3-Substituted 1-Naphthamidomethyl-C-galactosyls Interact with Two Unique Sub-Sites for High-Affinity and High-Selectivity Inhibition of Galectin-3

 ,
,  , and
, and
Abstract
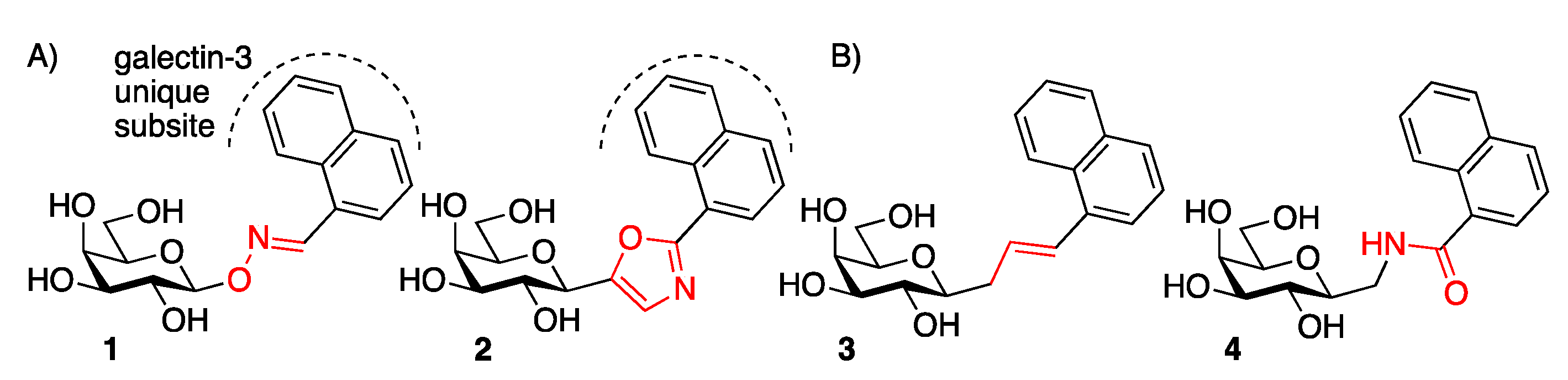
:1. Introduction
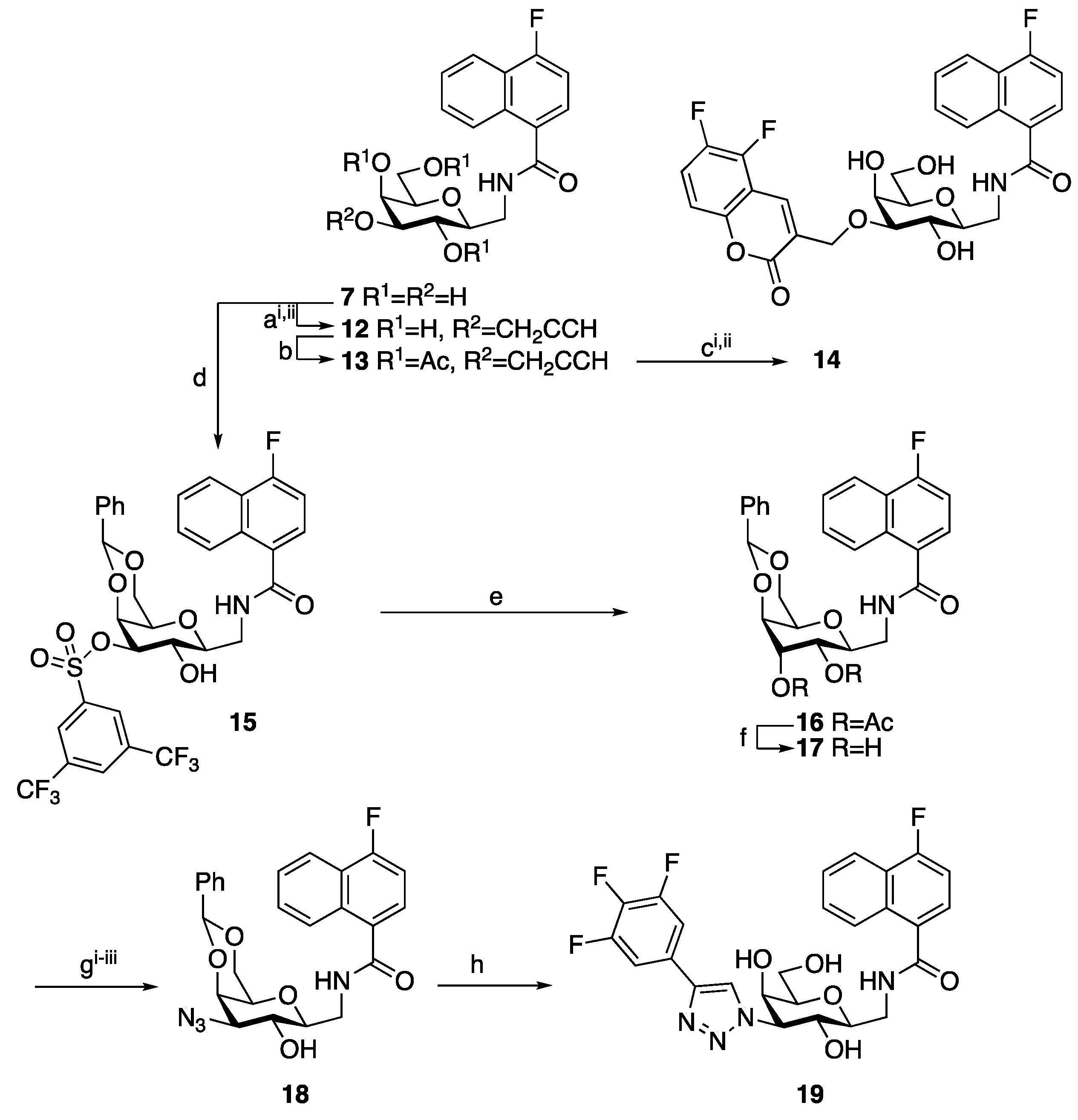
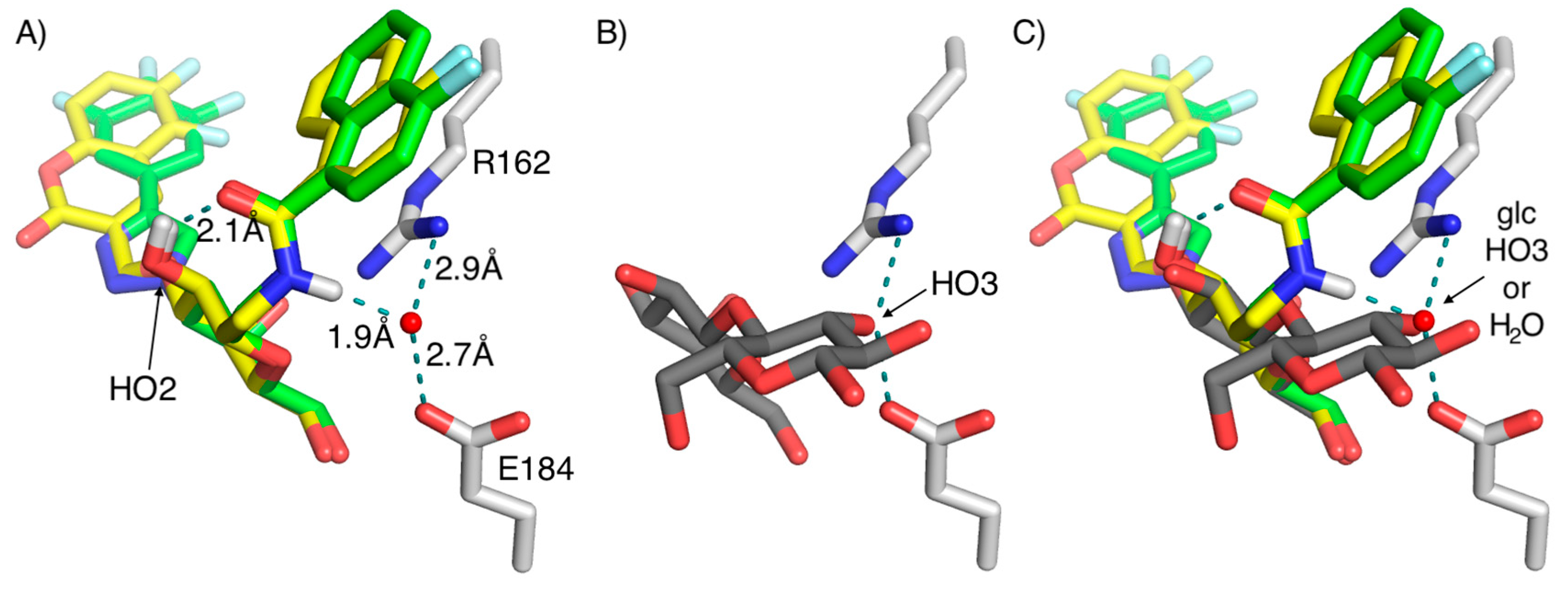
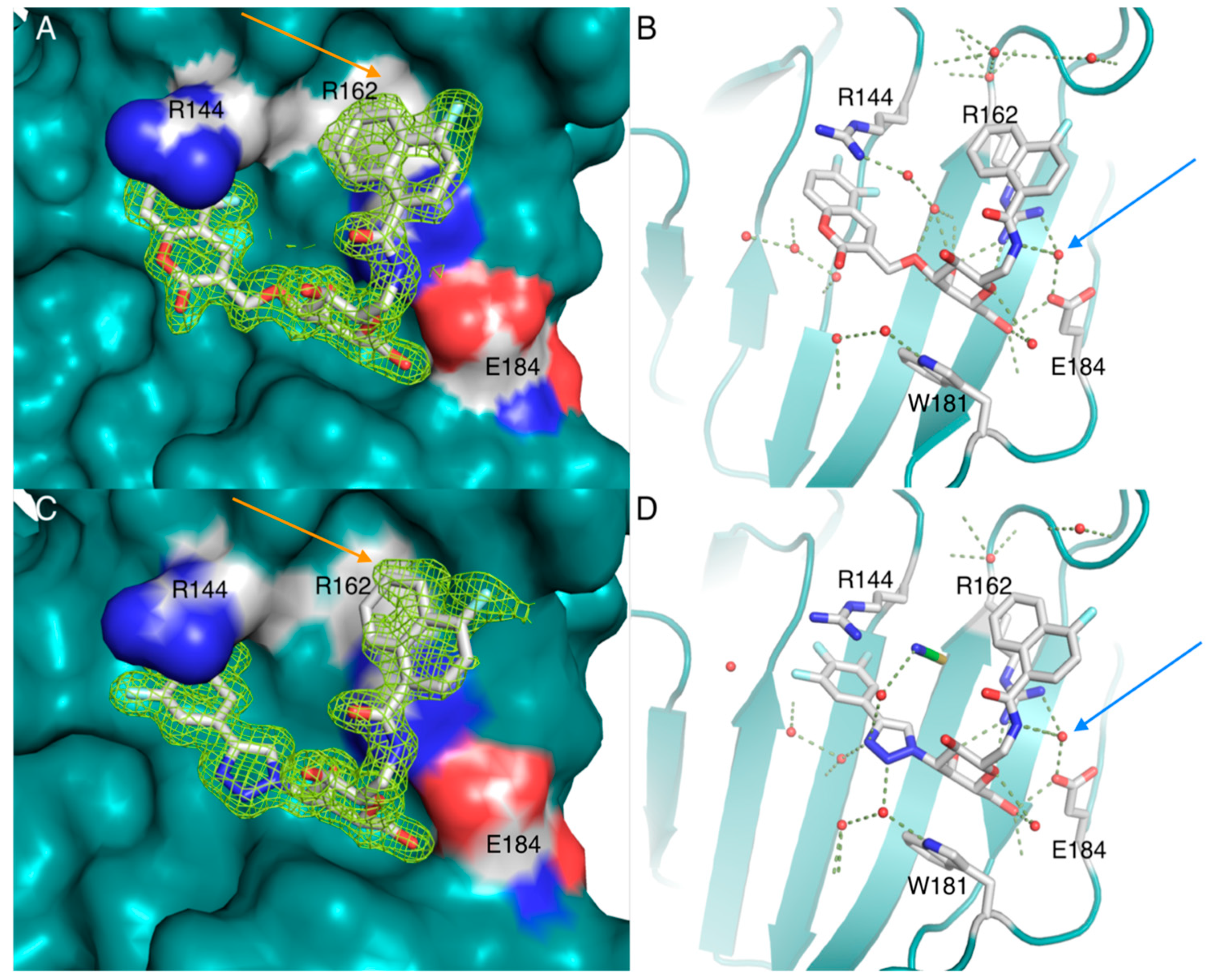
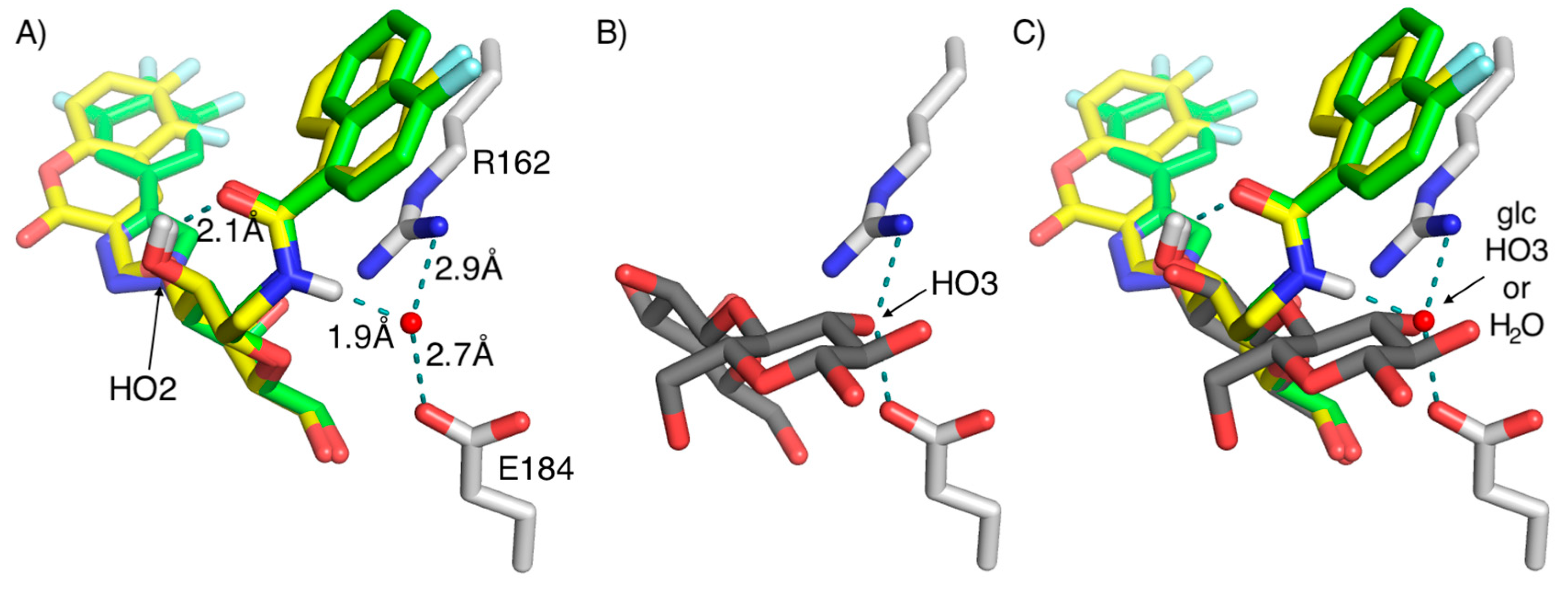
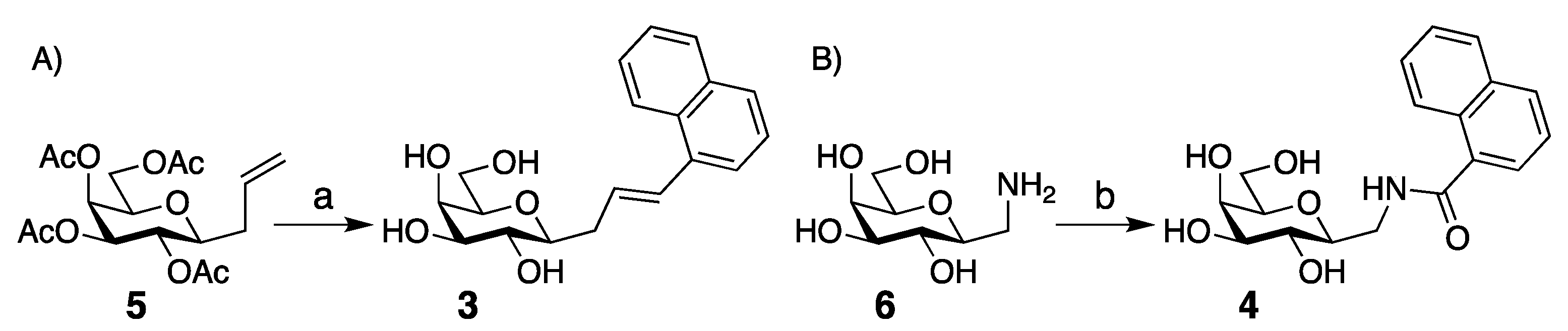
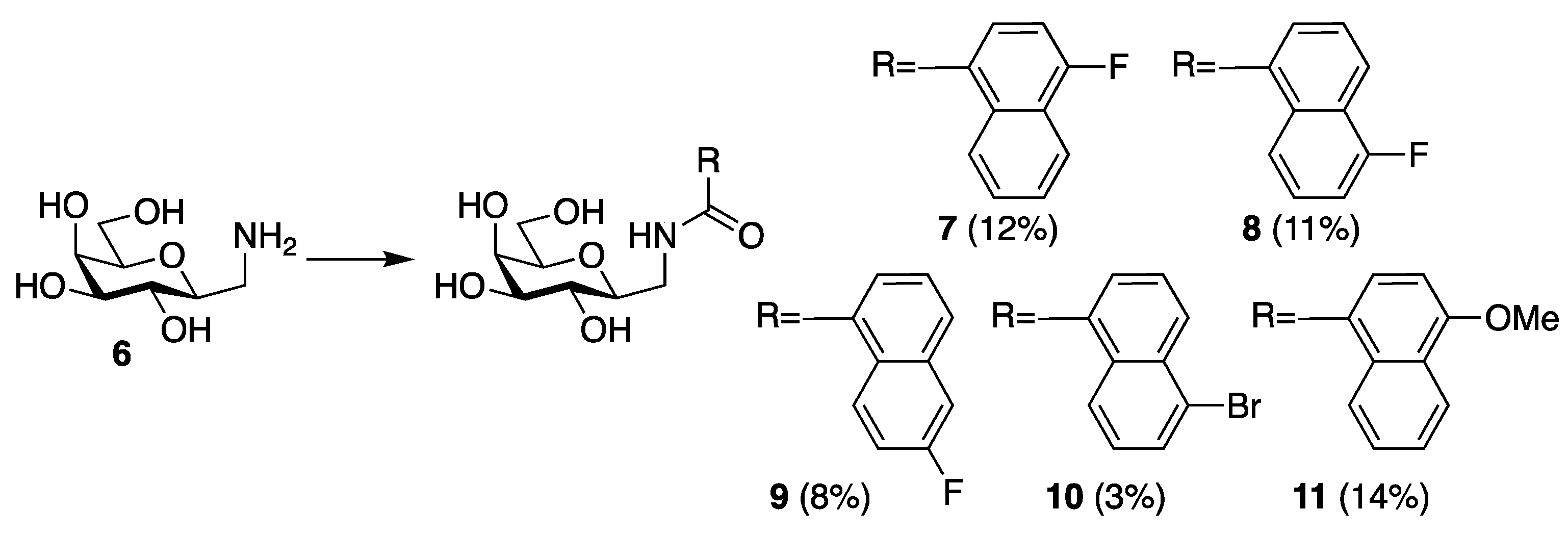
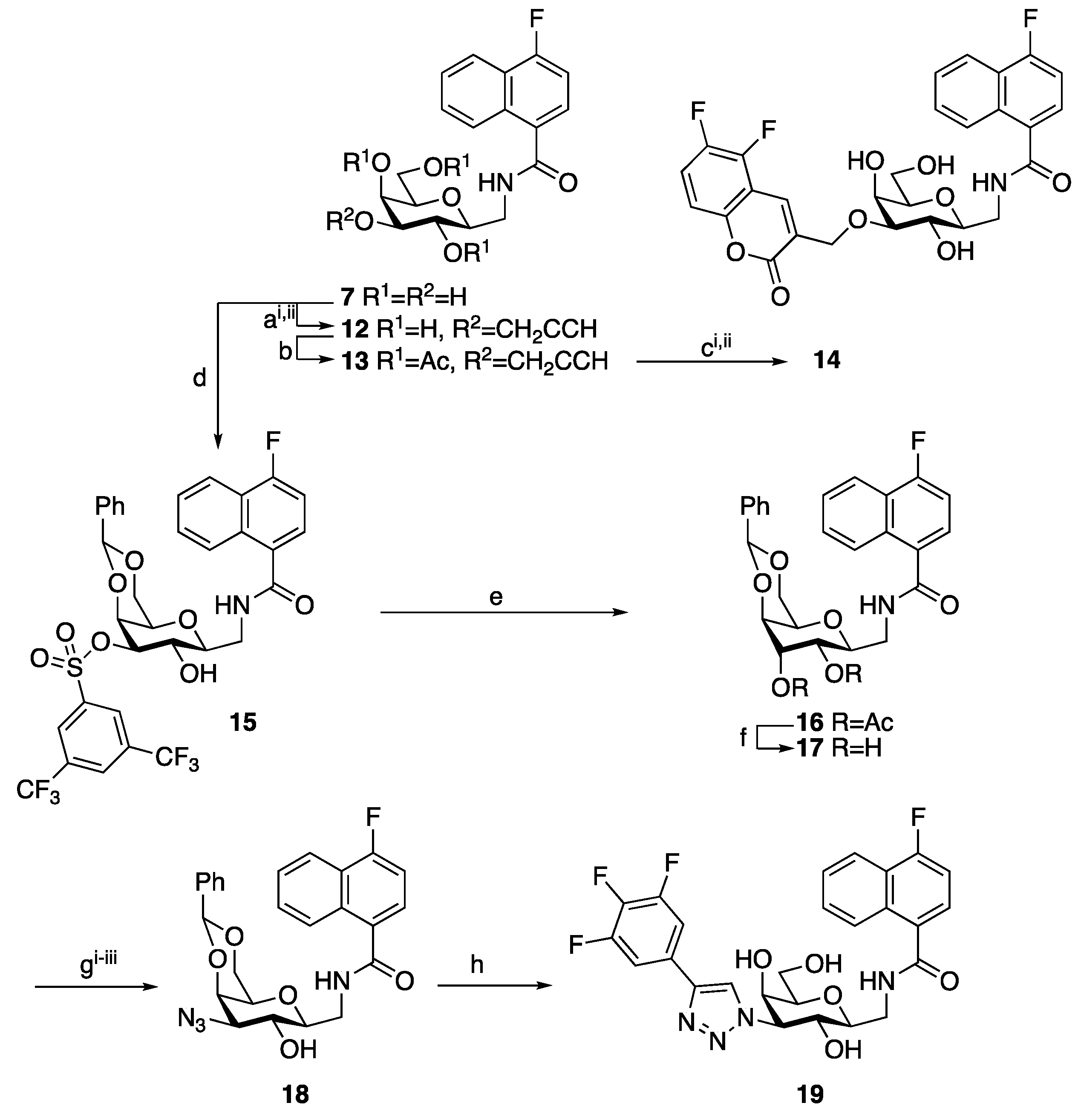
2. Results and Discussion
3. Conclusions
4. Materials and Methods
General
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-T.; Rabinovich, G.A. Galectins: Regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 2010, 1183, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Nabi, I.R.; Shankar, J.; Dennis, J.W. The galectin lattice at a glance. J. Cell. Sci 2015, 128, 2213–2219. [Google Scholar] [CrossRef] [Green Version]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell. Sci 2018, 131, jcs208884. [Google Scholar] [CrossRef] [Green Version]
- Thiemann, S.; Baum, L.G. Galectins and Immune Responses—Just How Do They Do Those Things They Do? Annu. Rev. Immunol. 2016, 34, 243–264. [Google Scholar] [CrossRef]
- Li, Y.-S.; Li, X.-T.; Yu, L.-G.; Wang, L.; Shi, Z.-Y.; Guo, X-L. Guo Roles of galectin-3 in metabolic disorders and tumor cell metabolism. Int. J. Biol. Macromol. 2019, in press. [Google Scholar] [CrossRef]
