An Overview of Significant Achievements in Ruthenium-Based Molecular Water Oxidation Catalysis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. A Brief History and Mini-review of Ru-Based Homogeneous Water Oxidation Catalysis
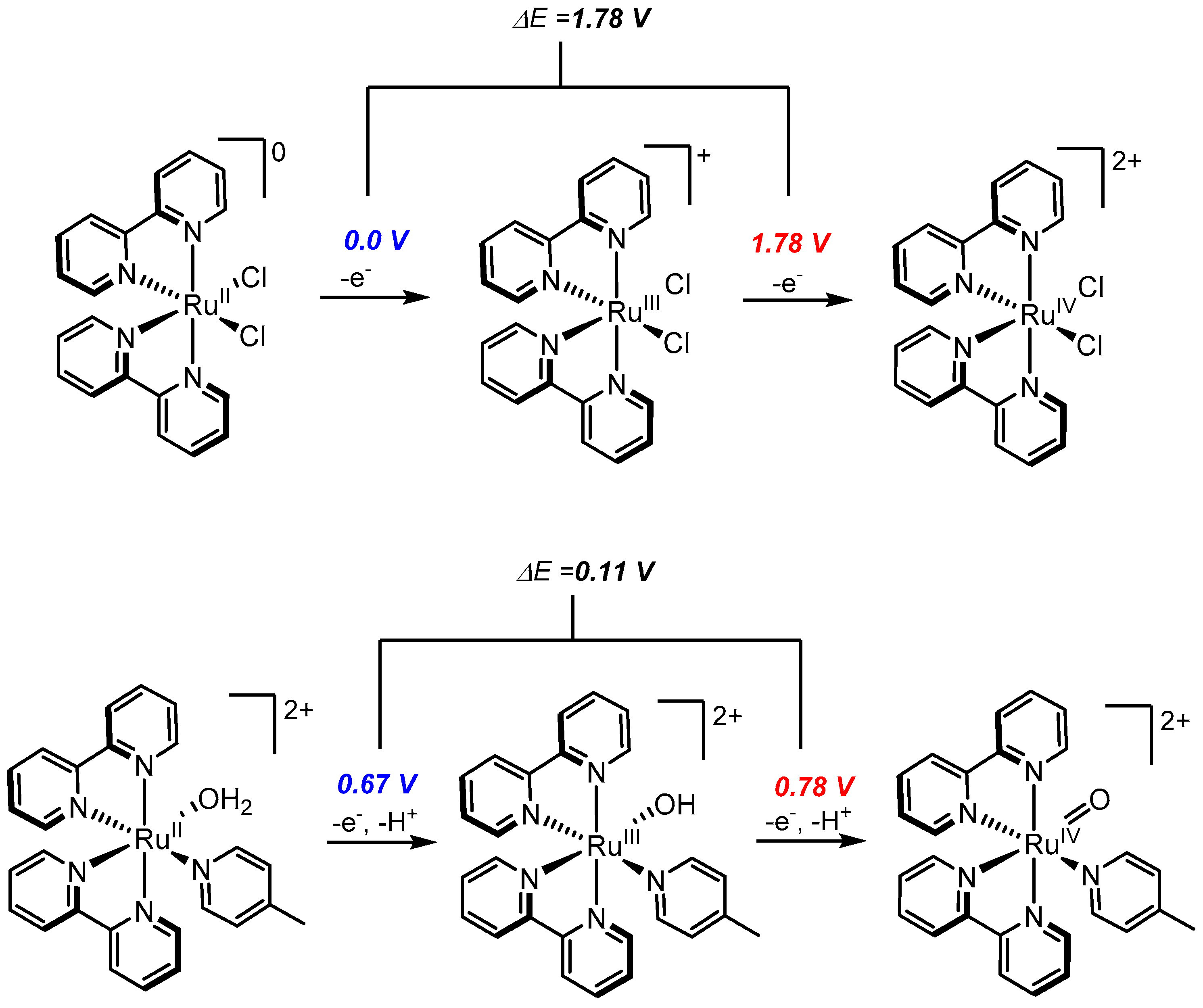
2.1. Ru-Polypyridyl Chemistry
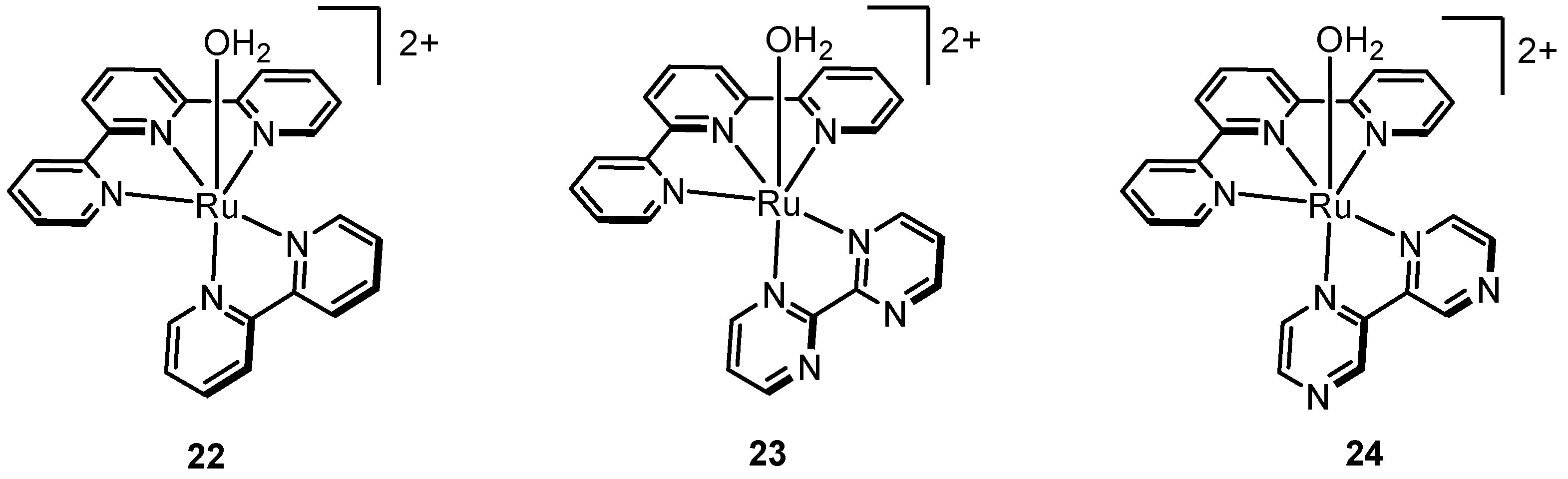
