Shining a Spotlight on DNA: Single-Molecule Methods to Visualise DNA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
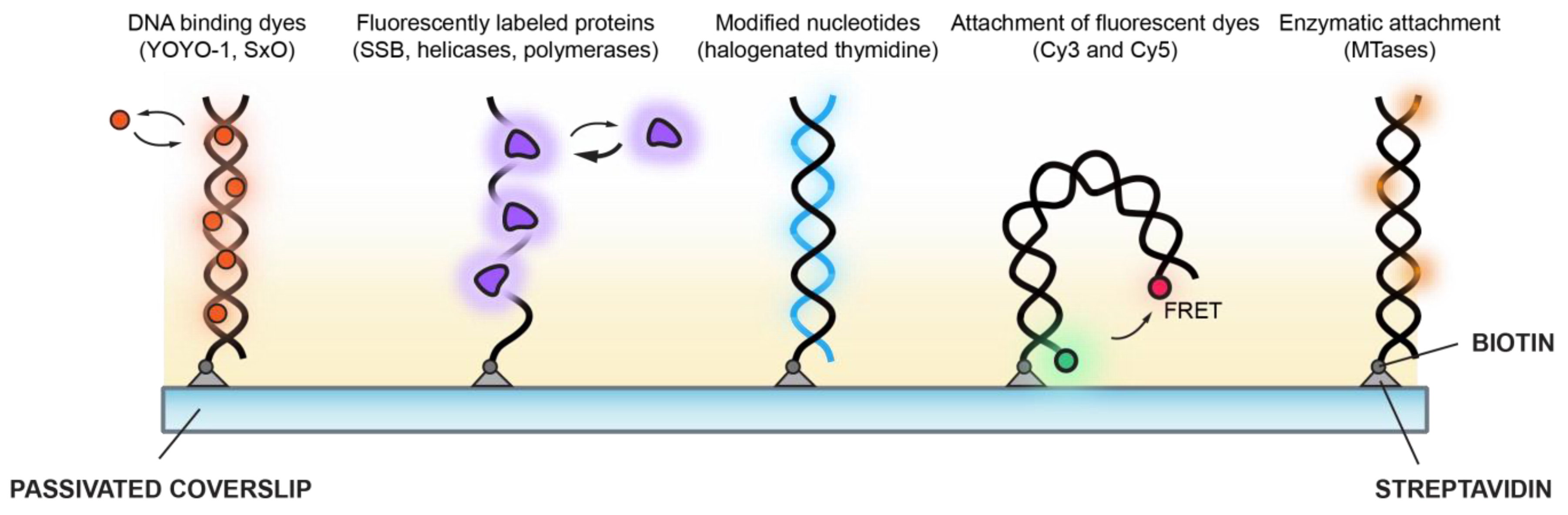
2. Visualisation of DNA Dynamics and Topological Intermediates
2.1. YOYO-1
2.2. SYTOX Dyes
2.3. Polymer Physics with Single DNA Molecules
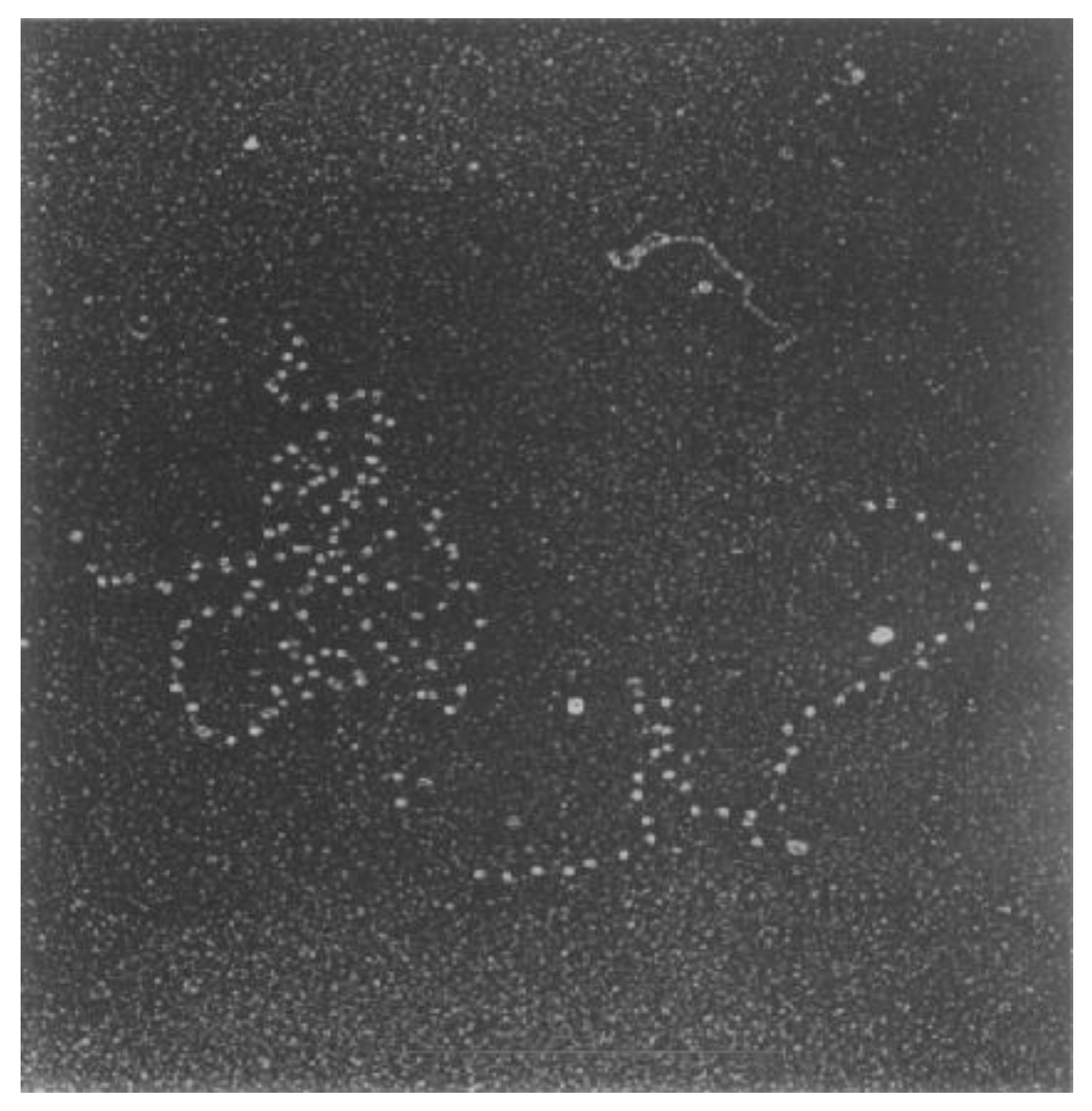
2.4. Knotted DNA
2.5. DNA Supercoiling
2.6. DNA Looping
3. Visualisation of Single-Stranded DNA
4. Studying Biological Processes on DNA: DNA Replication
5. Internal Site-Specific Labelling on DNA: Visualisation of Long DNA Molecules
5.1. Molecular DNA Combing
5.2. Optical Mapping
6. Commercial Applications of Single-Molecule Sequencing
7. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Dufrêne, Y.F.; Ando, T.; Garcia, R.; Alsteens, D.; Martinez-Martin, D.; Engel, A.; Gerber, C.; Müller, D.J. Imaging modes of atomic force microscopy for application in molecular and cell biology. Nat. Nanotechnol. 2017, 12, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Vilfan, I.; Lipfert, J.; Koster, D.; Lemay, S.; Dekker, N. Magnetic tweezers for single-molecule experiments. In Handbook of Single-Molecule Biophysics; Hinterdorfer, P., van Oijen, A., Eds.; Springer: New York, NY, USA, 2009; pp. 371–395. [Google Scholar]
- Ha, T.; Tinnefeld, P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu. Rev. Phys. Chem. 2012, 63, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Van Oijen, A.M. Single-molecule approaches to characterizing kinetics of biomolecular interactions. Curr. Opin. Biotechnol. 2011, 22, 75–80. [Google Scholar] [CrossRef]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Monachino, E.; Spenkelink, L.M.; van Oijen, A.M. Watching cellular machinery in action, one molecule at a time. J. Cell. Biol. 2017, 216, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.E.; Gosling, R.G. Molecular configuration in sodium thymonucleate. Nature 1953, 171, 740–741. [Google Scholar] [CrossRef]
- Williams, R.C.; Wyckoff, R.W. Applications of metallic shadow-casting to microscopy. J. App. Phys. 1946, 17, 23–33. [Google Scholar] [CrossRef]
- Hall, C.E. Method for the observation of macromolecules with the electron microscope illustrated with micrographs of DNA. J. Biophys. Biochem. Cytol. 1956, 2, 625–628. [Google Scholar] [CrossRef]
- Griffith, J.; Huberman, J.A.; Kornberg, A. Electron microscopy of DNA polymerase bound to DNA. J. Mol. Biol. 1971, 55, 209–214. [Google Scholar] [CrossRef]
- Morikawa, K.; Yanagida, M. Visualization of individual DNA molecules in solution by light microscopy: DAPI staining method. J. Biochem. 1981, 89, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, S.; Wells, K.S.; Johnson, I.D.; Bustamante, C. Direct visualization of individual DNA molecules by fluorescence microscopy: Characterization of the factors affecting signal/background and optimization of imaging conditions using YOYO. Anal. Biochem. 1997, 249, 44–53. [Google Scholar] [CrossRef]
- Reuter, M.; Dryden, D.T. The kinetics of YOYO-1 intercalation into single molecules of double-stranded DNA. Biochem. Biophys. Res. Commun. 2010, 403, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Petty, J.T.; Bordelon, J.A.; Robertson, M.E. Thermodynamic characterization of the association of cyanine dyes with DNA. J. Phys. Chem. B 2000, 104, 7221–7227. [Google Scholar] [CrossRef]