- Tejler, J.; Leffler, H.; Nilsson, U.J. Synthesis of O-galactosyl aldoximes as potent LacNAc-mimetic galectin-3 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2343–2345. [Google Scholar] [CrossRef] [Green Version]
- Tejler, J.; Salameh, B.; Leffler, H.; Nilsson, U.J. Fragment-based development of triazole-substituted O-galactosyl aldoximes with fragment-induced affinity and selectivity for galectin-3. Org. Biomol. Chem. 2009, 7, 3982–3990. [Google Scholar] [CrossRef]
- Dahlqvist, A.; Leffler, H.; Nilsson, U.J. C1-Galactopyranosyl Heterocycle Structure Guides Selectivity: Triazoles Prefer Galectin-1 and Oxazoles Prefer Galectin-3. ACS Omega 2019, 4, 7047–7053. [Google Scholar] [CrossRef]
- Uchiyama, T.; Woltering, T.J.; Wong, W.; Lin, C.C.; Kajimoto, T.; Takebayashi, M.; Weitz-Schmidt, G.; Asakura, T.; Noda, M.; Wong, C.-H. Design and synthesis of C-linked fucosides as inhibitors of E-selectin. Bioorg. Med. Chem. Lett. 1996, 4, 1149–1165. [Google Scholar] [CrossRef]
- Coxon, B.; Fletcher, H.G. The Structure of 2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl Cyanide and Some Derivatives Therefrom. Synthesis of 1-Deoxy-D- galacto-heptulose. J. Am. Chem. Soc. 1964, 86, 922–926. [Google Scholar] [CrossRef]
- Sörme, P.; Kahl-Knutsson, B.; Huflejt, M.; Nilsson, U.J.; Leffler, H. Fluorescence polarization as an analytical tool to evaluate galectin–ligand interactions. Anal. Biochem. 2004, 334, 36–47. [Google Scholar] [CrossRef]