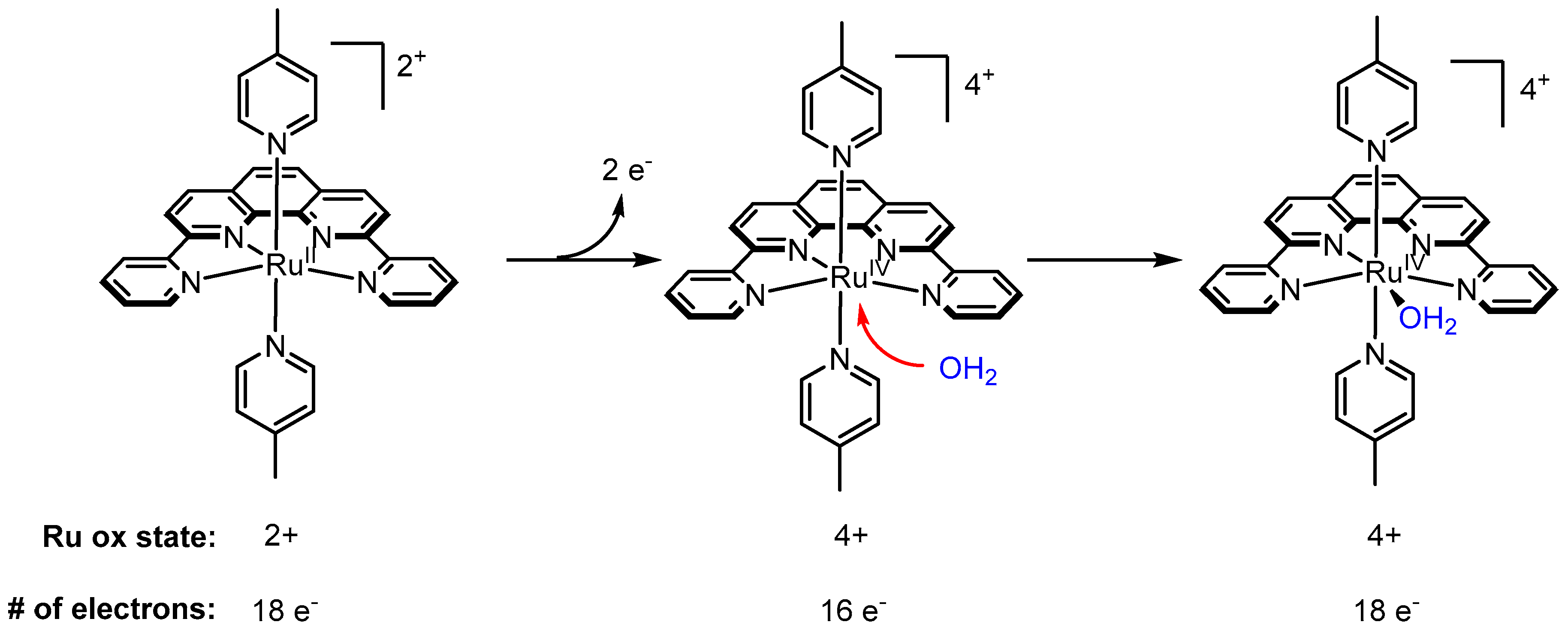
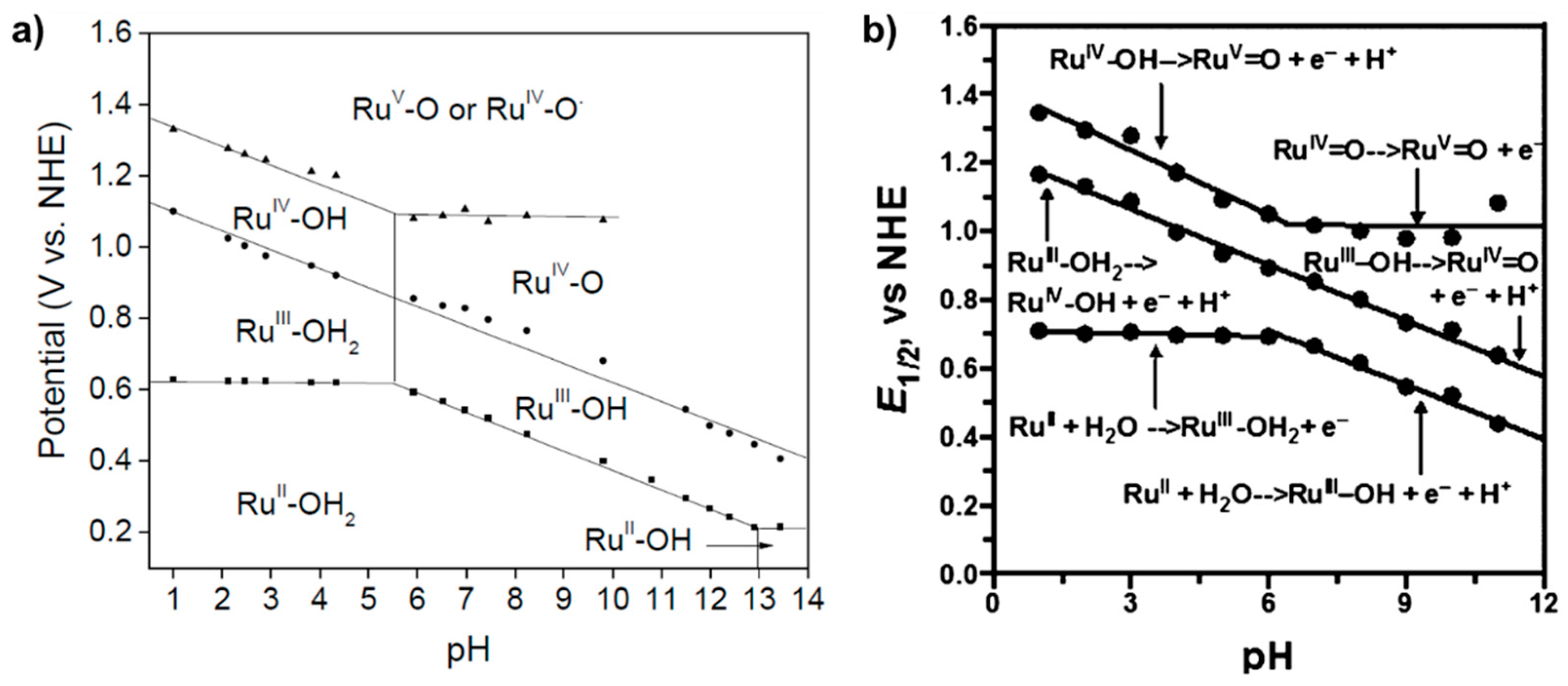
2.2. A Stable Ru-Oxo Species Derived from a Ru-Aquo Complex: RuII(bpy)2(py)(H2O)2+
