 2017, 12, 548–557. [Google Scholar] [CrossRef]
- Lang, A.; Polnick, S.; Nicke, T.; William, P.; Patallo, E.P.; Naismith, J.H.; van Pee, K.H. Changing the Regioselectivity of the Tryptophan 7-Halogenase PrnA by Site-Directed Mutagenesis. Angew. Chem. Int. Ed. 2011, 50, 2951–2953. [Google Scholar] [CrossRef]
- Zeng, J.; Zhan, J. Characterization of a tryptophan 6-halogenase from Streptomyces toxytricini. Biotechnol. Lett. 2011, 33, 1607–1613. [Google Scholar] [CrossRef]
- Hölzer, M.; Burd, W.; Reißig, H.U.; Pée, K.H.V. Substrate specificity and regioselectivity of tryptophan 7-halogenase from Pseudomonas fluorescens BL915. Adv. Synth. Catal. 2001, 343, 591–595. [Google Scholar] [CrossRef]
- Hammer, P.E.; Hill, D.S.; Lam, S.T.; Van Pée, K.-H.; Ligon, J.M. Four genes from Pseudomonas fluorescens that encode the biosynthesis of pyrrolnitrin. Appl. Environ. Microb. 1997, 63, 2147–2154. [Google Scholar]
- Magarvey, N.A.; Beck, Z.Q.; Golakoti, T.; Ding, Y.; Huber, U.; Hemscheidt, T.K.; Abelson, D.; Moore, R.E.; Sherman, D.H. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from Nostoc cyanobionts. ACS Chem. Biol. 2006, 1, 766–779. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.S.; Walsh, C.T.; Kay, R.R. A flavin-dependent halogenase catalyzes the chlorination step in the biosynthesis of Dictyostelium differentiation-inducing factor 1. Proc. Natl. Acad. Sci. USA 2010, 107, 5798–5803. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; El Gamal, A.A.; Yamanaka, K.; Poth, D.; Kersten, R.D.; Schorn, M.; Allen, E.E.; Moore, B.S. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014, 10, 640. [Google Scholar] [CrossRef]
- Zeng, J.; Zhan, J. A Novel Fungal Flavin-Dependent Halogenase for Natural Product Biosynthesis. ChemBioChem 2010, 11, 2119–2123. [Google Scholar] [CrossRef]
- Chinnan Velmurugan, K. Halogenases for Biosynthesis and Biocatalysis. Ph.D. Thesis, University of Manchester, Manchester, UK, 2014. [Google Scholar]
- Andorfer, M.C.; Grob, J.E.; Hajdin, C.E.; Chael, J.R.; Siuti, P.; Lilly, J.; Tan, K.L.; Lewis, J.C. Understanding Flavin-Dependent Halogenase Reactivity via Substrate Activity Profiling. ACS Catal. 2017, 7, 1897–1904. [Google Scholar] [CrossRef]
- Fujimori, D.G.; Hrvatin, S.; Neumann, C.S.; Strieker, M.; Marahiel, M.A.; Walsh, C.T. Cloning and characterization of the biosynthetic gene cluster for kutznerides. Proc. Nat. Acad. Sci. USA 2007, 104, 16498–16503. [Google Scholar] [CrossRef] [Green Version]
- Rachid, S.; Krug, D.; Kunze, B.; Kochems, I.; Scharfe, M.; Zabriskie, T.M.; Blöcker, H.; Müller, R. Molecular and biochemical studies of chondramide formation—Highly cytotoxic natural products from Chondromyces crocatus Cm c5. Chem. Biol. 2006, 13, 667–681. [Google Scholar] [CrossRef]
- Seibold, C.; Schnerr, H.; Rumpf, J.; Kunzendorf, A.; Hatscher, C.; Wage, T.; Ernyei, A.J.; Dong, C.; Naismith, J.H.; Van Pée, K.-H. A flavin-dependent tryptophan 6-halogenase and its use in modification of pyrrolnitrin biosynthesis. Biocatal. Biotransform. 2006, 24, 401–408. [Google Scholar] [CrossRef]
- Foulston, L.C.; Bibb, M.J. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Nat. Acad. Sci. USA 2010, 107, 13461–13466. [Google Scholar] [CrossRef] [Green Version]