- Delaine, T.; Collins, P.; Mackinnon, A.; Sharma, G.; Stegmayr, J.; Rajput, V.K.; Mandal, S.; Cumpstey, I.; Larumbe, A.; Salameh, B.A.; et al. Galectin-3-Binding Glycomimetics that Strongly Reduce Bleomycin-Induced Lung Fibrosis and Modulate Intracellular Glycan Recognition. ChemBioChem 2016, 17, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Cumpstey, I.; Carlsson, S.; Leffler, H.; Nilsson, U. Synthesis of a phenyl thio-beta-D-galactopyranoside library from 1,5-difluoro-2,4-dinitrobenzene: Discovery of effcient and selective monosaccharide inhibitors of galectin-7. Org. Biomol. Chem. 2005, 3, 1922–1932. [Google Scholar] [CrossRef]
- Rajput, V.K.; Mackinnon, A.; Mandal, S.; Collins, P.; Blanchard, H.; Leffler, H.; Sethi, T.; Schambye, H.; Mukhopadhyay, B.; Nilsson, U.J. A Selective Galactose–Coumarin-Derived Galectin-3 Inhibitor Demonstrates Involvement of Galectin-3-glycan Interactions in a Pulmonary Fibrosis Model. J. Med. Chem. 2016, 59, 8141–8147. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, U.J.; Leffler, H.; Mukhopadhyay, B.; Rajput, V. Galectoside inhibitors of galectins. U.S. Patent US9353141B2, 31 May 2016. [Google Scholar]
- Peterson, K.; Kumar, R.; Verma, P.; Verma, P.R.; Zetterberg, F.; Akke, M. Systematic Tuning of Fluoro-galectin-3 Interactions Provides Thiodigalactoside Derivatives with Single-Digit nM Affinity and High Selectivity. J. Med. Chem. 2018, 61, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, F.R.; Peterson, K.; Johnsson, R.E.; Brimert, T.; Håkansson, M.; Logan, D.T.; Leffler, H.; Nilsson, U.J. Monosaccharide Derivatives with Low-Nanomolar Lectin Affinity and High Selectivity Based on Combined Fluorine-Amide, Phenyl-Arginine, Sulfur-π, and Halogen Bond Interactions. ChemMedChem 2018, 27, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.