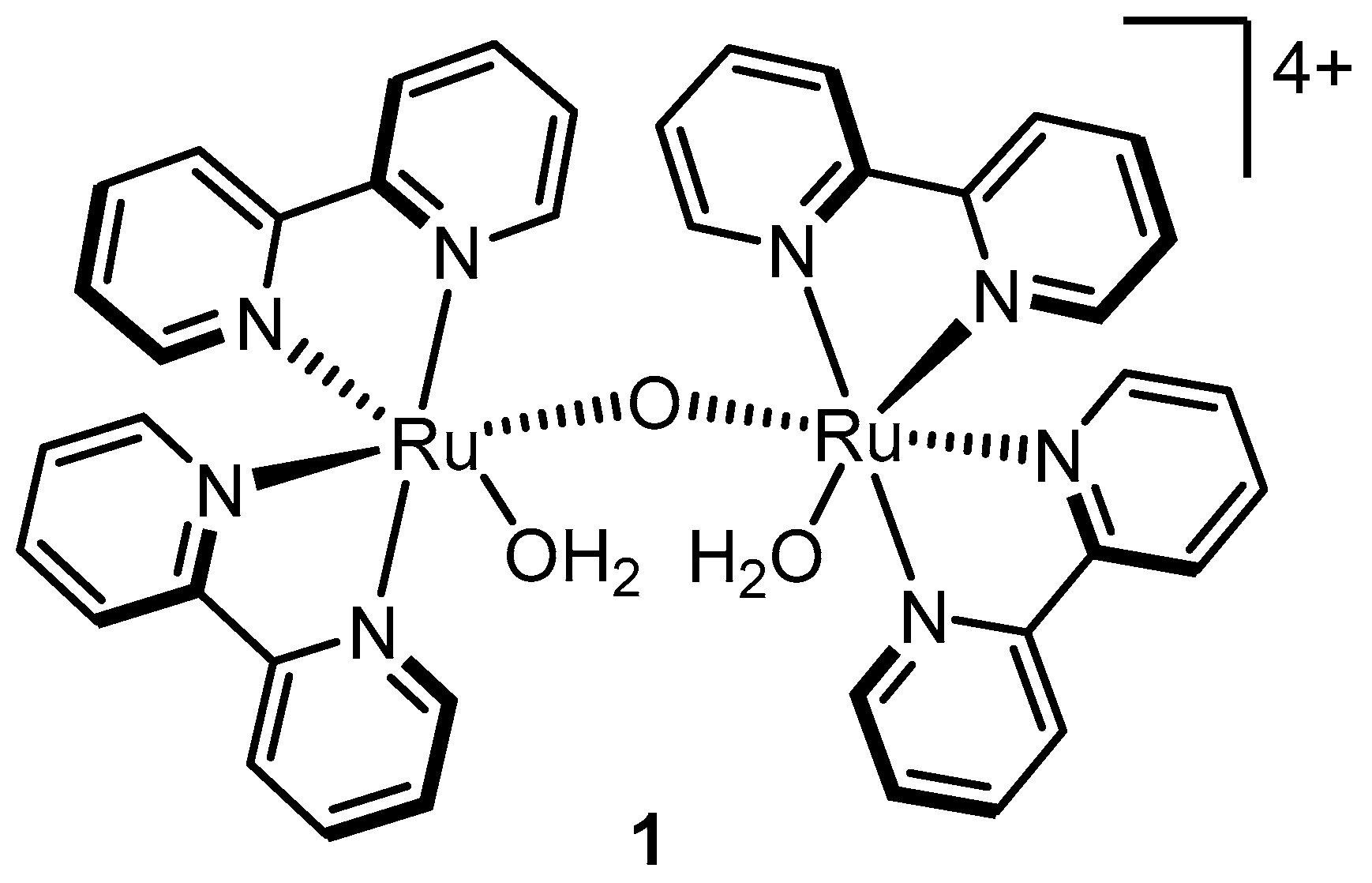
2.3. The Blue Dimer–the First Homogeneous Ruthenium Water Oxidation Catalyst
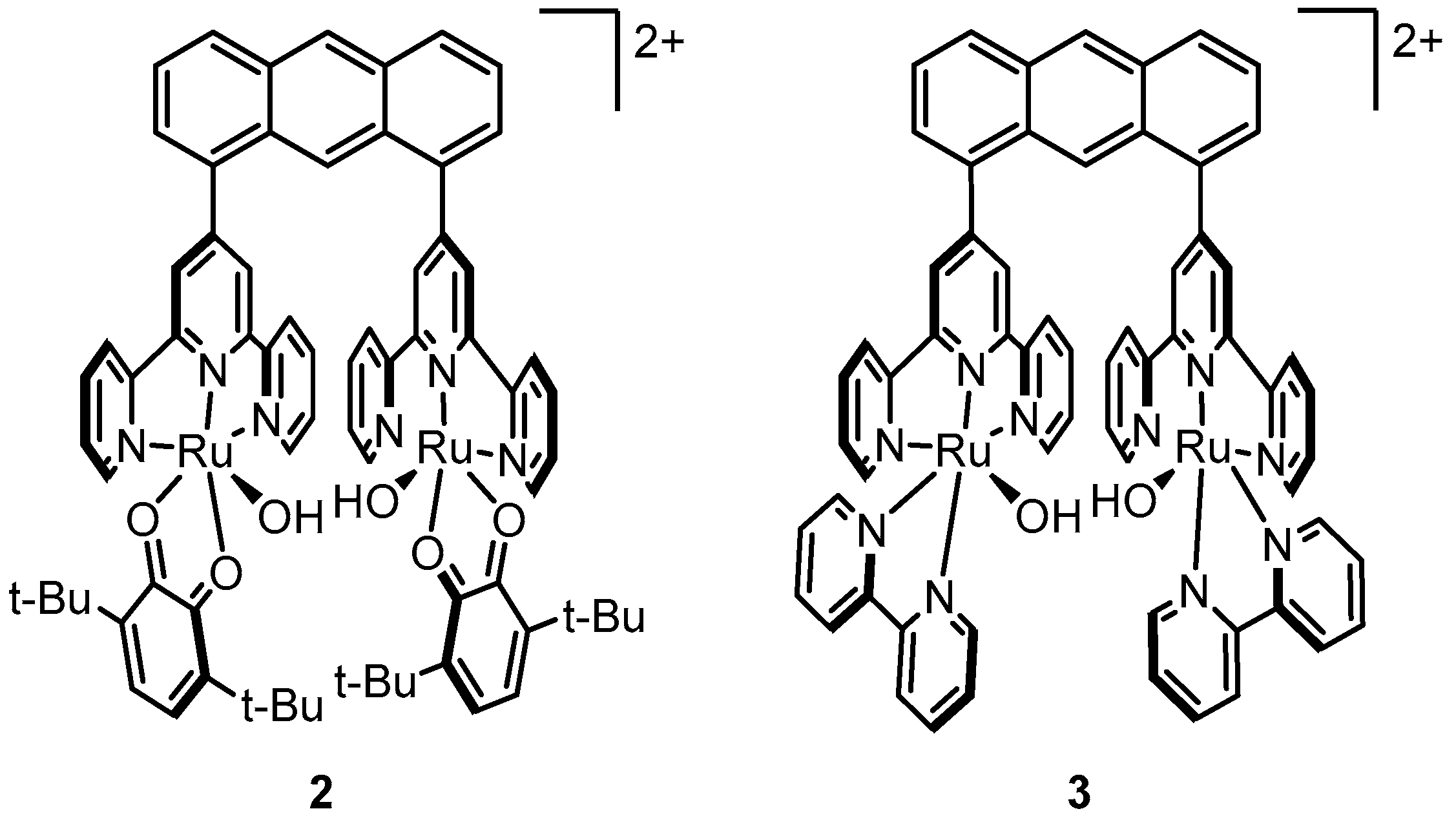
