
Selectivity of Terpyridine Platinum Anticancer Drugs for G-quadruplex DNA


 ,
,
Abstract
:1. Introduction
2. Results
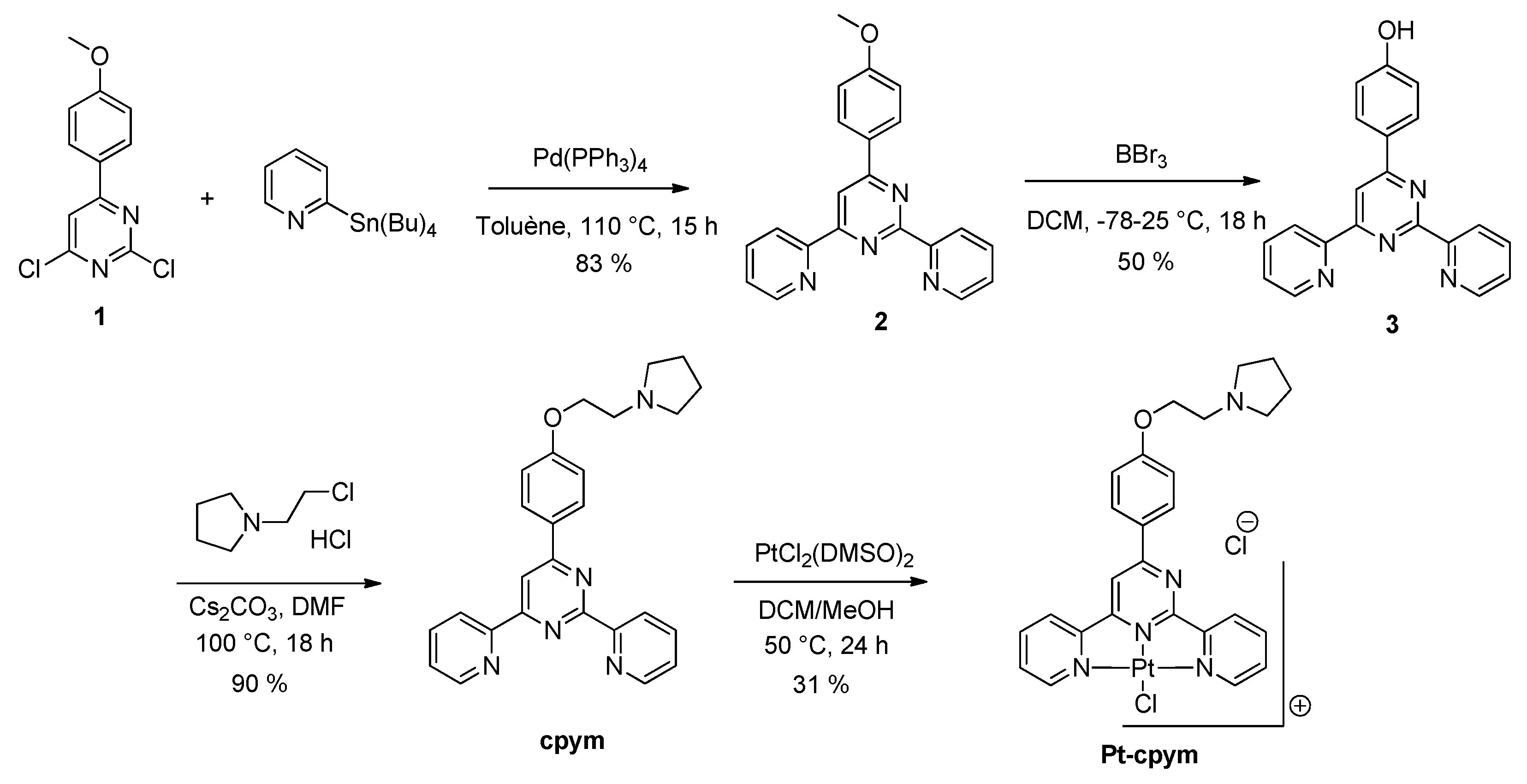
2.1. Panel of Platinum Complexes
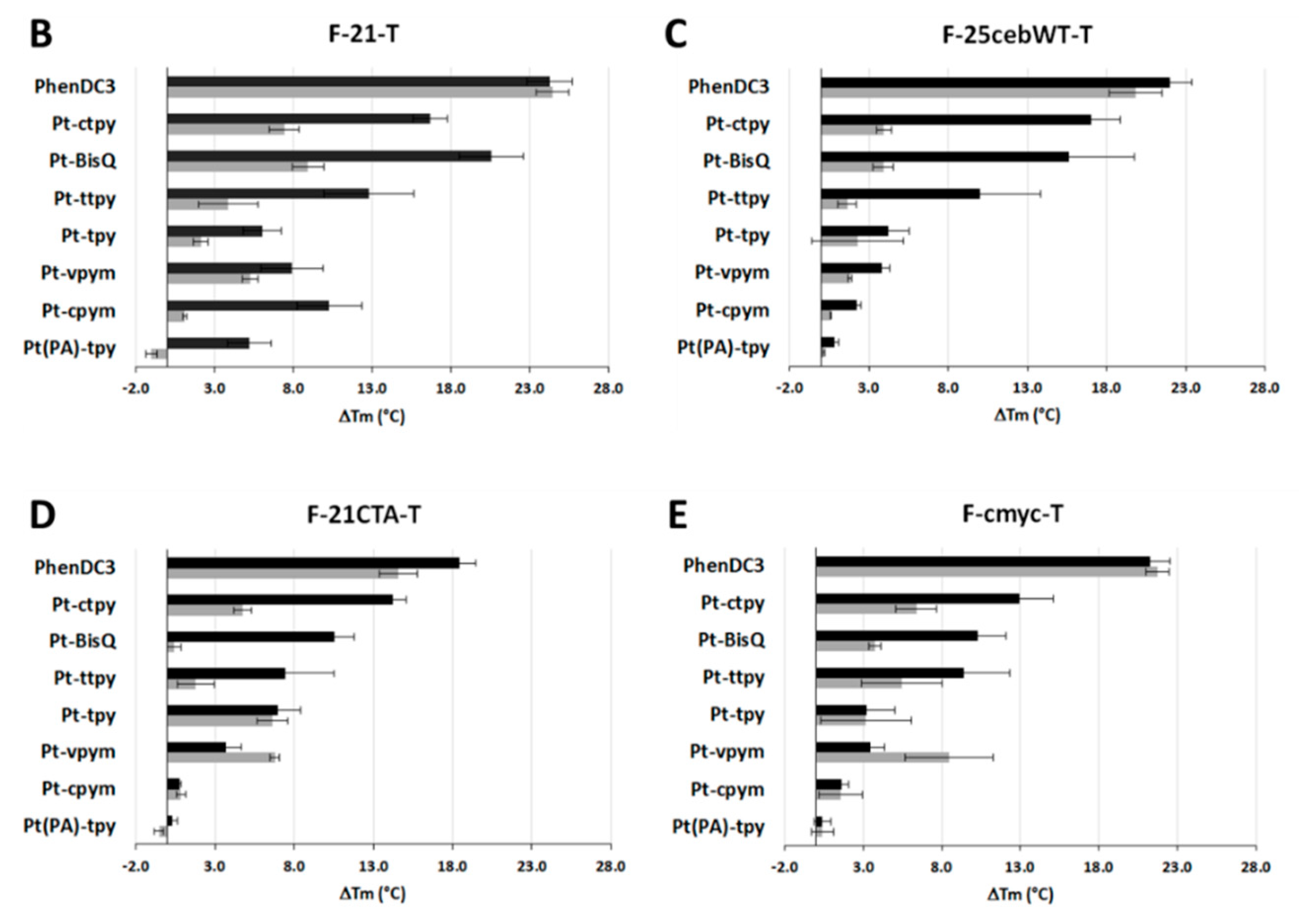
2.2. Interaction Measurements
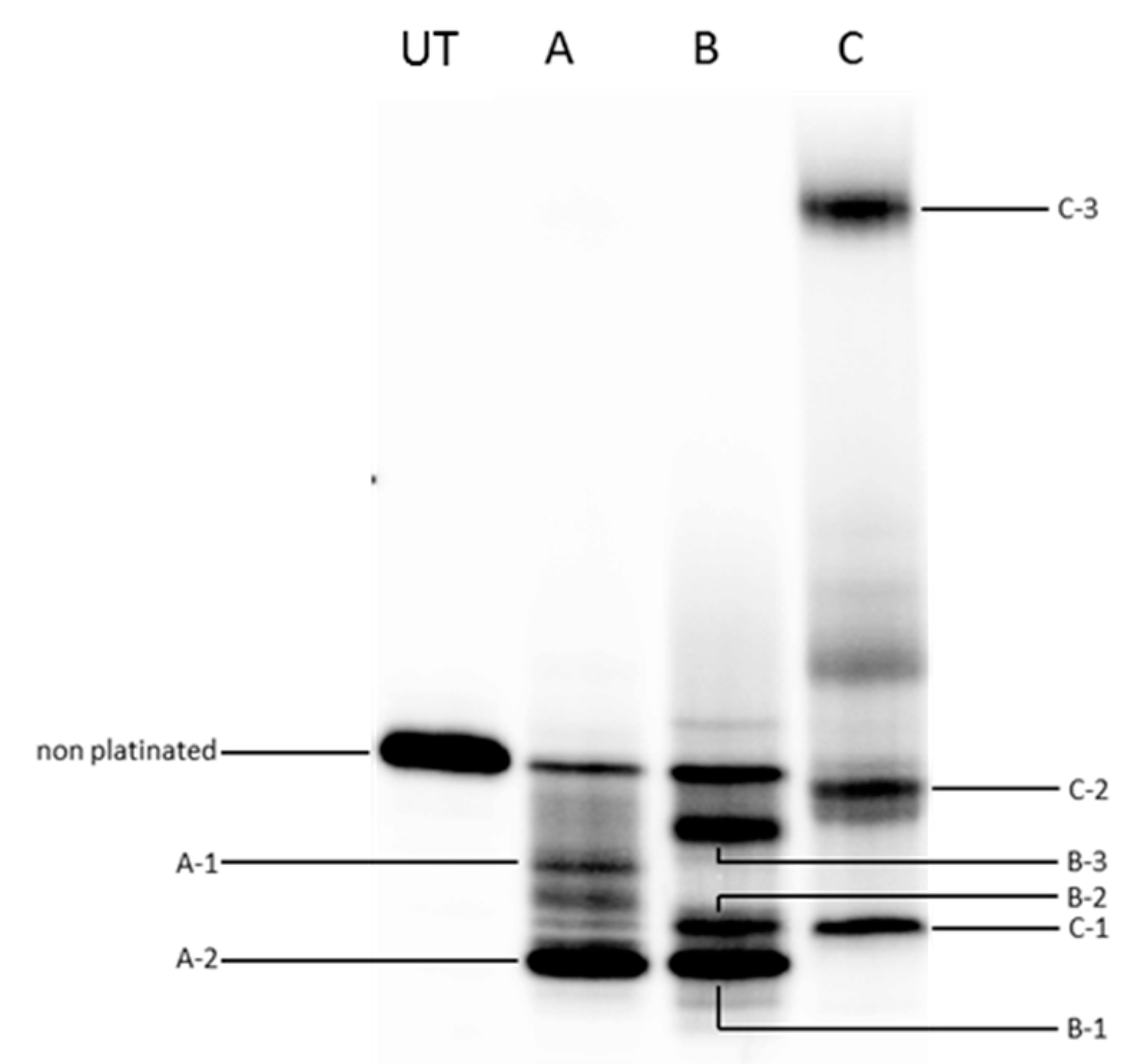
2.3. Quadruplex Platination
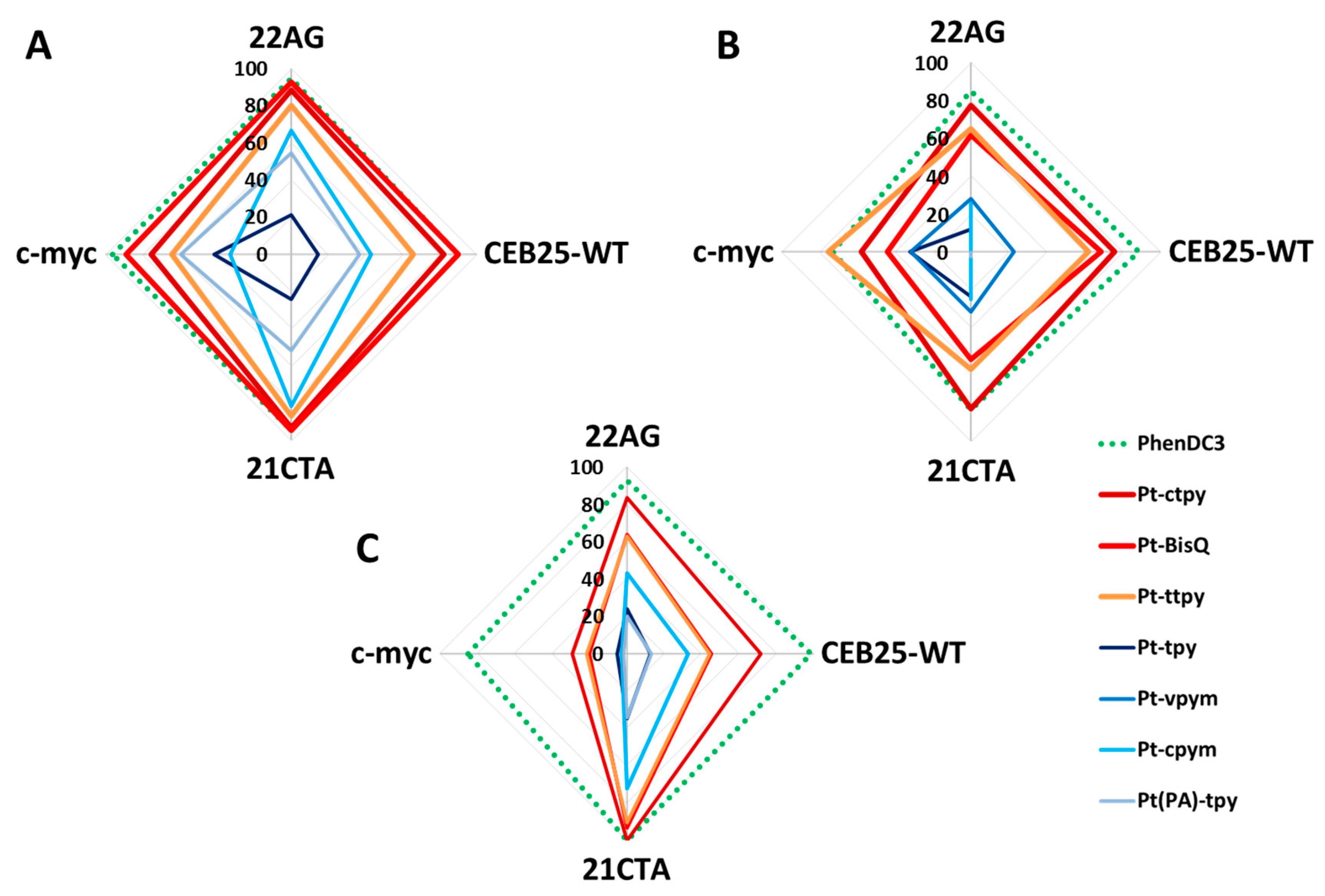
2.4. Kinetics and Selectivity Studies
2.5. In Vitro Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Organic Synthesis
4.3. Oligonucleotides
| 22AG | 5’-A GGGTTAGGGTTAGGGTTAGGG-3’ |
| c-myc (myc22) | 5’-TGAGGGTGGGTAGGGTGGGTAA-3’ |
| ds26 | 5’-CAATCGGATCGAATTCGATCCGATTG-3’ |
| 21CTA | AGGGCTAGGGCTAGGGCTAGGG |
| CEB25-WT | AAGGGTGGGTGTAAGTGTGGGTGGGT |
| F-21-T | 5’-FAM-GGG TTA GGG TTA GGG TTA GGG-TAMRA-3’ |
| F-myc-T | 5’-FAM-TGA GGG T GGG TA GGG T GGG TAA-TAMRA-3’ |
| F-21CTA-T | 5’-FAM-AGGGCTAGGGCTAGGGCTAGGG- TAMRA-3’ |
| F-CEB25-WT-T | 5’-FAM-AGGGTGGGTGTAAGTGTGGGTGGGT- TAMRA-3’ |
4.4. Preparation of Oligonucleotides
4.5. FRET-Melting Experiments
4.6. HT-G4-FID Assay
4.7. Fluorimetric Titrations for Affinity Constant Evaluation Reported in Table 1
4.8. Gel Electrophoresis
4.9. Cell Culture
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, J. New clues for platinum antitumor chemistry: Kinetically controlled metal binding to DNA. Proc. Natl. Acad. Sci. USA 2003, 100, 3611–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Lippard, S.J. Direct cellular responses to Platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N. G4-associated human diseases. EMBO Rep. 2015, 16, 910–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Bedrat, A.; Lacroix, L.; Mergny, J.L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef] [Green Version]