- Rye, H.S.; Yue, S.; Wemmer, D.E.; Quesada, M.A.; Haugland, R.P.; Mathies, R.A.; Glazer, A.N. Stable fluorescent complexes of double-stranded DNA with bis-intercalating asymmetric cyanine dyes: Properties and applications. Nucleic Acids Res. 1992, 20, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Houseal, T.; Bustamante, C.; Stump, R.; Maestre, M. Real-time imaging of single DNA molecules with fluorescence microscopy. Biophys. J. 1989, 56, 507–516. [Google Scholar] [CrossRef]
- Gurrieri, S.; Smith, S.B.; Wells, K.S.; Johnson, I.D.; Bustamante, C. Real-time imaging of the reorientation mechanisms of YOYO-labelled DNA molecules during 90 and 120 pulsed field gel electrophoresis. Nucleic Acids Res. 1996, 24, 4759–4767. [Google Scholar] [CrossRef]
- Schwartz, D.C.; Li, X.; Hernandez, L.I.; Ramnarain, S.P.; Huff, E.J.; Wang, Y.-K. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 1993, 262, 110–114. [Google Scholar] [CrossRef]
- Carisson, C.; Johnson, M.; Åkerman, B. Double bands in DNA gel elctrophoresis caused by bis-intercalating dyes. Nucleic Acids Res. 1995, 23, 2413–2420. [Google Scholar] [CrossRef]
- Tycon, M.A.; Dial, C.F.; Faison, K.; Melvin, W.; Fecko, C.J. Quantification of dye-mediated photodamage during single-molecule DNA imaging. Anal. Biochem. 2012, 426, 13–21. [Google Scholar] [CrossRef]
- Åkerman, B.; Tuite, E. Single-and double-strand photocleavage of DNA by YO, YOYO and TOTO. Nucleic Acids Res. 1996, 24, 1080–1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanner, N.A.; Loparo, J.J.; Hamdan, S.M.; Jergic, S.; Dixon, N.E.; van Oijen, A.M. Real-time single-molecule observation of rolling-circle DNA replication. Nucleic Acids Res. 2009, 37, 27. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Cattoni, D.I.; Nöllmann, M. The fluorescence properties and binding mechanism of SYTOX green, a bright, low photo-damage DNA intercalating agent. Eur. Biophys. J. 2015, 44, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Lim, J.; Ha, T. Acidification of the oxygen scavenging system in single-molecule fluorescence studies: In situ sensing with a ratiometric dual-emission probe. Anal. Chem. 2010, 82, 6132–6138. [Google Scholar] [CrossRef] [PubMed]
- Aitken, C.E.; Marshall, R.A.; Puglisi, J.D. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008, 94, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, M.; Henig, J.; Cheng, H.-M.; Brugger, D.; Haltrich, D.; Plumeré, N.; Schlierf, M. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano 2012, 6, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.D.; Spence, M.T.Z. Molecular Probes Handbook: A Guide to Fluorescent Probes and Labelling Technologies; Life Technologies Corporation: Eugene, OR, USA, 2010. [Google Scholar]
- Schroeder, C.M.; Babcock, H.P.; Shaqfeh, E.S.; Chu, S. Observation of polymer conformation hysteresis in extensional flow. Science 2003, 301, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Loparo, J.J.; Takahashi, M.; Richardson, C.C.; van Oijen, A.M. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 2009, 457, 336–339. [Google Scholar] [CrossRef]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 1997, 63, 2421–2431. [Google Scholar]
- Yan, X.; Habbersett, R.C.; Cordek, J.M.; Nolan, J.P.; Yoshida, T.M.; Jett, J.H.; Marrone, B.L. Development of a mechanism-based, DNA staining protocol using SYTOX orange nucleic acid stain and DNA fragment sizing flow cytometry. Anal. Biochem. 2000, 286, 138–148. [Google Scholar] [CrossRef]
- Yan, X.; Habbersett, R.C.; Yoshida, T.M.; Nolan, J.P.; Jett, J.H.; Marrone, B.L. Probing the kinetics of SYTOX Orange stain binding to double-stranded DNA with implications for DNA analysis. Anal. Chem. 2005, 77, 3554–3562. [Google Scholar] [CrossRef] [PubMed]