- Zehner, S.; Kotzsch, A.; Bister, B.; Süssmuth, R.D.; Méndez, C.; Salas, J.A.; van Peé, K.-H. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. [Google Scholar] [CrossRef]
- Ismail, M.; Frese, M.; Patschkowski, T.; Ortseifen, V.; Niehaus, K.; Sewald, N. Flavin-Dependent Halogenases from Xanthomonas campestris pv. campestris B100 Prefer Bromination over Chlorination. Adv. Synth. Catal. 2019, 361, 2475–2486. [Google Scholar] [CrossRef]
- Neubauer, P.R.; Widmann, C.; Wibberg, D.; Schroder, L.; Frese, M.; Kottke, T.; Kalinowski, J.; Niemann, H.H.; Sewald, N. A flavin-dependent halogenase from metagenomic analysis prefers bromination over chlorination. PLoS ONE 2018, 13, e0196797. [Google Scholar] [CrossRef]
- Kirner, S.; Hammer, P.E.; Hill, D.S.; Altmann, A.; Fischer, I.; Weislo, L.J.; Lanahan, M.; van Pée, K.-H.; Ligon, J.M. Functions encoded by pyrrolnitrin biosynthetic genes from Pseudomonas fluorescens. J. Bacteriol. 1998, 180, 1939–1943. [Google Scholar]
- Dairi, T.; Nakano, T.; Aisaka, K.; Katsumata, R.; Hasegawa, M. Cloning and nucleotide sequence of the gene responsible for chlorination of tetracycline. Biosci. Biotechnol. Biochem. 1995, 59, 1099–1106. [Google Scholar] [CrossRef]
- Dorrestein, P.C.; Yeh, E.; Garneau-Tsodikova, S.; Kelleher, N.L.; Walsh, C.T. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 13843–13848. [Google Scholar] [CrossRef]
- Zhang, X.; Parry, R.J. Cloning and characterization of the pyrrolomycin biosynthetic gene clusters from Actinosporangium vitaminophilum ATCC 31673 and Streptomyces sp. strain UC 11065. Antimicrob. Agents Chemother. 2007, 51, 946–957. [Google Scholar] [CrossRef]
- Qiao, Y.; Yan, J.; Jia, J.; Xue, J.; Qu, X.; Hu, Y.; Deng, Z.; Bi, H.; Zhu, D. Characterization of the Biosynthetic Gene Cluster for the Antibiotic Armeniaspirols in Streptomyces armeniacus. J. Nat. Prod. 2019, 82, 318–323. [Google Scholar] [CrossRef]
- Bayer, K.; Scheuermayer, M.; Fieseler, L.; Hentschel, U. Genomic mining for novel FADH 2-dependent halogenases in marine sponge-associated microbial consortia. Mar. Biotechnol. 2013, 15, 63–72. [Google Scholar] [CrossRef]
- Pelzer, S.; Süssmuth, R.; Heckmann, D.; Recktenwald, J.; Huber, P.; Jung, G.; Wohlleben, W. Identification and analysis of the balhimycin biosynthetic gene cluster and its use for manipulating glycopeptide biosynthesis in Amycolatopsis mediterranei DSM5908. Antimicrob. Agents Chemother. 1999, 43, 1565–1573. [Google Scholar] [CrossRef]
- Lin, S.; Van Lanen, S.G.; Shen, B. Regiospecific chlorination of (S)-β-tyrosyl-S-carrier protein catalyzed by SgcC3 in the biosynthesis of the enediyne antitumor antibiotic C-1027. J. Am. Chem. Soc. 2007, 129, 12432–12438. [Google Scholar] [CrossRef] [PubMed]
- Wynands, I.; van Pee, K.H. A novel halogenase gene from the pentachloropseudilin producer Actinoplanes sp. ATCC 33002 and detection of in vitro halogenase activity. FEMS Microbiol. Lett. 2004, 237, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Philmus, B.; Chang, J.H.; Loper, J.E. Novel mechanism of metabolic co-regulation coordinates the biosynthesis of secondary metabolites in Pseudomonas protegens. eLife 2017, 6, e22835. [Google Scholar] [CrossRef] [PubMed]