- Schaffitzel, C.; Berger, I.; Postberg, J.; Hanes, J.; Lipps, H.J.; Pluckthun, A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. USA 2001, 98, 8572–8577. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Yang, S.Y.; Wong, J.M.Y.; Monchaud, D. Cellular Detection of G-Quadruplexes by Optical Imaging Methods. Curr. Protoc. Cell Biol. 2017, 76, 4–33. [Google Scholar] [CrossRef] [PubMed]
- Shivalingam, A.; Vysniauskas, A.; Albrecht, T.; White, A.J.; Kuimova, M.K.; Vilar, R. Trianguleniums as optical probes for G-quadruplexes: A photophysical, electrochemical, and computational study. Chem. Eur. J. 2016, 22, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Granotier, C.; Pennarun, G.; Riou, L.; Hoffschir, F.; Gauthier, L.R.; De Cian, A.; Gomez, D.; Mandine, E.; Riou, J.F.; Mergny, J.L.; et al. Preferential binding of a G-quadruplex ligand to human chromosome ends. Nucleic Acids Res. 2005, 33, 4182–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, E.Y.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013, 4, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877. [Google Scholar] [CrossRef]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Rodriguez, R. G-quadruplex interacting small molecules and drugs: From bench toward bedside. Expert Rev. Clin. Pharmacol. 2014, 7, 663–679. [Google Scholar] [CrossRef]
- Hansel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Monchaud, D.; Teulade-Fichou, M.P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef]
- Georgiades, S.N.; Abd Karim, N.H.; Suntharalingam, K.; Vilar, R. Interaction of metal complexes with G-Quadruplex DNA. Angew. Chem. Int. Ed. Engl. 2010, 49, 4020–4034. [Google Scholar] [CrossRef]