- Biebricher, A.S.; Heller, I.; Roijmans, R.F.; Hoekstra, T.P.; Peterman, E.J.; Wuite, G.J. The impact of DNA intercalators on DNA and DNA-processing enzymes elucidated through force-dependent binding kinetics. Nat. Commun. 2015, 6, 7304. [Google Scholar] [CrossRef] [PubMed]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S. Dynamics of concentrated polymer systems. Part 1. Brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1789–1801. [Google Scholar] [CrossRef]
- Smith, D.E.; Perkins, T.T.; Chu, S. Self-diffusion of an entangled DNA molecule by reptation. Phys. Rev. Lett. 1995, 75, 4146–4149. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.T.; Smith, D.E.; Chu, S. Direct observation of tube-like motion of a single polymer chain. Science 1994, 264, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.T.; Smith, D.E.; Chu, S. Single polymer dynamics in an elongational flow. Science 1997, 276, 2016–2021. [Google Scholar] [CrossRef]
- Smith, D.E.; Chu, S. Response of flexible polymers to a sudden elongational flow. Science 1998, 281, 1335–1340. [Google Scholar] [CrossRef]
- Shaw, S.Y.; Wang, J.C. DNA knot formation in aqueous solutions. J. Knot Theor. Ramif. 1994, 3, 287–298. [Google Scholar] [CrossRef]
- Shaw, S.Y.; Wang, J.C. Knotting of a DNA chain during ring closure. Science 1993, 260, 533–536. [Google Scholar] [CrossRef]
- Rybenkov, V.V.; Cozzarelli, N.R.; Vologodskii, A.V. Probability of DNA knotting and the effective diameter of the DNA double helix. Proc. Natl. Acad. Sci. USA 1993, 90, 5307–5311. [Google Scholar] [CrossRef] [PubMed]
- Spengler, S.J.; Stasiak, A.; Cozzarelli, N.R. The stereostructure of knots and catenanes produced by phage λ integrative recombination: Implications for mechanism and DNA structure. Cell 1985, 42, 325–334. [Google Scholar] [CrossRef]
- Heichman, K.A.; Moskowitz, I.; Johnson, R.C. Configuration of DNA strands and mechanism of strand exchange in the Hin invertasome as revealed by analysis of recombinant knots. Genes Dev. 1991, 5, 1622–1634. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wasserman, S.A.; Cozzarelli, N.R. Supercoiled DNA-directed knotting by T4 topoisomerase. J. Biol. Chem. 1991, 266, 20567–20573. [Google Scholar] [PubMed]
- Sogo, J.M.; Stasiak, A.; Martínez-Robles, M.A.L.; Krimer, D.B.; Hernández, P.; Schvartzman, J.B. Formation of knots in partially replicated DNA molecules. J. Mol. Biol. 1999, 286, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Du, N.; Doyle, P.S. Compression and self-entanglement of single DNA molecules under uniform electric field. Proc. Natl. Acad. Sci. USA 2011, 108, 16153–16158. [Google Scholar] [CrossRef]
- Dean, F.B.; Stasiak, A.; Koller, T.; Cozzarelli, N.R. Duplex DNA knots produced by Escherichia coli topoisomerase I. Structure and requirements for formation. J. Biol. Chem. 1985, 260, 4975–4983. [Google Scholar] [PubMed]
- Wasserman, S.A.; Dungan, J.M.; Cozzarelli, N.R. Discovery of a predicted DNA knot substantiates a model for site-specific recombination. Science 1985, 229, 171–174. [Google Scholar] [CrossRef]
- Arai, Y.; Yasuda, R.; Akashi, K.-i.; Harada, Y.; Miyata, H.; Kinosita Jr, K.; Itoh, H. Tying a molecular knot with optical tweezers. Nature 1999, 399, 446–448. [Google Scholar]
- Bao, X.R.; Lee, H.J.; Quake, S.R. Behavior of complex knots in single DNA molecules. Phys. Rev. Lett. 2003, 91, 265506. [Google Scholar] [CrossRef]