- El Gamal, A.; Agarwal, V.; Diethelm, S.; Rahman, I.; Schorn, M.A.; Sneed, J.M.; Louie, G.V.; Whalen, K.E.; Mincer, T.J.; Noel, J.P. Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase–thioesterase enzyme pair. Proc. Nat. Acad. Sci. USA 2016, 113, 3797–3802. [Google Scholar] [CrossRef] [PubMed]
- Podzelinska, K.; Latimer, R.; Bhattacharya, A.; Vining, L.C.; Zechel, D.L.; Jia, Z. Chloramphenicol biosynthesis: The structure of CmlS, a flavin-dependent halogenase showing a covalent flavin–aspartate bond. J. Mol. Biol. 2010, 397, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Buedenbender, S.; Rachid, S.; Müller, R.; Schulz, G.E. Structure and action of the myxobacterial chondrochloren halogenase CndH: A new variant of FAD-dependent halogenases. J. Mol. Biol. 2009, 385, 520–530. [Google Scholar] [CrossRef]
- Menon, B.R.K.; Brandenburger, E.; Sharif, H.H.; Klemstein, U.; Shepherd, S.A.; Greaney, M.F.; Micklefield, J. RadH: A Versatile Halogenase for Integration into Synthetic Pathways. Angew. Chem. Int. Ed. 2017, 56, 11841–11845. [Google Scholar] [CrossRef]
- Zhou, H.; Qiao, K.; Gao, Z.; Vederas, J.C.; Tang, Y. Insights into radicicol biosynthesis via heterologous synthesis of intermediates and analogs. J. Biol. Chem. 2010, 285, 41412–41421. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, S.; Niu, S.; Ma, L.; Zhang, G.; Zhang, H.; Zhang, G.; Ju, J.; Zhang, C. Characterization of tiacumicin B biosynthetic gene cluster affording diversified tiacumicin analogues and revealing a tailoring dihalogenase. J. Am. Chem. Soc. 2010, 133, 1092–1105. [Google Scholar] [CrossRef]
- Wagner, C.; El Omari, M.; König, G.M. Biohalogenation: Nature’s way to synthesize halogenated metabolites. J. Nat. Prod. 2009, 72, 540–553. [Google Scholar] [CrossRef]
- Yeh, E.; Blasiak, L.C.; Koglin, A.; Drennan, C.L.; Walsh, C.T. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry 2007, 46, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Gkotsi, D.; Ludewig, H.; Sharma, S.; Connolly, J.; Dhaliwal, J.; Wang, Y.; Unsworth, W.; Taylor, R.; McLachlan, M.; Shanahan, S. A marine viral halogenase that iodinates diverse substrates. Nat. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cahn, J.K.B.; Wilson, M.C.; Leopold-Messer, S.; Piel, J. A novel class of flavin-dependent halogenase from an uncultivated microbe suggests convergent evolution. In Proceedings of the BIOTRANS 14th International symposium on Biocatalysis and Biotransformations, Poster Abstract, Groningen, The Netherlands, 7–11 July 2019; p. 132. [Google Scholar]
- Kemker, I.; Schnepel, C.; Schröder, D.C.C.; Marion, A.; Sewald, N. Cyclization of RGD peptides by Suzuki-Miyaura cross-coupling. J. Med. Chem. 2019, 62, 7417–7430. [Google Scholar] [CrossRef] [PubMed]
- Mantri, M.; Krojer, T.; Bagg, E.A.; Webby, C.J.; Butler, D.S.; Kochan, G.; Kavanagh, K.L.; Oppermann, U.; McDonough, M.A.; Schofield, C.J. Crystal structure of the 2-oxoglutarate-and Fe (II)-dependent lysyl hydroxylase JMJD6. J. Mol. Biol. 2010, 401, 211–222. [Google Scholar] [CrossRef]
- Blasiak, L.C.; Vaillancourt, F.H.; Walsh, C.T.; Drennan, C.L. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature 2006, 440, 368. [Google Scholar] [CrossRef]