-L.; Lin, X.-F.; Wang, Y.-G. Novel and Efficient Synthesis of Iminocoumarins via Copper-Catalyzed Multicomponent Reaction. Org. Lett. 2006, 8, 4517–4520. [Google Scholar] [CrossRef]
- Peterson, K.; Weymouth-Wilson, A.; Nilsson, U.J. Aryl Sulfonates in Inversions at Secondary Carbohydrate Hydroxyl Groups: A New and Improved Route Toward 3-Azido-3-deoxy-β-d-galactopyranosides. J. Carbohydr. Chem. 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Öberg, C.-T.; Blanchard, H.; Leffler, H.; Nilsson, U.J. Protein subtype-targeting through ligand epimerization: Talose-selectivity of galectin-4 and galectin-8. Bioorg. Med. Chem. Lett. 2008, 18, 3691–3694. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Rajput, V.K.; Sundin, A.P.; Leffler, H.; Mukhopadhyay, B.; Nilsson, U.J. Galactose-amidine derivatives as selective antagonists of galectin-9. Can. J. Chem. 2016, 94, 936–939. [Google Scholar] [CrossRef]
- Seetharaman, J.; Kanigsberg, A.; Slaaby, R.; Leffler, H.; Barondes, S.H.; Rini, J.M. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-A resolution. J. Biol. Chem. 1998, 273, 13047–13052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Ignjatović, M.M.; Peterson, K.; Olsson, M.; Leffler, H.; Ryde, U.; Nilsson, U.J.; Logan, D.T. Structure and Energetics of Ligand–Fluorine Interactions with Galectin-3 Backbone and Side-Chain Amides: Insight into Solvation Effects and Multipolar Interactions. ChemMedChem 2019, 73, 4345–4349. [Google Scholar] [CrossRef] [Green Version]