2.4. An Anthracene-Bridged Dinuclear System—Electronically Isolated Metal Centers
2.5. Dinuclear Catalysts with a more Rigid Backbone
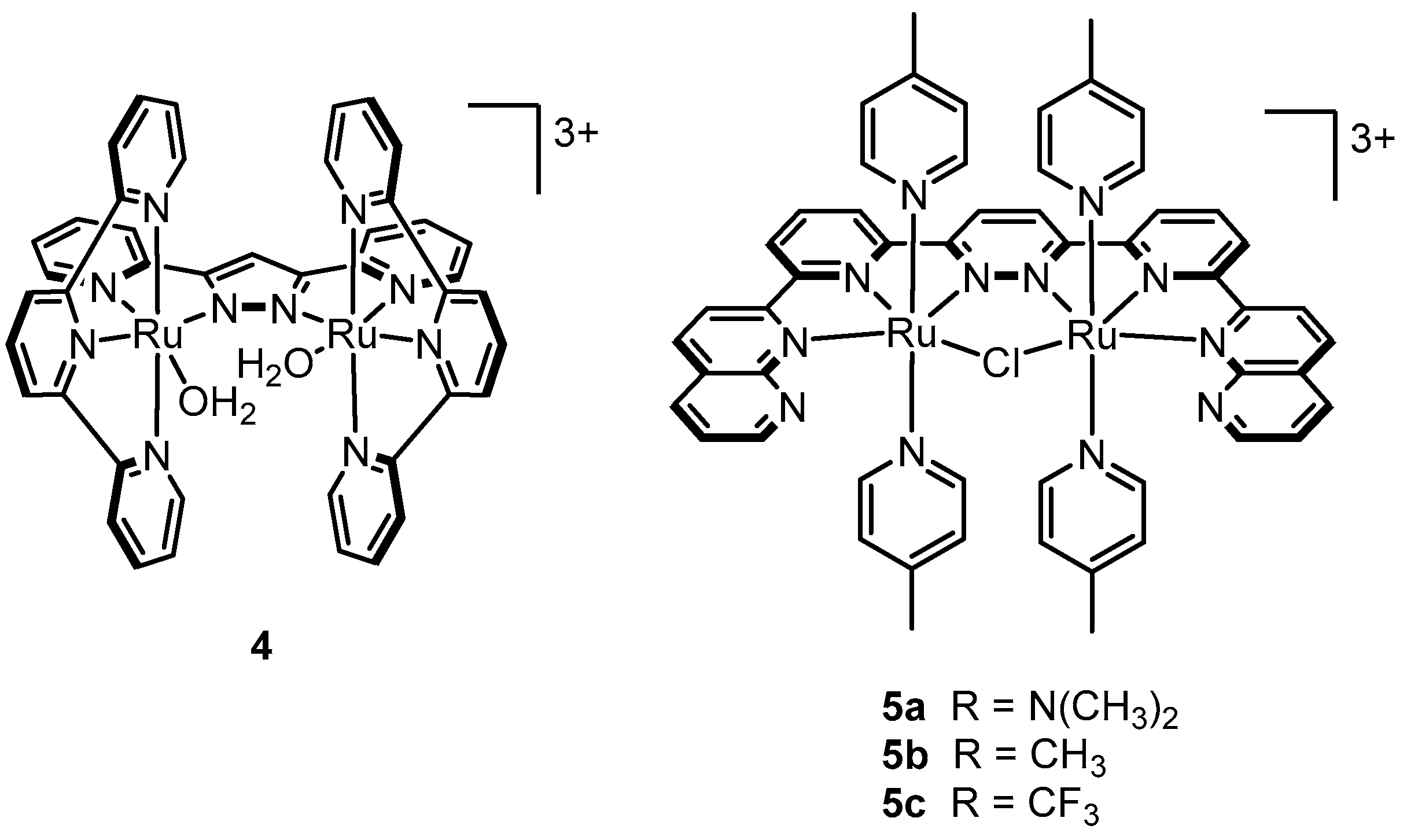
