
Insight into the Structure and Properties of Novel Imidazole-Based Salts of Salicylic Acid

 , , ,
, , , 
Abstract
1. Introduction
2. Results and Discussion
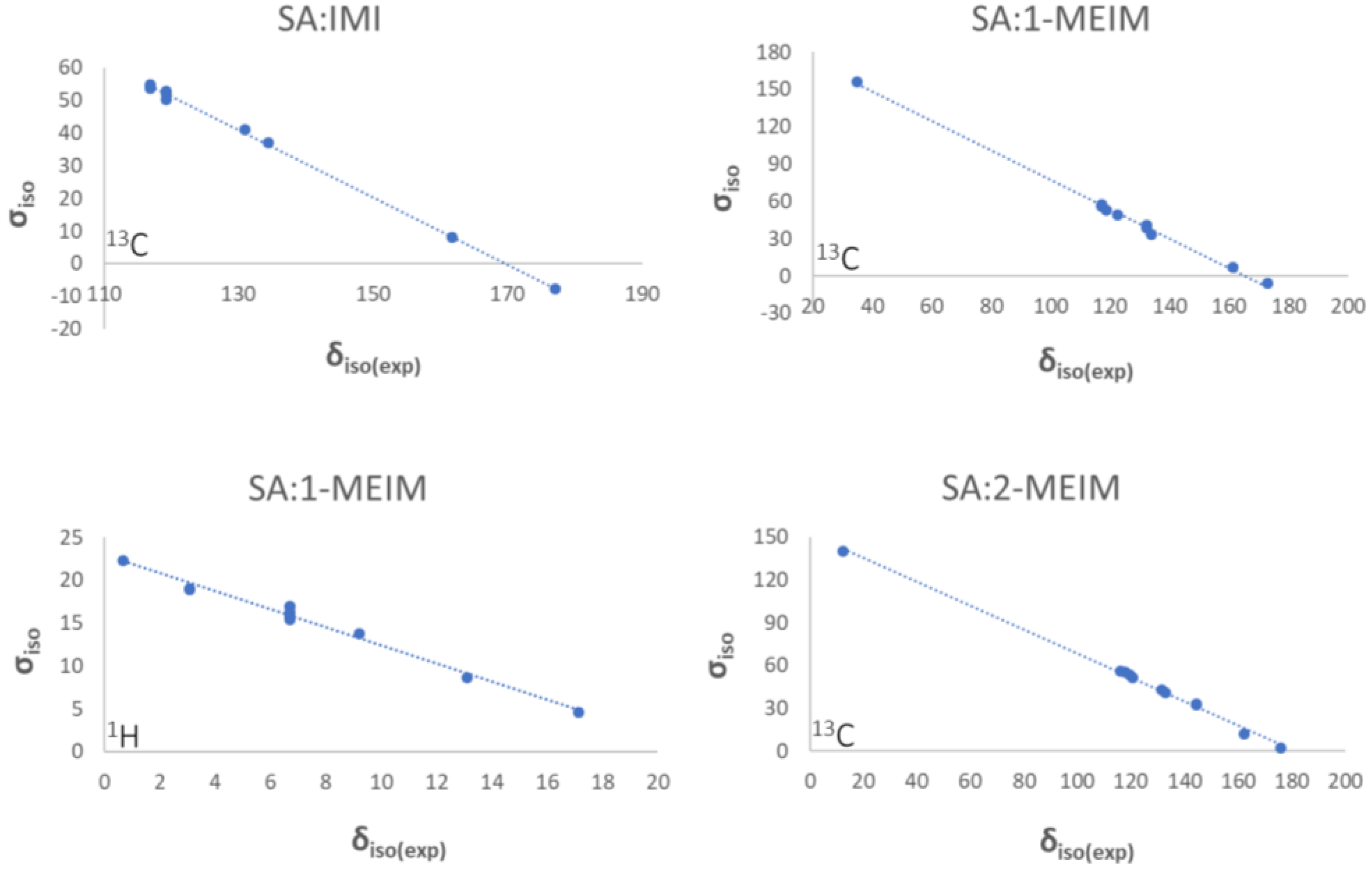
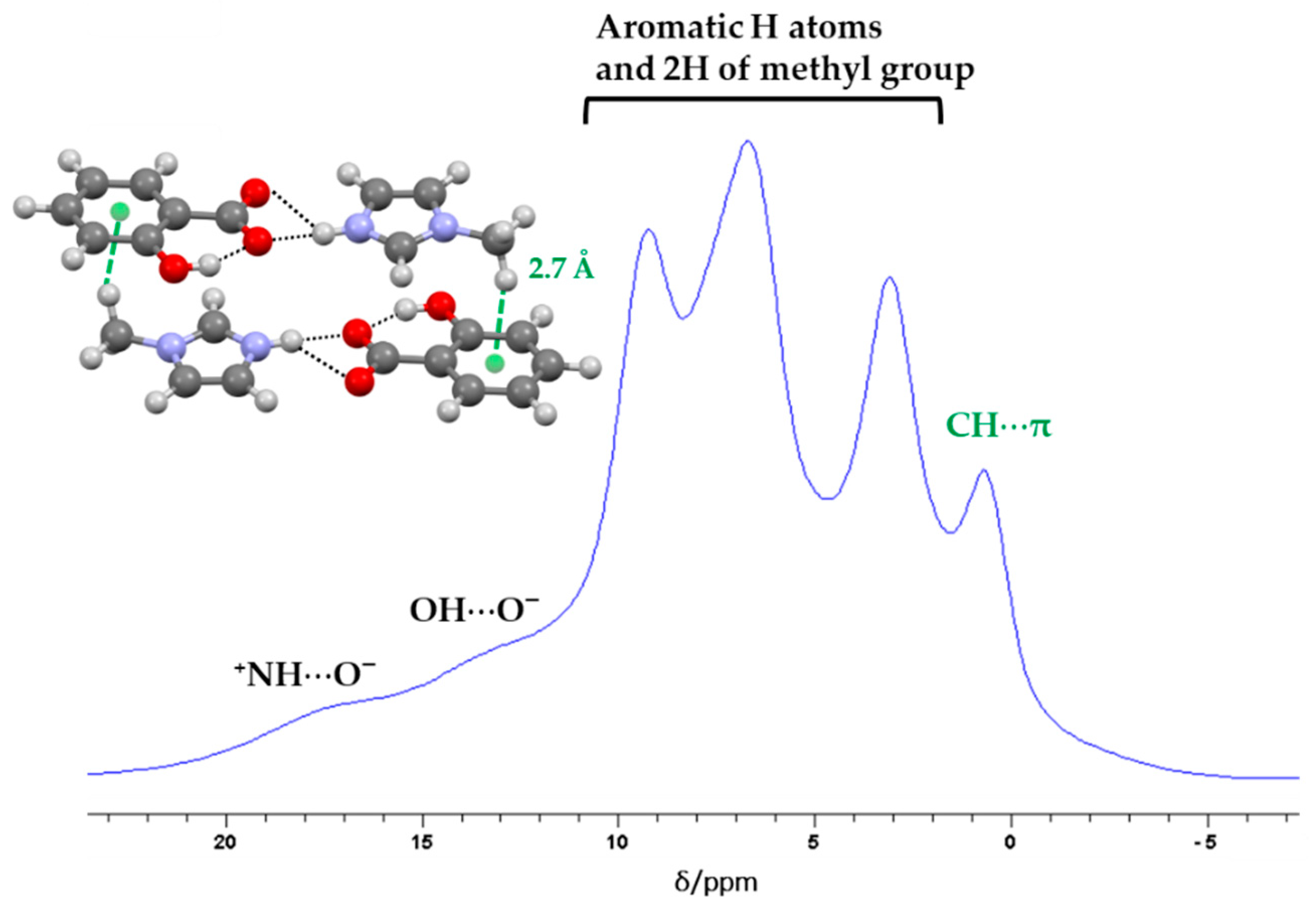
2.1. Structural Analysis
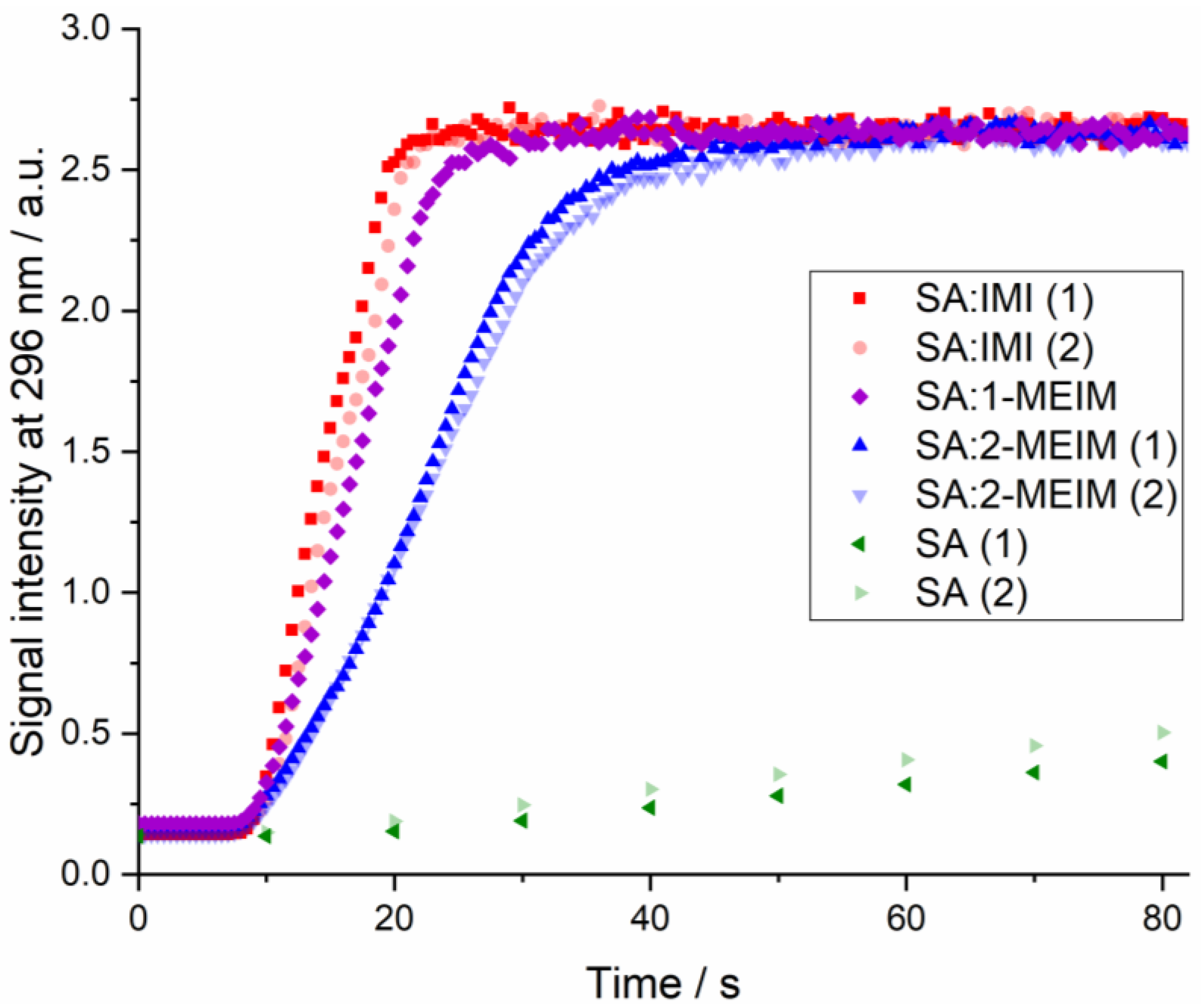
2.2. Structure-Property Relationships
3. Materials and Methods
3.1. Synthesis
3.2. Powder X-Ray Diffraction (PXRD)
3.3. Density Functional Theory (DFT) Calculations
3.4. Solid-State Nuclear Magnetic Resonance (ssNMR)
3.5. Thermal Stability Studies
3.6. Solubility Studies
3.7. Dissolution Rate Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martins, I.C.B.; Sardo, M.; Alig, E.; Fink, L.; Schmidt, M.U.; Mafra, L.; Duarte, M.T. Enhancing Adamantylamine Solubility through Salt Formation: Novel Products Studied by X-ray Diffraction and Solid-State NMR. Cryst. Growth Des. 2019, 19, 1860–1873. [Google Scholar] [CrossRef]
- Hodgson, J. ADMET - turning chemicals into drugs. Nat. Biotechnol. 2001, 19, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- ICH Harmonized Tripartite Guideline Q6A, 06 October 1999, ICH Harmonisation for Better Health. Available online: https://www.ich.org/page/quality-guidelines (accessed on 19 October 2019).