- Zhang, Z.; Ren, J.-S.; Harlos, K.; McKinnon, C.H.; Clifton, I.J.; Schofield, C.J. Crystal structure of a clavaminate synthase–Fe (II)–2-oxoglutarate–substrate–NO complex: Evidence for metal centred rearrangements. FEBS Lett. 2002, 517, 7–12. [Google Scholar] [CrossRef]
- Martinez, S.; Fellner, M.; Herr, C.Q.; Ritchie, A.; Hu, J.; Hausinger, R.P. Structures and mechanisms of the non-heme Fe (II)-and 2-oxoglutarate-dependent ethylene-forming enzyme: Substrate binding creates a twist. J. Am. Chem. Soc. 2017, 139, 11980–11988. [Google Scholar] [CrossRef]
- Wong, S.D.; Srnec, M.; Matthews, M.L.; Liu, L.V.; Kwak, Y.; Park, K.; Bell III, C.B.; Alp, E.E.; Zhao, J.; Yoda, Y. Elucidation of the Fe (IV)= O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 2013, 499, 320. [Google Scholar] [CrossRef]
- Matthews, M.L.; Neumann, C.S.; Miles, L.A.; Grove, T.L.; Booker, S.J.; Krebs, C.; Walsh, C.T.; Bollinger, J.M. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. USA 2009, 106, 17723–17728. [Google Scholar] [CrossRef] [Green Version]
- Vaillancourt, F.H.; Yin, J.; Walsh, C.T. SyrB2 in syringomycin E biosynthesis is a nonheme FeII α-ketoglutarate-and O2-dependent halogenase. Proc. Nat. Acad. Sci. USA 2005, 102, 10111–10116. [Google Scholar] [CrossRef]
- Wong, C.; Fujimori, D.G.; Walsh, C.T.; Drennan, C.L. Structural analysis of an open active site conformation of nonheme iron halogenase CytC3. J. Am. Chem. Soc. 2009, 131, 4872–4879. [Google Scholar] [CrossRef] [PubMed]
- Ueki, M.; Galonić, D.P.; Vaillancourt, F.H.; Garneau-Tsodikova, S.; Yeh, E.; Vosburg, D.A.; Schroeder, F.C.; Osada, H.; Walsh, C.T. Enzymatic generation of the antimetabolite γ, γ-dichloroaminobutyrate by NRPS and mononuclear iron halogenase action in a streptomycete. Chem. Biol. 2006, 13, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Zhu, Q.; Maggiolo, A.O.; Ananth, N.R.; Hillwig, M.L.; Liu, X.; Boal, A.K. Structural basis for halogenation by iron-and 2-oxo-glutarate-dependent enzyme WelO5. Nat. Chem. Biol. 2016, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Hillwig, M.L.; Liu, X. A new family of iron-dependent halogenases acts on freestanding substrates. Nat. Chem. Biol. 2014, 10, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Hillwig, M.L.; Zhu, Q.; Ittiamornkul, K.; Liu, X. Discovery of a Promiscuous Non-Heme Iron Halogenase in Ambiguine Alkaloid Biogenesis: Implication for an Evolvable Enzyme Family for Late-Stage Halogenation of Aliphatic Carbons in Small Molecules. Angew. Chem. Int. Ed. 2016, 55, 5780–5784. [Google Scholar] [CrossRef]
- Liu, X. In vitro Analysis of Cyanobacterial Nonheme Iron-Dependent Aliphatic Halogenases WelO5 and AmbO5. In Methods Enzymol; Elsevier: Amsterdam, The Netherlands, 2018; Volume 604, pp. 389–404. [Google Scholar]
- Zhu, Q.; Liu, X. Characterization of non-heme iron aliphatic halogenase WelO5 * from Hapalosiphon welwitschii IC-52-3: Identification of a minimal protein sequence motif that confers enzymatic chlorination specificity in the biosynthesis of welwitindolelinones. Beilstein J. Org. Chem. 2017, 13, 1168–1173. [Google Scholar] [CrossRef]