- Stafford, V.S.; Suntharalingam, K.; Shivalingam, A.; White, A.J.P.; Mann, D.J.; Vilar, R. Syntheses of polypyridyl metal complexes and studies of their interaction with quadruplex DNA. Dalton Trans. 2015, 44, 3686–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Li, Y.; Freisinger, E.; Qin, P.Z.; Sigel, R.K.O.; Mao, Z.-W. G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs. Inorg. Chem. Front. 2017, 4, 10–32. [Google Scholar] [CrossRef] [Green Version]
- Vilar, R. Nucleic Acid Quadruplexes and Metallo-Drugs. Met. Ions Life Sci. 2018, 18, 325. [Google Scholar] [CrossRef]
- Pradines, V.; Pratviel, G. Interaction of cationic manganese porphyrin with G-quadruplex nucleic acids probed by differential labeling of the two faces of the porphyrin. Angew. Chem. Int. Ed. Engl. 2013, 52, 2185–2188. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.E.; Neidle, S.; Vilar, R. Stabilisation of human telomeric quadruplex DNA and inhibition of telomerase by a platinum–phenanthroline complex. Chem. Commun. 2007, 42, 4366–4368. [Google Scholar] [CrossRef]
- Reed, J.E.; Arnal, A.A.; Neidle, S.; Vilar, R. Stabilization of G-Quadruplex DNA and Inhibition of Telomerase Activity by Square-Planar Nickel(II) Complexes. J. Am. Chem. Soc. 2006, 128, 5992–5993. [Google Scholar] [CrossRef]
- Leczkowska, A.; Gonzalez-Garcia, J.; Perez-Arnaiz, C.; Garcia, B.; White, A.J.P.; Vilar, R. Binding studies of metal-salphen and metal-bipyridine complexes towards G-Quadruplex DNA. Chem. Eur. J. 2018, 24, 11785–11794. [Google Scholar] [CrossRef]
- Bertrand, H.; Monchaud, D.; De Cian, A.; Guillot, R.; Mergny, J.-L.; Teulade-Fichou, M.-P. The importance of metal geometry in the recognition of G-quadruplex-DNA by metal-terpyridine complexes. Org. Biomol. Chem. 2007, 5, 2555–2559. [Google Scholar] [CrossRef]