- Elder, D.P.; Holm, R.; de Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Domingos, S.; Andre, V.; Quaresma, S.; Martins, I.C.B.; da Piedade, M.F.M.; Duarte, M.T. New forms of old drugs: Improving without changing. J. Pharm. Pharmacol. 2015, 67, 830–846. [Google Scholar] [CrossRef]
- Nangia, A. Supramolecular chemistry and crystal engineering. J. Chem. Sci. 2010, 122, 295–310. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: A brief overview. J. Chem. Sci. 2010, 122, 667–675. [Google Scholar] [CrossRef]
- Chan, F.C.; Anwar, J.; Cernik, R.; Barnes, P.; Wilson, R.M. Ab initio structure determination of sulfathiazole polymorph V from synchrotron X-ray powder diffraction data. J. Appl. Crystallogr. 1999, 32, 436–441. [Google Scholar] [CrossRef]
- Portalone, G.; Ballirano, P.; Maras, A. The crystal structure of 3-methyluracil from X-ray powder diffraction data. J. Mol. Struct. 2002, 608, 35–39. [Google Scholar] [CrossRef]
- Nowell, H.; Attfield, J.P.; Cole, J.C.; Cox, P.J.; Shankland, K.; Maginn, S.J.; Motherwell, W.D.S. Structure solution and refinement of tetracaine hydrochloride from X-ray powder diffraction data. New J. Chem. 2002, 26, 469–472. [Google Scholar] [CrossRef]
- Fischer, F.; Schmidt, M.U.; Greiser, S.; Emmerling, F. The challenging case of the theophylline-benzamide cocrystal. Acta Crystallogr. Sect. C-Struct. Chem. 2016, 72, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.; Schmidt, M.U. The determination of crystal structures of active pharmaceutical ingredients from X-ray powder diffraction data: A brief, practical introduction, with fexofenadine hydrochloride as example. J. Pharm. Pharmacol. 2015, 67, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Florence, A.J.; Shankland, N.; Shankland, K.; David, W.I.F.; Pidcock, E.; Xu, X.L.; Johnston, A.; Kennedy, A.R.; Cox, P.J.; Evans, J.S.O.; et al. Solving molecular crystal structures from laboratory X-ray powder diffraction data with DASH: The state of the art and challenges. J. Appl. Crystallogr. 2005, 38, 249–259. [Google Scholar] [CrossRef]
- Schlesinger, C.; Tapmeyer, L.; Gumbert, S.D.; Prill, D.; Bolte, M.; Schmidt, M.U.; Saal, C. Absolute Configuration of Pharmaceutical Research Compounds Determined by X-ray Powder Diffraction. Angew. Chem. -Int. Ed. 2018, 57, 9150–9153. [Google Scholar] [CrossRef]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Sardo, M.; Santos, S.M.; Femandes, A.; Antunes, A.; Andre, V.; Mafra, L.; Duarte, M.T. Packing Interactions and Physicochemical Properties of Novel Multicomponent Crystal Forms of the Anti-Inflammatory Azelaic Acid Studied by X-ray and Solid-State NMR. Cryst. Growth Des. 2016, 16, 154–166. [Google Scholar] [CrossRef]
- Mafra, L.; Santos, S.M.; Siegel, R.; Alves, I.; Almeida Paz, F.A.; Dudenko, D.; Spiess, H.W. Packing Interactions in Hydrated and Anhydrous Forms of the Antibiotic Ciprofloxacin: A Solid-State NMR, X-ray Diffraction, and Computer Simulation Study. J. Am. Chem. Soc. 2012, 134, 71–74. [Google Scholar] [CrossRef]
- Dempah, K.E.; Barich, D.H.; Kaushal, A.M.; Zong, Z.X.; Desai, S.D.; Suryanarayanan, R.; Kirsch, L.; Munson, E.J. Investigating Gabapentin Polymorphism Using Solid-State NMR Spectroscopy. Aaps Pharmscitech 2013, 14, 19–28. [Google Scholar] [CrossRef]
- Andre, V.; Martins, I.; Quaresma, S.; Martins, M.; Duarte, M.T. Transforming aspirin into novel molecular salts of salicylic acid. Struct. Chem. 2014, 25, 707–714. [Google Scholar] [CrossRef]