- Krasnow, M.A.; Stasiak, A.; Spengler, S.J.; Dean, F.; Koller, T.; Cozzarelli, N.R. Determination of the absolute handedness of knots and catenanes of DNA. Nature 1983, 304, 559. [Google Scholar] [CrossRef] [PubMed]
- Walba, D.M. Topological stereochemistry. Tetrahedron 1985, 41, 3161–3212. [Google Scholar] [CrossRef]
- Pierański, P.; Przybył, S.; Stasiak, A. Tight open knots. Eur. Phys. J. E 2001, 6, 123–128. [Google Scholar] [CrossRef]
- Klotz, A.R.; Soh, B.W.; Doyle, P.S. Motion of Knots in DNA Stretched by Elongational Fields. Phys. Rev. Lett. 2018, 120, 188003. [Google Scholar] [CrossRef] [PubMed]
- Narsimhan, V.; Renner, C.B.; Doyle, P.S. Jamming of knots along a tensioned chain. ACS Macro. Lett. 2016, 5, 123–127. [Google Scholar] [CrossRef]
- Huang, L.; Makarov, D.E. Langevin dynamics simulations of the diffusion of molecular knots in tensioned polymer chains. J. Phys. Chem. A 2007, 111, 10338–10344. [Google Scholar] [CrossRef]
- Metzler, R.; Reisner, W.; Riehn, R.; Austin, R.; Tegenfeldt, J.; Sokolov, I.M. Diffusion mechanisms of localised knots along a polymer. Europhys. Lett. 2006, 76, 696–702. [Google Scholar] [CrossRef]
- Plesa, C.; Verschueren, D.; Pud, S.; van der Torre, J.; Ruitenberg, J.W.; Witteveen, M.J.; Jonsson, M.P.; Grosberg, A.Y.; Rabin, Y.; Dekker, C. Direct observation of DNA knots using a solid-state nanopore. Nat. Nanotechnol. 2016, 11, 1093–1097. [Google Scholar] [CrossRef]
- Vinograd, J.; Lebowitz, J.; Radloff, R.; Watson, R.; Laipis, P. The twisted circular form of polyoma viral DNA. Proc. Natl. Acad. Sci. USA 1965, 53, 1104–1111. [Google Scholar] [CrossRef]
- Paleček, E. Local supercoil-stabilized DNA structure. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 151–226. [Google Scholar] [CrossRef]
- Liu, Y.; Bondarenko, V.; Ninfa, A.; Studitsky, V.M. DNA supercoiling allows enhancer action over a large distance. Proc. Natl. Acad. Sci. USA 2001, 98, 14883–14888. [Google Scholar] [CrossRef]
- Dunaway, M.; Ostrander, E.A. Local domains of supercoiling activate a eukaryotic promoter in vivo. Nature 1993, 361, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Strick, T.R.; Allemand, J.-F.; Bensimon, D.; Bensimon, A.; Croquette, V. The elasticity of a single supercoiled DNA molecule. Science 1996, 271, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, M.T.; de Grunt, M.; Dekker, C. Dynamics of DNA supercoils. Science 2012, 338, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Ganji, M.; Kim, S.H.; van der Torre, J.; Abbondanzieri, E.; Dekker, C. Intercalation-based single-molecule fluorescence assay to study DNA supercoil dynamics. Nano Lett. 2016, 16, 4699–4707. [Google Scholar] [CrossRef] [PubMed]
- Bellomy, G.R.; Record, M.T. Stable DNA loops in vivo and in vitro: Roles in gene regulation at a distance and in biophysical characterization of DNA. Prog. Nucleic Acid Res. Mol. Biol. 1990, 39, 81–128. [Google Scholar]
- De Vargas, L.M.; Landy, A. A switch in the formation of alternative DNA loops modulates lambda site-specific recombination. Proc. Natl. Acad. Sci. USA 1991, 88, 588–592. [Google Scholar] [CrossRef]
- Pandey, M.; Syed, S.; Donmez, I.; Patel, G.; Ha, T.; Patel, S.S. Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature 2009, 462, 940–943. [Google Scholar] [CrossRef]
- Aragon, L.; Martinez-Perez, E.; Merkenschlager, M. Condensin, cohesin and the control of chromatin states. Curr. Opin. Genet. Dev. 2013, 23, 204–211. [Google Scholar] [CrossRef][Green Version]