- Rizzo, A.; Iachettini, S.; Zizza, P.; Cingolani, C.; Porru, M.; Artuso, S.; Stevens, M.; Hummersone, M.; Biroccio, A.; Salvati, E.; et al. Identification of novel RHPS4-derivative ligands with improved toxicological profiles and telomere-targeting activities. J. Exp. Clin. Cancer Res. 2014, 33, 81. [Google Scholar] [CrossRef]
- Bertrand, H.; Bombard, S.; Monchaud, D.; Talbot, E.; Guédin, A.; Mergny, J.-L.; Grünert, R.; Bednarski, P.J.; Teulade-Fichou, M.-P. Exclusive platination of loop adenines in the human telomeric G-quadruplex. Org. Biomol. Chem. 2009, 7, 2864. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, H.; Bombard, S.; Monchaud, D.; Teulade-Fichou, M.P. A Platinum-Quinacridine Hybrid as G-Quadruplex Ligand. J. Biol. Inorg. Chem. 2007, 12, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Betzer, J.F.; Nuter, F.; Chtchigrovsky, M.; Hamon, F.; Kellermann, G.; Ali, S.; Calméjane, M.A.; Roque, S.; Poupon, J.; et al. Linking of antitumour trans NHC-Pt(II) complexes to G-quadruplex DNA ligand for telomeric targeting. Bioconjug. Chem. 2016, 27, 1456. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Bierbach, U. Kinetically favored platination of adenine in the g-rich human telomeric repeat. J. Am. Chem. Soc. 2007, 129, 15764–15765. [Google Scholar] [CrossRef] [PubMed]
- Ourliac-Garnier, I.; Elizondo-Riojas, M.A.; Redon, S.; Farrell, N.P.; Bombard, S. Cross-links of quadruplex structures from human telomeric DNA by dinuclear platinum complexes show the flexibility of both structures. Biochemistry 2005, 44, 10620–10634. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Hamon, F.; Rosu, F.; Gabelica, V.; De Pauw, E.; Guédin, A.; Mergny, J.-L.; Teulade-Fichou, M.-P. Tridentate N-donor Palladium(II) complexes as efficient coordinating quadruplex DNA binders. Chemistry 2011, 17, 13274–13283. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Carmena, M.; Liskovykh, M.; Peat, E.; Kim, J.H.; Oshimura, M.; Masumoto, H.; Teulade-Fichou, M.P.; Pommier, Y.; Earnshaw, W.C.; et al. Systematic analysis of compounds specifically targeting telomeres and telomerase for clinical implications in cancer therapy. Cancer Res. 2018, 78, 6282–6296. [Google Scholar] [CrossRef]