- Zürcher, M.; Diederich, F. Structure-based drug design: Exploring the proper filling of apolar pockets at enzyme active sites. J. Org. Chem. 2008, 73, 4345–4361. [Google Scholar] [CrossRef]
- Sörme, P.; Arnoux, P.; Kahl-Knutsson, B.; Leffler, H.; Rini, J.M.; Nilsson, U.J. Structural and thermodynamic studies on cation-Pi interactions in lectin-ligand complexes: High-affinity galectin-3 inhibitors through fine-tuning of an arginine-arene interaction. J. Am. Chem. Soc. 2005, 127, 1737–1743. [Google Scholar] [CrossRef]
- Noresson, A.L.; Aurelius, O.; Oberg, C.T.; Engström, O.; Sundin, A.P.; Håkansson, M.; Stenström, O.; Akke, M.; Logan, D.T.; Leffler, H.; et al. Designing interactions by control of protein–ligand complex conformation: Tuning arginine–arene interaction geometry for enhanced electrostatic protein–ligand interactions. Chem. Sci. 2018, 9, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Peterson, K.; Misini Ignjatović, M.; Leffler, H.; Ryde, U.; Nilsson, U.J.; Logan, D.T. Substituted polyfluoroaryl interactions with an arginine side chain in galectin-3 are governed by steric-, desolvation and electronic conjugation effects. Org. Biomol. Chem. 2019, 17, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Saraboji, K.; Håkansson, M.; Genheden, S.; Diehl, C.; Qvist, J.; Weininger, U.; Nilsson, U.J.; Leffler, H.; Ryde, U.; Akke, M.; et al. The carbohydrate-binding site in galectin-3 is preorganized to recognize a sugarlike framework of oxygens: Ultra-high-resolution structures and water dynamics. Biochemistry 2012, 51, 296–306. [Google Scholar] [CrossRef]