- Hayashi, T.; Ligibel, M.; Sager, E.; Voss, M.; Hunziker, J.; Schroer, K.; Snajdrova, R.; Buller, R. Evolved Aliphatic Halogenases Enable Regiocomplementary C-H Functionalization of an Added-Value Chemical. Angew. Chem. Int. Ed. 2019, (in press). [Google Scholar] [CrossRef]
- Flatt, P.M.; O’Connell, S.J.; McPhail, K.L.; Zeller, G.; Willis, C.L.; Sherman, D.H.; Gerwick, W.H. Characterization of the initial enzymatic steps of barbamide biosynthesis. J. Nat. Prod. 2006, 69, 938–944. [Google Scholar] [CrossRef]
- Moosmann, P.; Ueoka, R.; Gugger, M.; Piel, J. Aranazoles: Extensively Chlorinated Nonribosomal Peptide–Polyketide Hybrids from the Cyanobacterium Fischerella sp. PCC 9339. Org. Lett. 2018, 20, 5238–5241. [Google Scholar] [CrossRef]
- Matthews, M.L.; Krest, C.M.; Barr, E.W.; Vaillancourt, F.H.; Walsh, C.T.; Green, M.T.; Krebs, C.; Bollinger, J.M., Jr. Substrate-triggered formation and remarkable stability of the C− H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. [Google Scholar] [CrossRef]
- Welch, K.D.; Davis, T.Z.; Aust, S.D. Iron autoxidation and free radical generation: Effects of buffers, ligands, and chelators. Arch. Biochem. Biophys. 2002, 397, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.L.; Chang, W.-c.; Layne, A.P.; Miles, L.A.; Krebs, C.; Bollinger, J.M., Jr. Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat. Chem. Biol. 2014, 10, 209–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Hillwig, M.L.; Doi, Y.; Liu, X. Aliphatic Halogenase Enables Late-Stage C− H Functionalization: Selective Synthesis of a Brominated Fischerindole Alkaloid with Enhanced Antibacterial Activity. ChemBioChem 2016, 17, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Dunham, N.P.; Bergman, J.A.; Wang, B.; Zhu, Q.; Chang, W.-c.; Liu, X.; Boal, A.K. Structure-guided reprogramming of a hydroxylase to halogenate its small molecule substrate. Biochemistry 2017, 56, 441–444. [Google Scholar] [CrossRef]
- Khare, D.; Wang, B.; Gu, L.; Razelun, J.; Sherman, D.H.; Gerwick, W.H.; Håkansson, K.; Smith, J.L. Conformational switch triggered by α-ketoglutarate in a halogenase of curacin A biosynthesis. Proc. Nat. Acad. Sci. USA 2010, 107, 14099–14104. [Google Scholar] [CrossRef]
- Vaillancourt, F.H.; Yeh, E.; Vosburg, D.A.; O’Connor, S.E.; Walsh, C.T. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature 2005, 436, 1191. [Google Scholar] [CrossRef]
- Schaffrath, C.; Deng, H.; O’Hagan, D. Isolation and characterisation of 5′-fluorodeoxyadenosine synthase, a fluorination enzyme from Streptomyces cattleya. FEBS Lett. 2003, 547, 111–114. [Google Scholar] [CrossRef]
- O’Hagan, D.; Deng, H. Enzymatic fluorination and biotechnological developments of the fluorinase. Chem. Rev. 2014, 115, 634–649. [Google Scholar] [CrossRef]
- Cadicamo, C.D.; Courtieu, J.; Deng, H.; Meddour, A.; O’Hagan, D. Enzymatic fluorination in Streptomyces cattleya takes place with an inversion of configuration consistent with an SN2 reaction mechanism. ChemBioChem 2004, 5, 685–690. [Google Scholar] [CrossRef]
- Dong, C.; Huang, F.; Deng, H.; Schaffrath, C.; Spencer, J.B.; O’Hagan, D.; Naismith, J.H. Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature 2004, 427, 561–565. [Google Scholar] [CrossRef]