- Charif, R.; Granotier-Beckers, C.; Bertrand, H.C.; Poupon, J.; Ségal-Bendirdjian, E.; Teulade-Fichou, M.P.; Boussin, F.D.; Bombard, S. Association of a platinum complex to a G-quadruplex ligand enhances the telomere disruption. Chem. Res. Tox. 2017, 38, 1629–1640. [Google Scholar] [CrossRef]
- Saker, L.; Ali, S.; Masserot, C.; Kellermann, G.; Poupon, J.; Teulade-Fichou, M.P.; Segal-Bendirdjian, E.; Bombard, S. Platinum complexes can bind to telomeres by coordination. Int. J. Mol. Sci. 2018, 19, 1951. [Google Scholar] [CrossRef]
- Wei, Z.Z.; Qin, Q.P.; Meng, T.; Deng, C.X.; Liang, H.; Chen, Z.F. 5-Bromo-oxoisoaporphine platinum(II) complexes exhibit tumor cell cytotoxcicity via inhibition of telomerase activity and disruption of c-myc G-quadruplex DNA and mitochondrial functions. Eur. J. Med. Chem. 2018, 145, 360–369. [Google Scholar] [CrossRef]
- Qin, Q.P.; Qin, J.L.; Chen, M.; Li, Y.L.; Meng, T.; Zhou, J.; Liang, H.; Chen, Z.F. Chiral platinum (II)-4-(2,3-dihydroxypropyl)- formamide oxo-aporphine (FOA) complexes promote tumor cells apoptosis by directly targeting G-quadruplex DNA in vitro and in vivo. Oncotarget 2017, 8, 61982–61997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovski, M.; Morel, E.; Hamon, F.; Bombard, S.; Teulade-Fichou, M.-P.; Plavec, J. Interactions of Pt-ttpy with G-quadruplexes originating from promoter region of the c-myc Gene deciphered by NMR and gel electrophoresis analysis. Chemistry 2015, 21, 7798–7807. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Poyer, F.; Vaslin, L.; Bombard, S.; Teulade-Fichou, M.P. Photoactivatable platinum(II) terpyridine derivatives for G-quadruplex DNA double anchoring. Inorg. Chim. Acta 2016, 452, 152–158. [Google Scholar] [CrossRef]