- Öberg, C.T.; Carlsson, S.; Fillion, E.; Leffler, H.; Nilsson, U.J. Efficient and Expedient Two-Step Pyranose-Retaining Fluorescein Conjugation of Complex Reducing Oligosaccharides: Galectin Oligosaccharide Specificity Studies in a Fluorescence Polarization Assay. Bioconj. Chem. 2003, 14, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.B.; Mahanti, M.; Leffelr, H.; Nilsson, U.J. A Galactoside-Binding Protein Tricked into Binding Unnatural Pyranose Derivatives: 3-Deoxy-3-Methyl-Gulosides Selectively Inhibit Galectin-1. Int. J. Mol. Sci. 2019, 20, 3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, C.; Engström, O.; Delaine, T.; Håkansson, M.; Genheden, S.; Modig, K.; Leffler, H.; Ryde, U.; Nilsson, U.J.; Akke, M. Protein Flexibility and Conformational Entropy in Ligand Design Targeting the Carbohydrate Recognition Domain of Galectin-3. J. Am. Chem. Soc. 2010, 132, 14577–14589. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Cryst. Sec. D 2010, D66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. Sec. D 2012, D68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. Sec. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. Sec. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Me β-gal | 1 | 2 | 3 | 4 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|
| 4400 | 370 [7,8] | 90 [9] | 1600 ± 90 1 | 39 ± 2.3 | 22 ± 0.75 | 79 ± 7.8 | 43 ± 0.38 | 28 ± 0.81 | 22 ± 0.79 |