- Hreiche, R.; Plante, I.; Drolet, B.; Morissette, P.; Turgeon, J. Pharmacokinetics, Pharmacodynamics and drug metabolism. J. Pharm. Sci. 2011, 100, 2469–2481. [Google Scholar] [CrossRef]
- Pozniak, B.; Grabowski, T.; Motykiewicz-Pers, K.; Bobrek, K.; Rak, L.; Bobusia, K.; Gawel, A.; Switala, M. Pharmacokinetics of Repeated Sodium Salicylate Administration to Laying Hens: Evidence for Time Dependent Increase in Drug Elimination from Plasma and Eggs. PLoS ONE 2015, 10, e0123526. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Saito, M.; Takenaka, H. Improvement of solubility and dissolution rate of poorly water-soluble salicylic acid by a spray-drying technique. J. Pharm. Pharmacol. 1975, 27, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Seitkalieva, M.M.; Posvyatenko, A.V.; Khrustalev, V.N.; Ananikov, V.P. Cytotoxic Activity of Salicylic Acid-Containing Drug Models with Ionic and Covalent Binding. Acs Med. Chem. Lett. 2015, 6, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Skovsgaard, S.; Bond, A.D. Co-crystallisation of benzoic acid derivatives with N-containing bases in solution and by mechanical grinding: Stoichiometric variants, polymorphism and twinning. CrystEngComm 2009, 11, 444–453. [Google Scholar] [CrossRef]
- Hathwar, V.R.; Pal, R.; Guru Row, T.N. Charge Density Analysis of Crystals of Nicotinamide with Salicylic Acid and Oxalic Acid: An Insight into the Salt to Cocrystal Continuum. Cryst. Growth Des. 2010, 10, 3306–3310. [Google Scholar] [CrossRef]
- Elbagerma, M.A.; Edwards, H.G.M.; Munshi, T.; Scowen, I.J. Identification of a new co-crystal of salicylic acid and benzamide of pharmaceutical relevance. Anal. Bioanal. Chem. 2010, 397, 137–146. [Google Scholar] [CrossRef]
- Cheney, M.L.; Weyna, D.R.; Shan, N.; Hanna, M.; Wojtas, L.; Zaworotko, M.J. Supramolecular Architectures of Meloxicam Carboxylic Acid Cocrystals, a Crystal Engineering Case Study. Cryst. Growth Des. 2010, 10, 4401–4413. [Google Scholar] [CrossRef]
- Babu, N.J.; Sanphui, P.; Nangia, A. Crystal Engineering of Stable Temozolomide Cocrystals. Chem. -Asian J. 2012, 7, 2274–2285. [Google Scholar] [CrossRef]
- Elbagerma, M.A.; Edwards, H.G.M.; Munshi, T.; Hargreaves, M.D.; Matousek, P.; Scowen, I.J. Characterization of New Cocrystals by Raman Spectroscopy, Powder X-ray Diffraction, Differential Scanning Calorimetry, and Transmission Raman Spectroscopy. Cryst. Growth Des. 2010, 10, 2360–2371. [Google Scholar] [CrossRef]
- Huang, N.; Rodríguez-Hornedo, N. Effect of Micellar Solubilization on Cocrystal Solubility and Stability. Cryst. Growth Des. 2010, 10, 2050–2053. [Google Scholar] [CrossRef]
- Childs, S.L.; Wood, P.A.; Rodríguez-Hornedo, N.; Reddy, L.S.; Hardcastle, K.I. Analysis of 50 Crystal Structures Containing Carbamazepine Using the Materials Module of Mercury CSD. Cryst. Growth Des. 2009, 9, 1869–1888. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The Salt−Cocrystal Continuum: The Influence of Crystal Structure on Ionization State. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Rodriguez-Hornedo, N.; Suryanarayanan, R. A rapid thermal method for cocrystal screening. CrystEngComm 2008, 10, 665–668. [Google Scholar] [CrossRef]
- Bučar, D.-K.; Henry, R.F.; Lou, X.; Duerst, R.W.; MacGillivray, L.R.; Zhang, G.G.Z. Cocrystals of Caffeine and Hydroxybenzoic Acids Composed of Multiple Supramolecular Heterosynthons: Screening via Solution-Mediated Phase Transformation and Structural Characterization. Cryst. Growth Des. 2009, 9, 1932–1943. [Google Scholar] [CrossRef]