- Merle, P.; Gueugneau, M.; Teulade-Fichou, M.-P.; Müller-Barthélémy, M.; Amiard, S.; Chautard, E.; Guetta, C.; Dedieu, V.; Communal, Y.; Mergny, J.-L.; et al. Highly efficient radiosensitization of human glioblastoma and lung cancer cells by a G-quadruplex DNA binding compound. Sci. Rep. 2015, 5, 16255. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; De Cian, A.; Teulade-Fichou, M.-P.; Mergny, J.-L.; Monchaud, D. Engineering Bisquinolinium/Thiazole Orange Conjugates for Fluorescent Sensing of G-Quadruplex DNA. Angew. Chem. Int. Ed. Engl. 2009, 48, 2188–2191. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.J. Molecular probes: Handbook of fluorescent probes and research chemicals: By R P Haugland. pp 390. Interchim (Molecular Probes Inc, PO Box 22010 Eugene, OR 97402-0414, USA, or 15 rue des Champs, 92600 Asnieres, Paris). 1992–1994. $15. Biochem. Educ. 1994, 22, 83. [Google Scholar] [CrossRef]
- Beauvineau, C.; Guetta, C.; Teulade-Fichou, M.P.; Mahuteau-Betzer, F. PhenDV, a turn-off fluorescent quadruplex DNA probe for improving the sensitivity of drug screening assays. Org. Biomol. Chem. 2017, 15, 7117–7121. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Hamon, F.; Teulade-Fichou, M.-P. Development of a high-throughput G4-FID assay for screening and evaluation of small molecules binding quadruplex nucleic acid structures. Anal. Bioanal. Chem. 2011, 400, 3419–3427. [Google Scholar] [CrossRef]
- Peng, Z.-H.; Journet, M.; Humphrey, G. A highly regioselective amination of 6-Aryl-2,4-dichloropyrimidine. Org. Lett. 2006, 8, 395–398. [Google Scholar] [CrossRef]