| Galectin | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 4N 1 | 4C 2 | 7 | 8N 1 | 9N 1 | 9C 2 | |
| 14 | 220 ± 19 3 | 2.0 ± 0.18 | 870 ± 170 | 100 ± 43 | ND 4 | 1300 ± 150 | >2000 | ND |
| 19 | 23 ± 1.4 | 0.54 ± 0.060 | 31 ± 0.80 | 1.3 ± 0.039 | >500 | 1000 ± 270 | 14 ± 0.89 | 130 ± 6 |
| Me β-gal | >10,000 [14] | 4400 [14] | 6600 [23] | 10,000 [23] | 4800 [14] | 6300 [14] | 3300 [14] | 8600 [24] |
| Compound | 14 | 19 |
|---|---|---|
| PDB code | 6TF6 | 6TF7 |
| station | I911-3 | I911-2 |
| wavelength (Å) | 1.000 | 1.03841 |
| unit cell (Å) | a = 35.85 b = 57.34 c = 62.04 | a = 36.14 b = 58.16 c = 62.84 |
| Space group | P212121 | P212121 |
| resolution range (Å) | 31.0–1.5 (1.54–1.50) | 29.1–1.4 (1.44–1.4) |
| completeness (%) | 98.8 (96.3) | 99.6 (96.8) |
| Total reflections unique reflections | 74,804 (6175) 20,755 (1522) | 155,192 (7866) 26,601 (1923) |
| multiplicity | 3.6 (3.4) | 5.8 (4.2) |
| Rmerge (%) | 0.073 (0.725) | 0.048 (0.519) |
| mean I/σ(I) | 12.6 (1.8) | 24.0 (2.8) |
| Wilson B-factor (Å2) | 15.6 | 14.6 |
| refinement program | Refmac5 | Refmac5 |
| Rmodel (F; %) | 0.116 (0.173) | 0.135 (0.240) |
| Rfree (F; %) | 0.174 (0.275) | 0.187 (0.324) |
| reflections used in refinement (for Rfree) | 19,717 (1374) 1038 (72) | 25,272 (1767) 1330 (93) |
| average B-factors (Å2) | protein: 14.2 ligand: 21.5 solvent: 33.4 | protein: 13.5 ligand: 19.4 solvent: 28.9 |
| Ramachandran outliers (%) | 1 | 0 |
| rotamer outliers (no. and %) MolProbity clash score | 1, 0.6% 2.48 | 0, 0.0% 2.12 |