- Lohman, D.C.; Edwards, D.R.; Wolfenden, R. Catalysis by desolvation: The catalytic prowess of SAM-dependent halide-alkylating enzymes. J. Am. Chem. Soc. 2013, 135, 14473–14475. [Google Scholar] [CrossRef] [PubMed]
- Eustáquio, A.S.; O’Hagan, D.; Moore, B.S. Engineering fluorometabolite production: Fluorinase expression in Salinispora tropica yields fluorosalinosporamide. J. Nat. Prod. 2010, 73, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Cobb, S.L.; McEwan, A.R.; McGlinchey, R.P.; Naismith, J.H.; O’Hagan, D.; Robinson, D.A.; Spencer, J.B. The fluorinase from Streptomyces cattleya is also a chlorinase. Angew. Chem. Int. Ed. 2006, 45, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Lowe, P.T.; Dall’Angelo, S.; Mulder-Krieger, T.; IJzerman, A.P.; Zanda, M.; O’Hagan, D. A New Class of Fluorinated A2A Adenosine Receptor Agonist with Application to Last-Step Enzymatic [18F] Fluorination for PET Imaging. ChemBioChem 2017, 18, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Lowe, P.T.; Dall’Angelo, S.; Fleming, I.N.; Piras, M.; Zanda, M.; O’Hagan, D. Enzymatic radiosynthesis of a 18 F-Glu-Ureido-Lys ligand for the prostate-specific membrane antigen (PSMA). Org. Biomol. Chem. 2019, 17, 1480–1486. [Google Scholar] [CrossRef]
- Lowe, P.T.; Dall’Angelo, S.; Devine, A.; Zanda, M.; O’Hagan, D. Enzymatic Fluorination of Biotin and Tetrazine Conjugates for Pretargeting Approaches to Positron Emission Tomography Imaging. ChemBioChem 2018, 19, 1969–1978. [Google Scholar] [CrossRef]
- Deng, H.; Ma, L.; Bandaranayaka, N.; Qin, Z.; Mann, G.; Kyeremeh, K.; Yu, Y.; Shepherd, T.; Naismith, J.H.; O’Hagan, D. Identification of fluorinases from Streptomyces sp. MA37, Norcardia brasiliensis, and Actinoplanes sp. N902-109 by Genome Mining. ChemBioChem 2014, 15, 364–368. [Google Scholar] [CrossRef]
- Ma, L.; Li, Y.; Meng, L.; Deng, H.; Li, Y.; Zhang, Q.; Diao, A. Biological fluorination from the sea: Discovery of a SAM-dependent nucleophilic fluorinating enzyme from the marine-derived bacterium Streptomyces xinghaiensis NRRL B24674. RSC Adv. 2016, 6, 27047–27051. [Google Scholar] [CrossRef]
- Sun, H.; Yeo, W.L.; Lim, Y.H.; Chew, X.; Smith, D.J.; Xue, B.; Chan, K.P.; Robinson, R.C.; Robins, E.G.; Zhao, H. Directed Evolution of a Fluorinase for Improved Fluorination Efficiency with a Non-native Substrate. Angew. Chem. 2016, 128, 14489–14492. [Google Scholar] [CrossRef]
- Yeo, W.; Chew, X.; Smith, D.; Chan, K.; Sun, H.; Zhao, H.; Lim, Y.; Ang, E. Probing the molecular determinants of fluorinase specificity. Chem. Commun. 2017, 53, 2559–2562. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.; McMahon, S.A.; Naismith, J.H.; O’Hagan, D. Exploration of a potential difluoromethyl-nucleoside substrate with the fluorinase enzyme. Bioorg. Chem. 2016, 64, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Zhao, H.; Ang, E. A coupled chlorinase–fluorinase system with a high efficiency of trans-halogenation and a shared substrate tolerance. Chem. Commun. 2018, 54, 9458–9461. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Expression | Kinetic Parameters | Substrates | |
|---|---|---|---|---|
| Host | Yield [mg/L] | |||
| Cf-CPO | C. fumago * | 430 [67] | 0.78 mm h−1 [59] | aromatic, alkenes |