- Hadad, C.; Achelle, S.; García-Martinez, J.C.; Rodríguez-López, J. 4-Arylvinyl-2,6-di(pyridin-2-yl)pyrimidines: Synthesis and optical properties. J. Org. Chem. 2011, 76, 3837–3845. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Maurizot, J.-C. Fluorescence resonance energy transfer as a probe for G-quartet formation by a telomeric repeat. ChemBioChem 2001, 2, 124–132. [Google Scholar] [CrossRef]
- Heringova, P.; Kasparkova, J.; Brabec, V. DNA adducts of antitumor cisplatin preclude telomeric sequences from forming G quadruplexes. J. Biol. Inorg. Chem. 2009, 14, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.D. Platinum complexes of terpyridine: Interaction and reactivity with biomolecules. Coord. Chem. Rev. 2009, 253, 1495–1516. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Chtchigrovsky, M.; Eloy, L.; Jullien, H.; Saker, L.; Segal-Bendirdjian, E.; Poupon, J.; Bombard, S.; Cresteil, T.; Retailleau, P.; Marinetti, A. Antitumor trans-N-heterocyclic carbene-amine-Pt(II) complexes: Synthesis of dinuclear species and exploratory investigations of DNA binding and cytotoxicity mechanisms. J. Med. Chem. 2013, 56, 2074–2086. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.; Bats, J.W.; Ihmels, H.; Granzhan, A.; Uebach, S.; Patrick, B.O. Polycyclic azoniahetarenes: Assessing the binding parameters of complexes between unsubstituted ligands and G-quadruplex DNA. Chem. Eur. J. 2012, 18, 10903–10915. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA | Sequence (5’ to 3’) | PhenDV | TO | ||
|---|---|---|---|---|---|
| KA (M−1) | Dye:DNA | KA (M−1) | Dye:DNA | ||
| 22AG | AG3T2AG3T2AG3T2AG3 | 3 × 106 (a) | 2:1 | 1 × 106 (a) | 1:1 |
| c-myc | TGAG3TG3TAG3TG3TA2 | 6 × 107 | 4:1 | 5 × 106 (b) | 1:1 |
| 21CTA | AG3CTAG3CTAG3CTAG3 | 1 × 106 | 3:1 | 1 × 106 (b) | 1:1 |