2.6. One Metal Site Is Enough
2.7. Emergence of Tetradentate Motif
2.8. Anionic Ligands Stabilize Higher Oxidation States
2.9. Catalysts Bearing 2,2’-Bipyridine-6,6’-Dicarboxylate as Tetradentate Ligands
2.10. External Bases Accelerate Catalysis
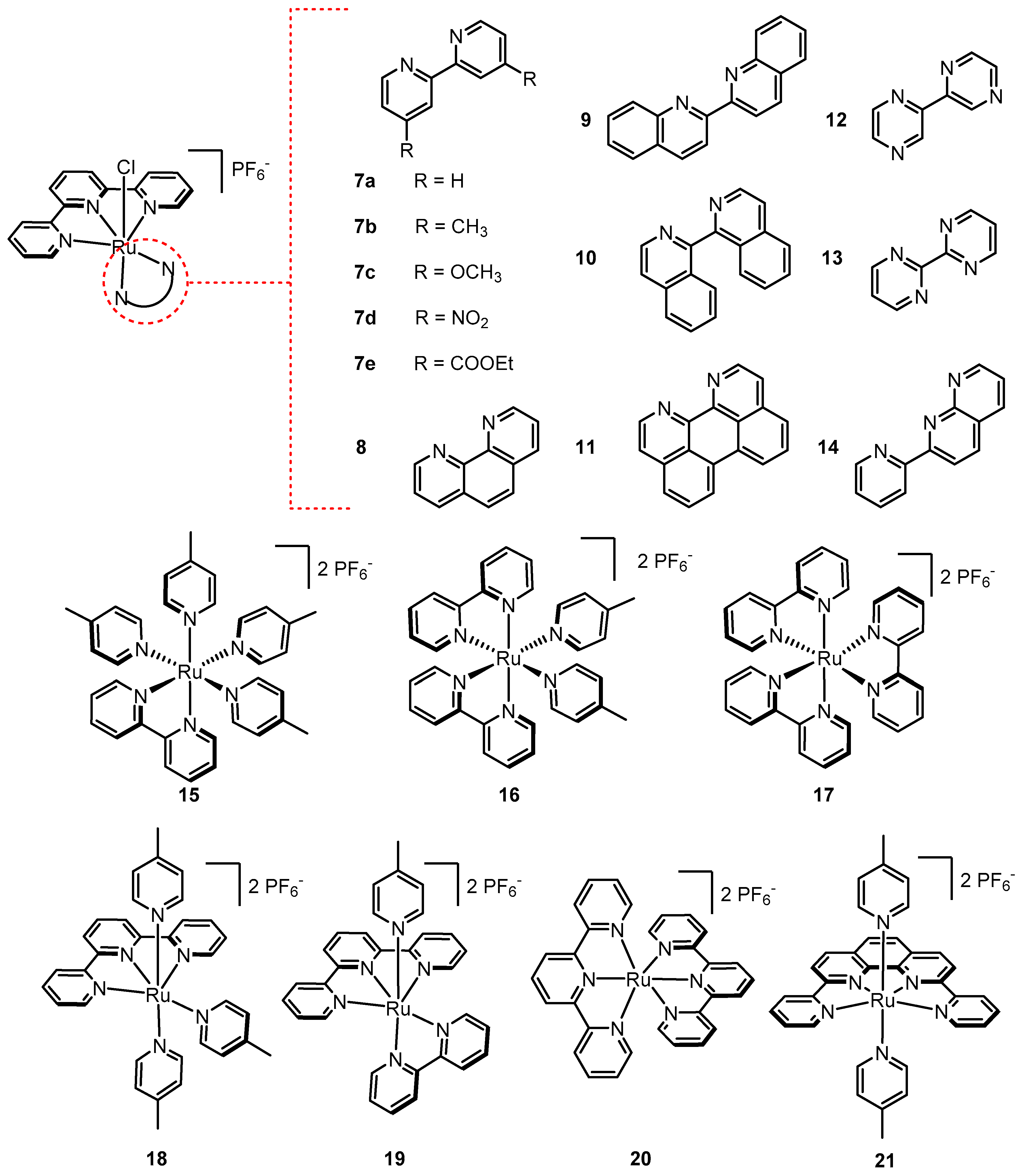
2.11. Internal Bases Accelerate Catalysis