| bond length rmsd from ideal (Å) bond angle rmsd from ideal (°) | 0.020 2.1 | 0.019 2.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlqvist, A.; Mandal, S.; Peterson, K.; Håkansson, M.; Logan, D.T.; Zetterberg, F.R.; Leffler, H.; Nilsson, U.J. 3-Substituted 1-Naphthamidomethyl-C-galactosyls Interact with Two Unique Sub-Sites for High-Affinity and High-Selectivity Inhibition of Galectin-3. Molecules 2019, 24, 4554. https://doi.org/10.3390/molecules24244554
Dahlqvist A, Mandal S, Peterson K, Håkansson M, Logan DT, Zetterberg FR, Leffler H, Nilsson UJ. 3-Substituted 1-Naphthamidomethyl-C-galactosyls Interact with Two Unique Sub-Sites for High-Affinity and High-Selectivity Inhibition of Galectin-3. Molecules. 2019; 24(24):4554. https://doi.org/10.3390/molecules24244554
Chicago/Turabian StyleDahlqvist, Alexander, Santanu Mandal, Kristoffer Peterson, Maria Håkansson, Derek T. Logan, Fredrik R. Zetterberg, Hakon Leffler, and Ulf J. Nilsson. 2019. "3-Substituted 1-Naphthamidomethyl-C-galactosyls Interact with Two Unique Sub-Sites for High-Affinity and High-Selectivity Inhibition of Galectin-3" Molecules 24, no. 24: 4554. https://doi.org/10.3390/molecules24244554
APA StyleDahlqvist, A., Mandal, S., Peterson, K., Håkansson, M., Logan, D. T., Zetterberg, F. R., Leffler, H., & Nilsson, U. J. (2019). 3-Substituted 1-Naphthamidomethyl-C-galactosyls Interact with Two Unique Sub-Sites for High-Affinity and High-Selectivity Inhibition of Galectin-3. Molecules, 24(24), 4554. https://doi.org/10.3390/molecules24244554