- Goswami, S.; Jana, S.; Hazra, A.; Fun, H.K.; Anjum, S.; Atta ur, R. Recognition of creatinine by weak aromatic acids in solid phase along with their supramolecular network. CrystEngComm 2006, 8, 712–718. [Google Scholar] [CrossRef]
- Limmatvapirat, S.; Yamaguchi, K.; Yonemochi, E.; Oguchi, T.; Yamamoto, K. A 1:1 Deoxycholic Acid-Salicylic Acid Complex. Acta Crystallogr. Sect. C 1997, 5, 803–805. [Google Scholar] [CrossRef]
- Takata, N.; Shiraki, K.; Takano, R.; Hayashi, Y.; Terada, K. Cocrystal Screening of Stanolone and Mestanolone Using Slurry Crystallization. Cryst. Growth Des. 2008, 8, 3032–3037. [Google Scholar] [CrossRef]
- Singh, T.P.; Vijayan, M. Structural studies of analgesics and their interactions. II. The crystal structure of a 1:1 complex between antipyrine and salicyclic acid (salipyrine). Acta Crystallogr. Sect. B 1974, 30, 557–562. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, H.; Rasmuson, A.C. Thermodynamics and crystallization of a theophylline-salicylic acid cocrystal. CrystEngComm 2015, 17, 4125–4135. [Google Scholar] [CrossRef]
- Kelley, S.P.; Narita, A.; Holbrey, J.D.; Green, K.D.; Reichert, W.M.; Rogers, R.D. Understanding the effects of ionicity in salts, solvates, co-crystals, ionic co-crystals, and ionic liquids, rather than nomenclature, is critical to understanding their behavior. Cryst. Growth Des. 2013, 13, 965–975. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.H.; Chow, P.S.; Tan, R.B.H. Pharmaceutical cocrystals of ethenzamide: Structural, solubility and dissolution studies. CrystEngComm 2012, 14, 8515–8524. [Google Scholar] [CrossRef]
- Anderson, E.B.; Long, T.E. Imidazole- and imidazolium-containing polymers for biology and material science applications. Polymer 2010, 51, 2447–2454. [Google Scholar] [CrossRef]
- Riduan, S.N.; Zhang, Y.G. Imidazolium salts and their polymeric materials for biological applications. Chem. Soc. Rev. 2013, 42, 9055–9070. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Rietveld, H. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallogr. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Rietveld, H. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Hofmann, D. Fast estimation of crystal densities. Acta Crystallogr. Sect. B 2002, 58, 489–493. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Solubility. In Drug-Like Properties, 2nd ed.; Academic Press: Boston, MA, USA, 2016; pp. 61–93. [Google Scholar]
- Wurster, D.E.; Taylor, P.W. Dissolution rates. J. Pharm. Sci. 1965, 54, 169–175. [Google Scholar] [CrossRef]
- Cochran, W. The crystal and molecular structure of salicylic acid. Acta Crystallogr. 1953, 6, 260–268. [Google Scholar] [CrossRef]
- Martinez-Carrera, S. The crystal structure of imidazole at −150 °C. Acta Crystallogr. 1966, 20, 783–789. [Google Scholar] [CrossRef]
- Wang, A.; Craven, B.M. Crystal structure of 1:1 complex of barbital with 1-methylimidazole. J. Pharm. Sci. 1979, 68, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Hachuła, B.; Nowak, M.; Kusz, J. Crystal and Molecular structure analysis of 2-methylimidazole. J. Chem. Crystallogr. 2010, 40, 201–206. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 1–25. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Pseudopotentials. Available online: https://sites.google.com/site/dceresoli/pseudopotentials (accessed on 12 June 2019).
- Pseudopotentials named X.pbe-tm-gipaw. UPF (X = H, C, N, O, S). Available online: http://www.quantumespresso.org (accessed on 19 October 2019).