- Jeppsson, K.; Kanno, T.; Shirahige, K.; Sjögren, C. The maintenance of chromosome structure: Positioning and functioning of SMC complexes. Nat. Rev. Mol. Cell. Biol. 2014, 15, 601–614. [Google Scholar] [CrossRef]
- Ganji, M.; Shaltiel, I.A.; Bisht, S.; Kim, E.; Kalichava, A.; Haering, C.H.; Dekker, C. Real-time imaging of DNA loop extrusion by condensin. Science 2018, 360, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Vafabakhsh, R.; Ha, T. Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science 2012, 337, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Kim, H.D. Studying DNA looping by single-molecule FRET. J. Vis. Exp. 2014, 88, 51667. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Le, T.T.; Kim, H.D. Single-molecule fluorescence studies on DNA looping. Methods 2016, 105, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Bryant, Z.; Smith, S.B. Ten years of tension: Single-molecule DNA mechanics. Nature 2003, 421, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Plank, J.L.; Dombrowski, C.C.; Kowalczykowski, S.C. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 2012, 491, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Gibb, B.; Silverstein, T.D.; Finkelstein, I.J.; Greene, E.C. Single-stranded DNA curtains for real-time single-molecule visualization of protein–nucleic acid interactions. Anal. Chem. 2012, 84, 7607–7612. [Google Scholar] [CrossRef]
- De Tullio, L.; Kaniecki, K.; Kwon, Y.; Crickard, J.B.; Sung, P.; Greene, E.C. Yeast Srs2 helicase promotes redistribution of single-stranded DNA-bound RPA and Rad52 in homologous recombination regulation. Cell. Rep. 2017, 21, 570–577. [Google Scholar] [CrossRef]
- Kaniecki, K.; De Tullio, L.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell. Rep. 2017, 21, 3166–3177. [Google Scholar] [CrossRef]
- Tanner, N.A.; Hamdan, S.M.; Jergic, S.; Loscha, K.V.; Schaeffer, P.M.; Dixon, N.E.; van Oijen, A.M. Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol. 2008, 15, 170–176. [Google Scholar] [CrossRef]
- Jergic, S.; Horan, N.P.; Elshenawy, M.M.; Mason, C.E.; Urathamakul, T.; Ozawa, K.; Robinson, A.; Goudsmits, J.M.; Wang, Y.; Pan, X. A direct proofreader–clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 2013, 32, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Geertsema, H.J.; Duderstadt, K.E.; van Oijen, A.M. Single-molecule observation of prokaryotic DNA replication. Methods Mol. Biol. 2015, 1300, 219–238. [Google Scholar] [PubMed]
- Lewis, J.S.; Spenkelink, L.M.; Jergic, S.; Wood, E.A.; Monachino, E.; Horan, N.P.; Duderstadt, K.E.; Cox, M.M.; Robinson, A.; Dixon, N.E. Single-molecule visualization of fast polymerase turnover in the bacterial replisome. Elife 2017, 6, 23932. [Google Scholar] [CrossRef] [PubMed]
- Monachino, E.; Ghodke, H.; Spinks, R.R.; Hoatson, B.S.; Jergic, S.; Xu, Z.-Q.; Dixon, N.E.; van Oijen, A.M. Design of DNA rolling-circle templates with controlled fork topology to study mechanisms of DNA replication. Anal. Biochem. 2018, 557, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.Y.; Georgescu, R.E.; Finkelstein, J.; O’Donnell, M.E. Single-molecule analysis reveals that the lagging strand increases replisome processivity but slows replication fork progression. Proc. Natl. Acad. Sci. USA 2009, 106, 13236–13241. [Google Scholar] [CrossRef] [PubMed]
- Debyser, Z.; Tabor, S.; Richardson, C.C. Coordination of leading and lagging strand DNA synthesis at the replication fork of bacteriophage T7. Cell 1994, 77, 157–166. [Google Scholar] [CrossRef]