| E. coli BL21(DE3) | n.a. [68] | |||
| Aspergillus niger | 10 [68] | |||
| BPO | Pseudomonas aureofaciens [65] | n.a. | n.a. partial diastereo-selectivity [69] | aromatic, N-hetero-cycles, alkenes |
| Penicillus capitatus [64,65] | n.a. | |||
| Enzyme | Expression | Kinetic Parameters | Substrates | |
|---|---|---|---|---|
| Host | Yield [mg/L] | |||
| Ci-VClPO | C. inaequalis | 10 [70] | 5.1 × 107 M−1 s−1 for Br− [75] | aromatic, alkenes |
| E. coli BL21(DE3) | 15 [76] | |||
| S. cerevisiae | 100 [75] | |||
| Co-VBrPO | C. officinalis | 200 U/mg for MCD [83] | aromatic, N-hetero-cycles, alkenes | |
| E. coli BL21(DE3) (insoluble) | 40 | |||
| Enzyme | Origin | Heterologous Expression Host 1 | Product | Miscellaneous |
|---|---|---|---|---|
| PrnA [96] | Pseudomonas fluorescens BL915 | E. coli ArcticExpress (DE3) | 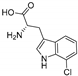 | |
| RebH [92] | Lechevalieria aerocolonigenes strain 39243 | E. coli BL21(DE3) | 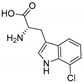 | |
| KtzQ [110] | Kutzneria sp. 744 | 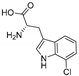 | ||
| KtzR [110] | Kutzneria sp. 744 |  | ||
| CmdE [111] | Chondromyces crocatus Cm c5 |  | Post-NRPS (non-ribosomal peptides) | |
| SSTH [101] | Streptomyces toxytricini NRRL 15443 | E. coli BL21 CodonPlus (DE3)-RIL |  | |
| Thal [112] | Streptomyces albogriseolus | P. fluorescens BL915 DORF1 and P. chlororaphis ACN | 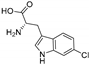 | |
| MibH [99,113] | Microbispora coralline NRRL 30420 | 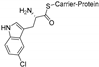 | NRPS-dependent | |
| PyrH [96,114] | Streptomyces rugosporus LL-42D005 | E. coli ArcticExpress (DE3); Pseudomonas fluorescens BL915 ΔORF1 | 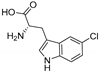 | |
| Xcc-B B100XXXX [115] | Xanthomonas campestris pv. campestris strain B100 | E. coli BL21(DE3) with pGro7 plasmid (Takara) for chaperone co-expression | Various substituted indoles and thereby differing regio-selectivity | |
| BrvH [116] | Brevundimonas BAL3 | E. coli BL21(DE3) with pGro7 plasmid (Takara) for chaperone co-expression | 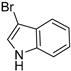 | |
| PrnC [93,117] | Pseudomonas fluorescens BL915 | 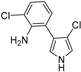 | ||
| Chl [118] | Streptomyces aureofaciens | 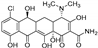 | ||
| ChlA [105] | Dictyostelium discoideum |  | ||
| PltA [119] | Pseudomonas fluorescens Pf-5 | E. coli BL21(DE3) |  | |
| Pyr29 [120] | Actinosporangium vitaminophilum ATCC 31673 and Streptomyces sp. Strain UC 11065 | 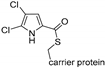 | ||
| Arm21 [121] | Streptomyces armeniacus | 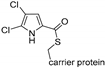 | ||
| CrpH [104] | Nostoc Cyanobionts |  | NRPS-dependent | |
| BhaA [122,123] | Amycolatopsis mediterranei DSM5908 | balhimycin | ||
| SgcC3 [124] | Streptomyces globisporus | E. coli BL21(DE3) pET-30Xa/LIC | 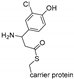 | |
| HalB [125] | Actinoplanes sp. ATCC 33002 | Pseudomonas aureofaciens ACN |  | |
| PltM [126] | Pseudomonas fluorescens Pf-5 | E. coli BL21 (DE3) |  | |
| Bmp5 [106] | Pseudoalteromonas luteoviolacea | 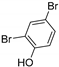 | ||