Sample Availability: Samples of compounds SA:IMI, SA:1-MEIM and SA:2-MEIM are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SA:IMI | SA:1-MEIM | SA:2-MEIM | |
|---|---|---|---|
| Chemical formula | C10H10N2O3 | C11H12N2O3 | C11H12N2O3 |
| Formula weight/g·mol−1 | 206.2 | 220.23 | 220.23 |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group | Pbca | Pbca | Pbcn |
| a/Å | 16.6736(3) | 24.9283(3) | 10.8972(6) |
| b/Å | 11.08182(19) | 8.00291(6) | 9.2671(5) |
| c/Å | 10.9319(2) | 10.98695(11) | 22.1726(11) |
| V/Å3 | 2019.92(6) | 2191.89(4) | 2239.10(2) |
| Z | 8 | 8 | 8 |
| Rp,R’p/%1 | 1.097, 10.065 | 1.179, 7.251 | 1.569, 7.136 |
| Rwp,R’wp/%1 | 1.514, 6.427 | 1.584, 5.495 | 2.056, 6.371 |
| Rexp,R’exp/%1 | 1.126, 4.779 | 1.067, 3.701 | 1.105, 3.234 |
| RBragg | 0.70378 | 0.57544 | 0.81733 |
| Gof | 1.345 | 1.485 | 1.752 |
| Structure | sym op | A | d(D-H) (Å) | A) (Å) | A) (Å) | DHA (deg) |
|---|---|---|---|---|---|---|
| SA:IMI | x, y, z x, 3/2 − y, −1/2 + z 2 − x, 1 − y, 1 − z 2 − x, 1 − y, 1 − z | O-H(SA)O− (SA) N-H(IMI)O− (SA) +N-H(IMI)O− (SA) +N-H(IMI)O− (SA) | 0.91(7) 0.94(7) 0.94(7) 0.94(7) | 1.76(8) 1.78(7) 2.35(7) 1.82(7) | 2.525(9) 2.698 (12) 3.093(10) 2.733(11) | 140(7) 163(6) 135(5) 161(6) |
| SA:1-MEIM | x, y, z x, y, z x, y, z | O-H(SA)O− (SA) N-H(IMI)O− (SA) +N-H (1-MEIM)O− (SA) | 0.93(8) 0.93(7) 0.93(7) | 1.65(7) 2.53(7) 1.72(7) | 2.523(10) 3.128(13) 2.649(14) | 154(7) 122(6) 176(8) |
| SA:2-MEIM | x, y, z 1/2 − x, 1/2 + y, z 1 + x, y, z | O-H(SA)O− (SA) N-H (IMI)O− (SA) +N-H (IMI)O− (SA) | 0.93(7) 0.94(6) 0.94(7) | 1.74(8) 1.78(6) 1.72(7) | 2.551(10) 2.725(12) 2.654(10) | 144(7) 176(8) 172(7) |
| Compound | Solubility (mg/mL) |
|---|---|
| SA | 2.38 |
| SA:IMI | 1304.3 |
| SA:1-MEIM | 2195.0 |
| SA:2-MEIM | 1162.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, I.C.B.; Al-Sabbagh, D.; Meyer, K.; Maiwald, M.; Scholz, G.; Emmerling, F. Insight into the Structure and Properties of Novel Imidazole-Based Salts of Salicylic Acid. Molecules 2019, 24, 4144. https://doi.org/10.3390/molecules24224144
Martins ICB, Al-Sabbagh D, Meyer K, Maiwald M, Scholz G, Emmerling F. Insight into the Structure and Properties of Novel Imidazole-Based Salts of Salicylic Acid. Molecules. 2019; 24(22):4144. https://doi.org/10.3390/molecules24224144
Chicago/Turabian StyleMartins, Inês C. B., Dominik Al-Sabbagh, Klas Meyer, Michael Maiwald, Gudrun Scholz, and Franziska Emmerling. 2019. "Insight into the Structure and Properties of Novel Imidazole-Based Salts of Salicylic Acid" Molecules 24, no. 22: 4144. https://doi.org/10.3390/molecules24224144
APA StyleMartins, I. C. B., Al-Sabbagh, D., Meyer, K., Maiwald, M., Scholz, G., & Emmerling, F. (2019). Insight into the Structure and Properties of Novel Imidazole-Based Salts of Salicylic Acid. Molecules, 24(22), 4144. https://doi.org/10.3390/molecules24224144