- Yang, J.; Zhuang, Z.; Roccasecca, R.M.; Trakselis, M.A.; Benkovic, S.J. The dynamic processivity of the T4 DNA polymerase during replication. Proc. Natl. Acad. Sci. USA 2004, 101, 8289–8294. [Google Scholar] [CrossRef]
- Loparo, J.J.; Kulczyk, A.W.; Richardson, C.C.; van Oijen, A.M. Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc. Natl. Acad. Sci. USA 2011, 3584–3589. [Google Scholar] [CrossRef]
- Geertsema, H.J.; Kulczyk, A.W.; Richardson, C.C.; van Oijen, A.M. Single-molecule studies of polymerase dynamics and stoichiometry at the bacteriophage T7 replication machinery. Proc. Natl. Acad. Sci. USA 2014, 111, 4073–4078. [Google Scholar] [CrossRef]
- Beattie, T.R.; Kapadia, N.; Nicolas, E.; Uphoff, S.; Wollman, A.J.; Leake, M.C.; Reyes-Lamothe, R. Frequent exchange of the DNA polymerase during bacterial chromosome replication. Elife 2017, 6, 21763. [Google Scholar] [CrossRef]
- Liao, Y.; Li, Y.; Schroeder, J.W.; Simmons, L.A.; Biteen, J.S. Single-molecule DNA polymerase dynamics at a bacterial replisome in live cells. Biophys. J. 2016, 111, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chastain II, P.D.; Kusakabe, T.; Griffith, J.D.; Richardson, C.C. Coordinated leading and lagging strand DNA synthesis on a minicircular template. Mol. Cell. 1998, 1, 1001–1010. [Google Scholar] [CrossRef]
- Li, X.; Marians, K.J. Two distinct triggers for cycling of the lagging-strand polymerase at the replication fork. J. Biol. Chem. 2000, 275, 34757–34765. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chastain Ii, P.D.; Griffith, J.D.; Richardson, C.C. Lagging strand synthesis in coordinated DNA synthesis by bacteriophage T7 replication proteins1. J. Mol. Biol. 2002, 316, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-B.; Hite, R.K.; Hamdan, S.M.; Xie, X.S.; Richardson, C.C.; van Oijen, A.M. DNA primase acts as a molecular brake in DNA replication. Nature 2006, 439, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Dixon, N.E. DNA replication: Prime-time looping. Nature 2009, 462, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Geertsema, H.J.; Stratmann, S.A.; Punter, C.M.; Kulczyk, A.W.; Richardson, C.C.; van Oijen, A.M. Simultaneous real-time imaging of leading and lagging strand synthesis reveals the coordination dynamics of single replisomes. Mol. Cell. 2016, 64, 1035–1047. [Google Scholar] [CrossRef]
- Graham, J.E.; Marians, K.J.; Kowalczykowski, S.C. Independent and stochastic action of DNA polymerases in the replisome. Cell 2017, 169, 1201–1213. [Google Scholar] [CrossRef]
- Walter, J.; Newport, J.W. Regulation of replicon size in Xenopus egg extracts. Science 1997, 275, 993–995. [Google Scholar] [CrossRef]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef]
- Byun, T.S.; Pacek, M.; Yee, M.-c.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef]
- Fu, Y.V.; Yardimci, H.; Long, D.T.; Ho, T.V.; Guainazzi, A.; Bermudez, V.P.; Hurwitz, J.; van Oijen, A.; Schärer, O.D.; Walter, J.C. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 2011, 146, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Yardimci, H.; Loveland, A.B.; Habuchi, S.; van Oijen, A.M.; Walter, J.C. Uncoupling of sister replisomes during eukaryotic DNA replication. Mol. Cell. 2010, 40, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Loveland, A.B.; Habuchi, S.; Walter, J.C.; van Oijen, A.M. A general approach to break the concentration barrier in single-molecule imaging. Nat. Methods 2012, 9, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Ticau, S.; Friedman, L.J.; Ivica, N.A.; Gelles, J.; Bell, S.P. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 2015, 161, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cid, A.; Riera, A.; Tognetti, S.; Herrera, M.C.; Samel, S.; Evrin, C.; Winkler, C.; Gardenal, E.; Uhle, S.; Speck, C. An ORC/Cdc6/MCM2-7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol. Cell. 2013, 50, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Evrin, C.; Samel, S.A.; Fernández-Cid, A.; Riera, A.; Kawakami, H.; Stillman, B.; Speck, C.; Li, H. Cryo-EM structure of a helicase loading intermediate containing ORC–Cdc6–Cdt1–MCM2-7 bound to DNA. Nat. Struct. Mol. Biol. 2013, 20, 944–951. [Google Scholar] [CrossRef]
- Sun, J.; Fernandez-Cid, A.; Riera, A.; Tognetti, S.; Yuan, Z.; Stillman, B.; Speck, C.; Li, H. Structural and mechanistic insights into Mcm2–7 double-hexamer assembly and function. Genes Dev. 2014, 28, 2291–2303. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.-B.; Fishel, R. Stochastic Processes and Component Plasticity Governing DNA Mismatch Repair. J. Mol. Biol. 2018, 430, 4456–4468. [Google Scholar] [CrossRef]
- Kim, D.; Fishel, R.; Lee, J.-B. Coordinating Multi-Protein Mismatch Repair by Managing Diffusion Mechanics on the DNA. J. Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Zohar, H.; Muller, S.J. Labelling DNA for single-molecule experiments: Methods of labelling internal specific sequences on double-stranded DNA. Nanoscale 2011, 3, 3027–3039. [Google Scholar] [CrossRef]
- Bensimon, A.; Simon, A.; Chiffaudel, A.; Croquette, V.; Heslot, F.; Bensimon, D. Alignment and sensitive detection of DNA by a moving interface. Science 1994, 265, 2096–2098. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.K.; Arcangioli, B.; Baker, S.P.; Bensimon, A.; Rhind, N. DNA replication origins fire stochastically in fission yeast. Mol. Biol. Cell. 2006, 17, 308–316. [Google Scholar] [CrossRef]
- Calderano, S.G.; Drosopoulos, W.C.; Quaresma, M.M.; Marques, C.A.; Kosiyatrakul, S.; McCulloch, R.; Schildkraut, C.L.; Elias, M.C. Single molecule analysis of Trypanosoma brucei DNA replication dynamics. Nucleic Acids Res. 2015, 43, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, M.; Winzeler, E.A.; Collingwood, D.; Hunt, S.; Wodicka, L.; Conway, A.; Lockhart, D.J.; Davis, R.W.; Brewer, B.J.; Fangman, W.L. Replication dynamics of the yeast genome. Science 2001, 294, 115–121. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Liu, J.; Hamlin, J.L.; Shao, Z. DNA combing reveals intrinsic temporal disorder in the replication of yeast chromosome VI. J. Mol. Biol. 2008, 375, 12–19. [Google Scholar] [CrossRef]
- Michalet, X.; Ekong, R.; Fougerousse, F.; Rousseaux, S.; Schurra, C.; Hornigold, N.; van Slegtenhorst, M.; Wolfe, J.; Povey, S.; Beckmann, J.S. Dynamic molecular combing: Stretching the whole human genome for high-resolution studies. Science 1997, 277, 1518–1523. [Google Scholar] [CrossRef]
- Langer-Safer, P.R.; Levine, M.; Ward, D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 4381–4385. [Google Scholar] [CrossRef]
- Yadav, H.; Sharma, P. A simple and novel DNA combing methodology for Fiber-FISH and optical mapping. Genomics 2018. [Google Scholar] [CrossRef]
- Kaykov, A.; Taillefumier, T.; Bensimon, A.; Nurse, P. Molecular combing of single DNA molecules on the 10 megabase scale. Sci. Rep. 2016, 6, 19636. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J.; Bensimon, A. Single molecule analysis of DNA replication. Biochimie 1999, 81, 859–871. [Google Scholar] [CrossRef]