| Bmp2 [127] | Pseudoalteromonas luteoviolacea | 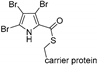 | NRPS-dependent | |
| (PltD) [13] | Pseudomonas fluorescens Pf-5 | n.a. | Not clear if FHal | |
| CmlS [128] | Streptomyces venezuelae |  | Flavin covalently bound to aspartate via CH3-Group | |
| CndH [129] | Chondromyces crocatus | 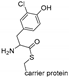 | NRPS-dependent | |
| RadH [107,130] | Chaetomium chiversii | E. coli Rosetta 2(DE3) | monocillin II | |
| Rdc2 [107,131] | Pochonia chlamydosporia | S. cerevisiae strain BJ5464-Npg E. coli BL21(DE3) | monocillin II | |
| TiaM [132] | Dactylosporangium aurantiacum NRRL 18085 | E. coli BL21(DE3) | tiacumicin B intermediate | NRPS-dependent |
| Enzyme | Origin/Expression Host and Yield | Features | Product/Biosynthesis |
|---|---|---|---|
| SyrB2 [139] | Pseudomonas syringae pv. syringae B301D/ E. coli strain B834(DE3) [139] n.a. | total turnover: 7 ± 2 [144] | 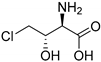 4-chloro-l-threonine/ syringomycin E |
| CytC3 [145] | Streptomyces sp./ E. coli BL21(DE3) [146] n.a. | n.a |  4,4-dichloro-l-valine/ dichloroaminobutyrate |
| WelO5 [147] | Hapalosiphon welwitschii/ E. coli C43(DE3) [148,149] or BL21(DE3) [150] 20 mg L−1 [151] | total turnover: 75 [148] kcat: 1.8 ± 0.2 min−1 [149] |  12-epi-fischerindole G/ fischerindole & hapalindole alkaloids |
| AmbO5 | Fischerella ambigua/ E. coli C43(DE3) [149] or BL21(DE3) [150] n.a. | kcat: 1.7 ± 0.1 min−1 [149] | 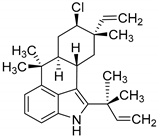 ambiguine A/ ambiguine, fischerindole and hapalindole alkaloids |
| WelO5* variant isoform of WelO5 (CB2) [152] | Hapalosiphon welwitschii IC-52-3/ E. coli BL21(DE3) n.a. | KM: 0.67 mM, kcat: 3.0 min−1 | 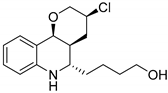 martinelline-derived fragment |
| Enzyme | Origin/Expression Host and Yield | Kinetic Parameters | Special Substrate Scope |
|---|---|---|---|
| FlA [165] | Streptomyces cattleya/ E. coli BL21(DE3) 50 mg L−1 [173] | KM: 29.4 ± 5.80 µm kcat: 0.084 ± 0.005 min−1 [173] | [169,170,171] |
| FlA1 | Streptomyces sp. MA37/ E. coli BL21(DE3) n.a. | KM: 8.36 ± 0.82 µm kcat: 0.13 min−1 [174] | |
| FlA4 | Streptomyces xinghaiensis/ E. coli BL21(DE3) n.a. | KM: 29.87 µm kcat: 0.69 ± 0.01 min−1 [177] | [177] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fejzagić, A.V.; Gebauer, J.; Huwa, N.; Classen, T. Halogenating Enzymes for Active Agent Synthesis: First Steps Are Done and Many Have to Follow. Molecules 2019, 24, 4008. https://doi.org/10.3390/molecules24214008
Fejzagić AV, Gebauer J, Huwa N, Classen T. Halogenating Enzymes for Active Agent Synthesis: First Steps Are Done and Many Have to Follow. Molecules. 2019; 24(21):4008. https://doi.org/10.3390/molecules24214008
Chicago/Turabian StyleFejzagić, Alexander Veljko, Jan Gebauer, Nikolai Huwa, and Thomas Classen. 2019. "Halogenating Enzymes for Active Agent Synthesis: First Steps Are Done and Many Have to Follow" Molecules 24, no. 21: 4008. https://doi.org/10.3390/molecules24214008
APA StyleFejzagić, A. V., Gebauer, J., Huwa, N., & Classen, T. (2019). Halogenating Enzymes for Active Agent Synthesis: First Steps Are Done and Many Have to Follow. Molecules, 24(21), 4008. https://doi.org/10.3390/molecules24214008