| CEB25-WT | AG3TG3TG3TG3T | 1 × 107 (a) | 4:1 | 1 × 106 (a) | 1:1 |
| A2780 | A2780 cis | Resistance Factor | CCD19Lu | |
|---|---|---|---|---|
| Pt-BisQ | 4.00 | 6.00 | 1.5 | 4.10 |
| Pt-ctpy | 4.60 | 5.00 | 1.1 | 5.20 |
| Pt-ttpy | 2.50 | 2.50 | 1.0 | 1.75 |
| Pt-cpym | 3.80 | 5.00 | 1.3 | 1.80 |
| Pt-vpym | 0.70 | 0.40 | 0.6 | 0.70 |
| Pt-tpy | 3.00 | 5.00 | 1.6 | 3.00 |
| Pt(PA)-tpy | 0.08 | 0.05 | 0.6 | 0.12 |
| cisplatin | 0.33 | 3.00 | 9.1 | 0.20 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morel, E.; Beauvineau, C.; Naud-Martin, D.; Landras-Guetta, C.; Verga, D.; Ghosh, D.; Achelle, S.; Mahuteau-Betzer, F.; Bombard, S.; Teulade-Fichou, M.-P. Selectivity of Terpyridine Platinum Anticancer Drugs for G-quadruplex DNA. Molecules 2019, 24, 404. https://doi.org/10.3390/molecules24030404
Morel E, Beauvineau C, Naud-Martin D, Landras-Guetta C, Verga D, Ghosh D, Achelle S, Mahuteau-Betzer F, Bombard S, Teulade-Fichou M-P. Selectivity of Terpyridine Platinum Anticancer Drugs for G-quadruplex DNA. Molecules. 2019; 24(3):404. https://doi.org/10.3390/molecules24030404
Chicago/Turabian StyleMorel, Elodie, Claire Beauvineau, Delphine Naud-Martin, Corinne Landras-Guetta, Daniela Verga, Deepanjan Ghosh, Sylvain Achelle, Florence Mahuteau-Betzer, Sophie Bombard, and Marie-Paule Teulade-Fichou. 2019. "Selectivity of Terpyridine Platinum Anticancer Drugs for G-quadruplex DNA" Molecules 24, no. 3: 404. https://doi.org/10.3390/molecules24030404
APA StyleMorel, E., Beauvineau, C., Naud-Martin, D., Landras-Guetta, C., Verga, D., Ghosh, D., Achelle, S., Mahuteau-Betzer, F., Bombard, S., & Teulade-Fichou, M. -P. (2019). Selectivity of Terpyridine Platinum Anticancer Drugs for G-quadruplex DNA. Molecules, 24(3), 404. https://doi.org/10.3390/molecules24030404