- Gueroui, Z.; Place, C.; Freyssingeas, E.; Berge, B. Observation by fluorescence microscopy of transcription on single combed DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 6005–6010. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.; Du, X.; Zhang, W.; Tosser-Klopp, G.; Wang, J.; Yang, S.; Liang, J. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2013, 31, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Chamala, S.; Chanderbali, A.S.; Der, J.P.; Lan, T.; Walts, B.; Albert, V.A.; Leebens-Mack, J.; Rounsley, S.; Schuster, S.C.; Wing, R.A. Assembly and validation of the genome of the nonmodel basal angiosperm Amborella. Science 2013, 342, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, G.; Howard, J.T.; Ward, J.M.; Li, J.; Li, B.; Li, Y.; Xiong, Y.; Zhang, Y.; Zhou, S.; Schwartz, D.C. High-coverage sequencing and annotated assemblies of the budgerigar genome. GigaScience 2014, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Neely, R.K.; Deen, J.; Hofkens, J. Optical mapping of DNA: Single-molecule-based methods for mapping genomes. Biopolymers 2011, 95, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Neely, R.K.; Dedecker, P.; Hotta, J.-i.; Urbanavičiūtė, G.; Klimašauskas, S.; Hofkens, J. DNA fluorocode: A single molecule, optical map of DNA with nanometre resolution. Chem. Sci. 2010, 1, 453–460. [Google Scholar] [CrossRef]
- Vranken, C.; Deen, J.; Dirix, L.; Stakenborg, T.; Dehaen, W.; Leen, V.; Hofkens, J.; Neely, R.K. Super-resolution optical DNA Mapping via DNA methyltransferase-directed click chemistry. Nucleic Acids Res. 2014, 42, 50. [Google Scholar] [CrossRef]
- Grunwald, A.; Dahan, M.; Giesbertz, A.; Nilsson, A.; Nyberg, L.K.; Weinhold, E.; Ambjörnsson, T.; Westerlund, F.; Ebenstein, Y. Bacteriophage strain typing by rapid single molecule analysis. Nucleic Acids Res. 2015, 43, 117. [Google Scholar] [CrossRef]
- Katsanis, S.H.; Katsanis, N. Molecular genetic testing and the future of clinical genomics. Nat. Rev. Genet. 2013, 14, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Vilfan, I.D.; Tsai, Y.-C.; Clark, T.A.; Wegener, J.; Dai, Q.; Yi, C.; Pan, T.; Turner, S.W.; Korlach, J. Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription. J. Nanobiotechnol. 2013, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Turner, S.; Craighead, H.G. Entropic trapping and escape of long DNA molecules at submicron size constriction. Phys. Rev. Lett. 1999, 83, 1688–1691. [Google Scholar] [CrossRef]
- Larkin, J.; Henley, R.Y.; Jadhav, V.; Korlach, J.; Wanunu, M. Length-independent DNA packing into nanopore zero-mode waveguides for low-input DNA sequencing. Nat. Nanotechnol. 2017, 12, 1169–1175. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, G.; Lewis, J.S.; van Oijen, A.M. Shining a Spotlight on DNA: Single-Molecule Methods to Visualise DNA. Molecules 2019, 24, 491. https://doi.org/10.3390/molecules24030491
Kaur G, Lewis JS, van Oijen AM. Shining a Spotlight on DNA: Single-Molecule Methods to Visualise DNA. Molecules. 2019; 24(3):491. https://doi.org/10.3390/molecules24030491
Chicago/Turabian StyleKaur, Gurleen, Jacob S. Lewis, and Antoine M. van Oijen. 2019. "Shining a Spotlight on DNA: Single-Molecule Methods to Visualise DNA" Molecules 24, no. 3: 491. https://doi.org/10.3390/molecules24030491
APA StyleKaur, G., Lewis, J. S., & van Oijen, A. M. (2019). Shining a Spotlight on DNA: Single-Molecule Methods to Visualise DNA. Molecules, 24(3), 491. https://doi.org/10.3390/molecules24030491