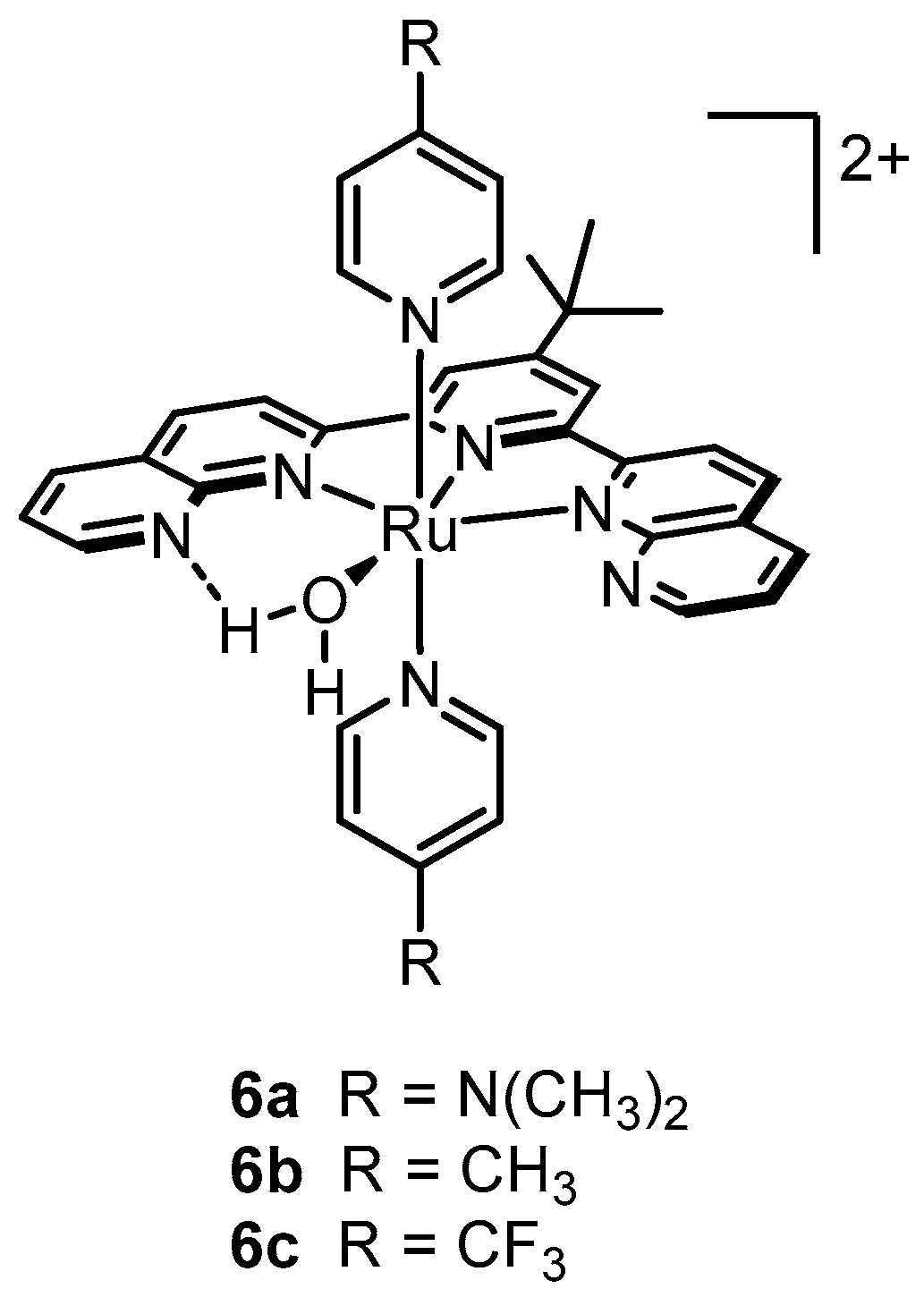
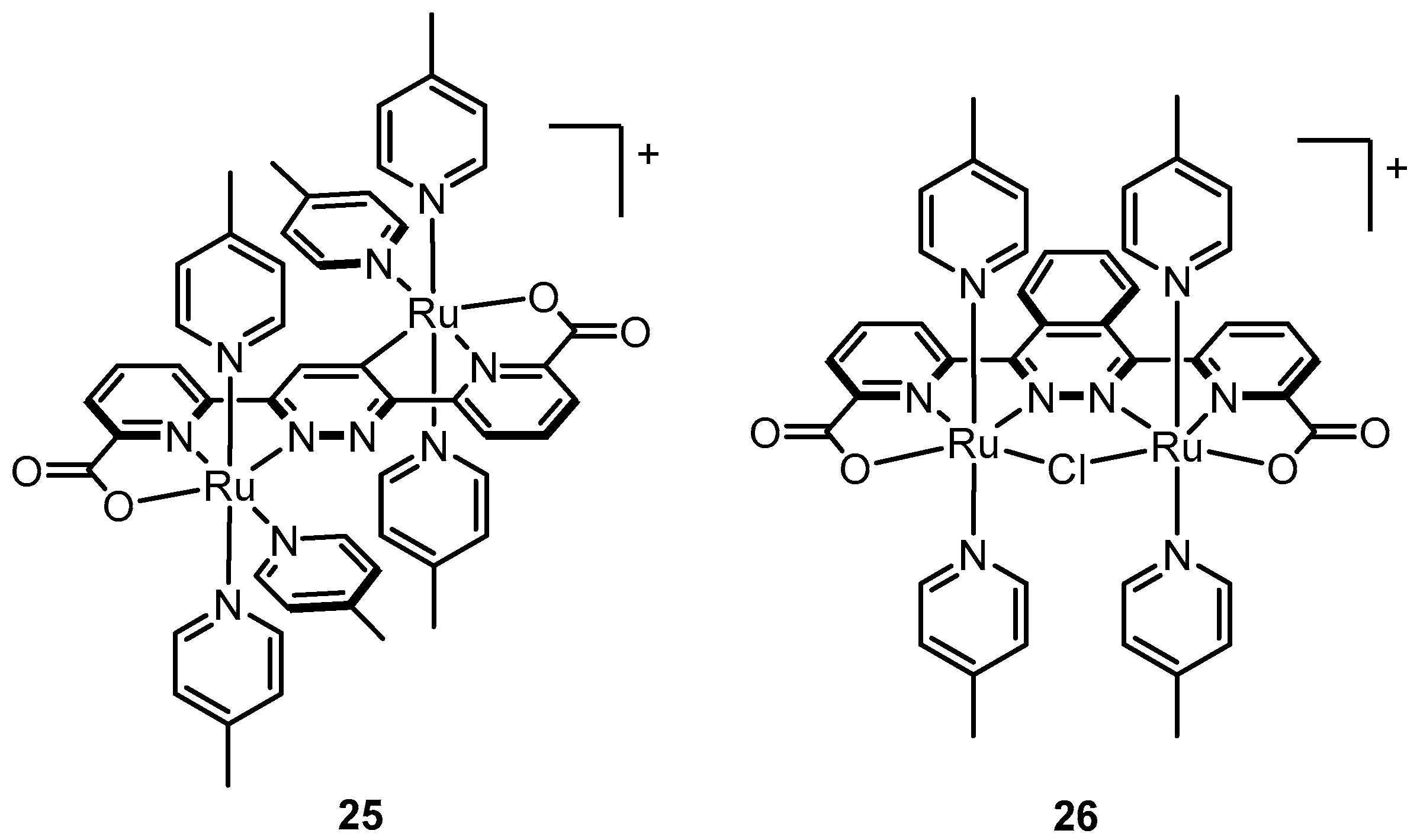
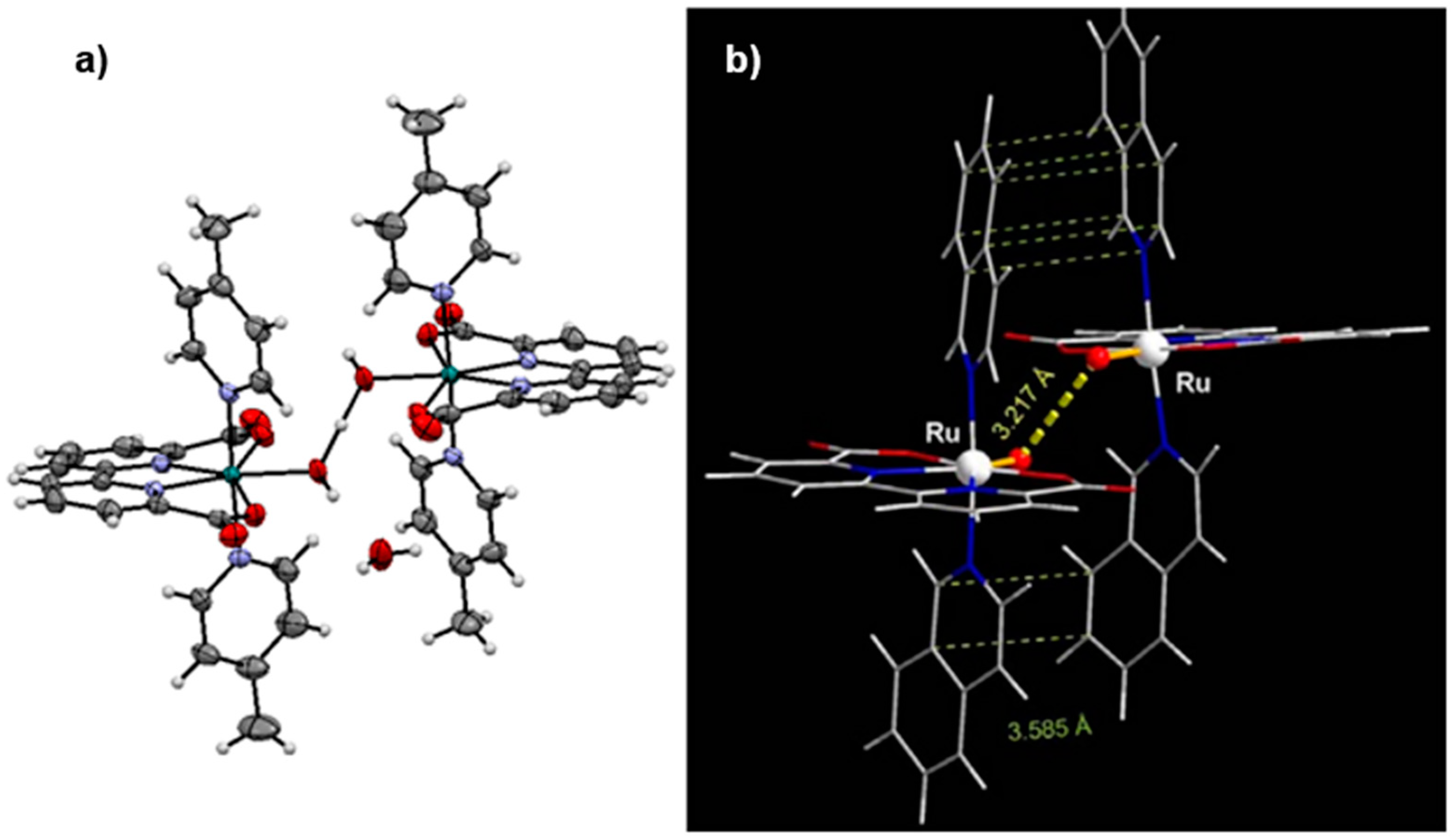
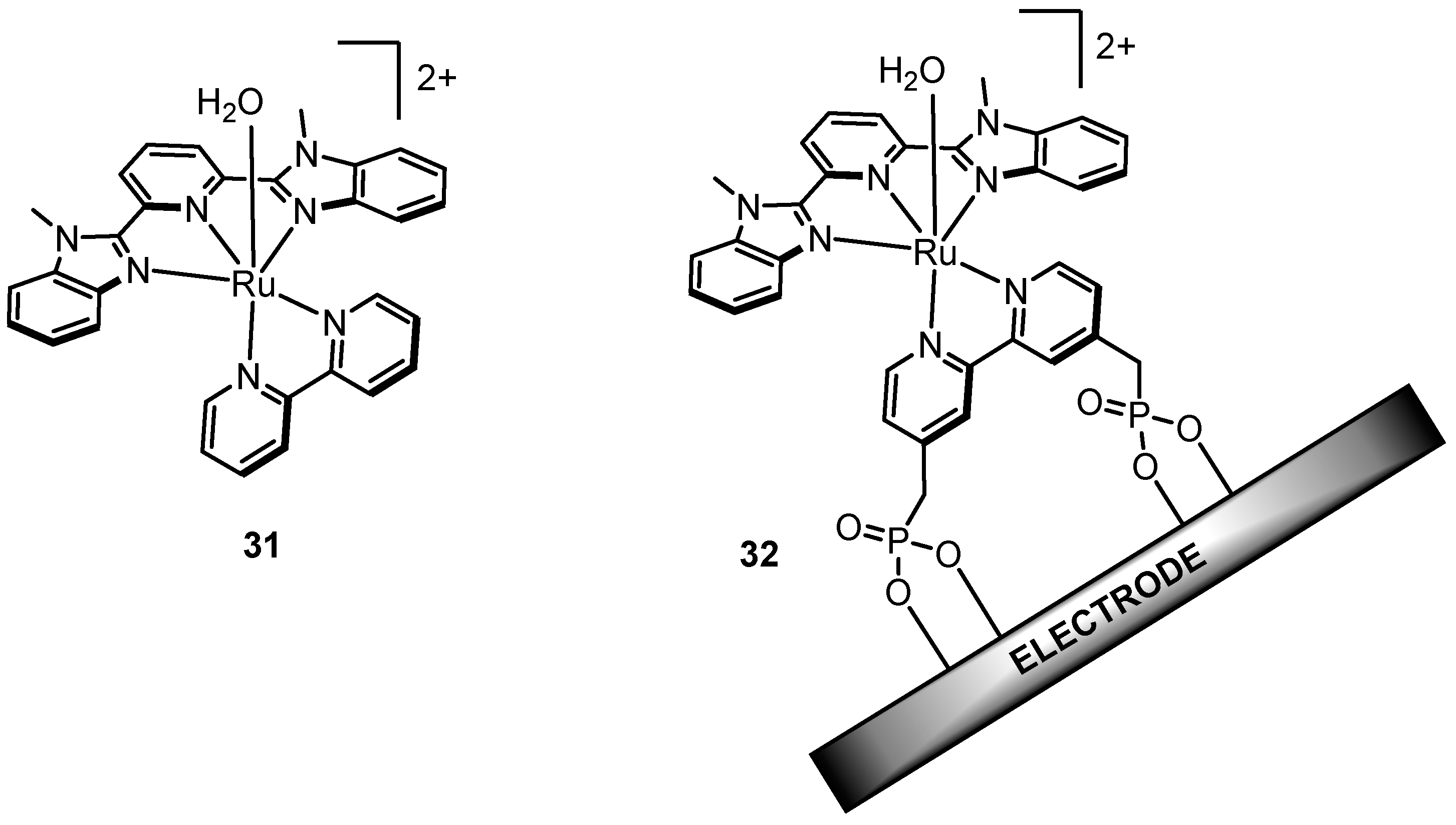
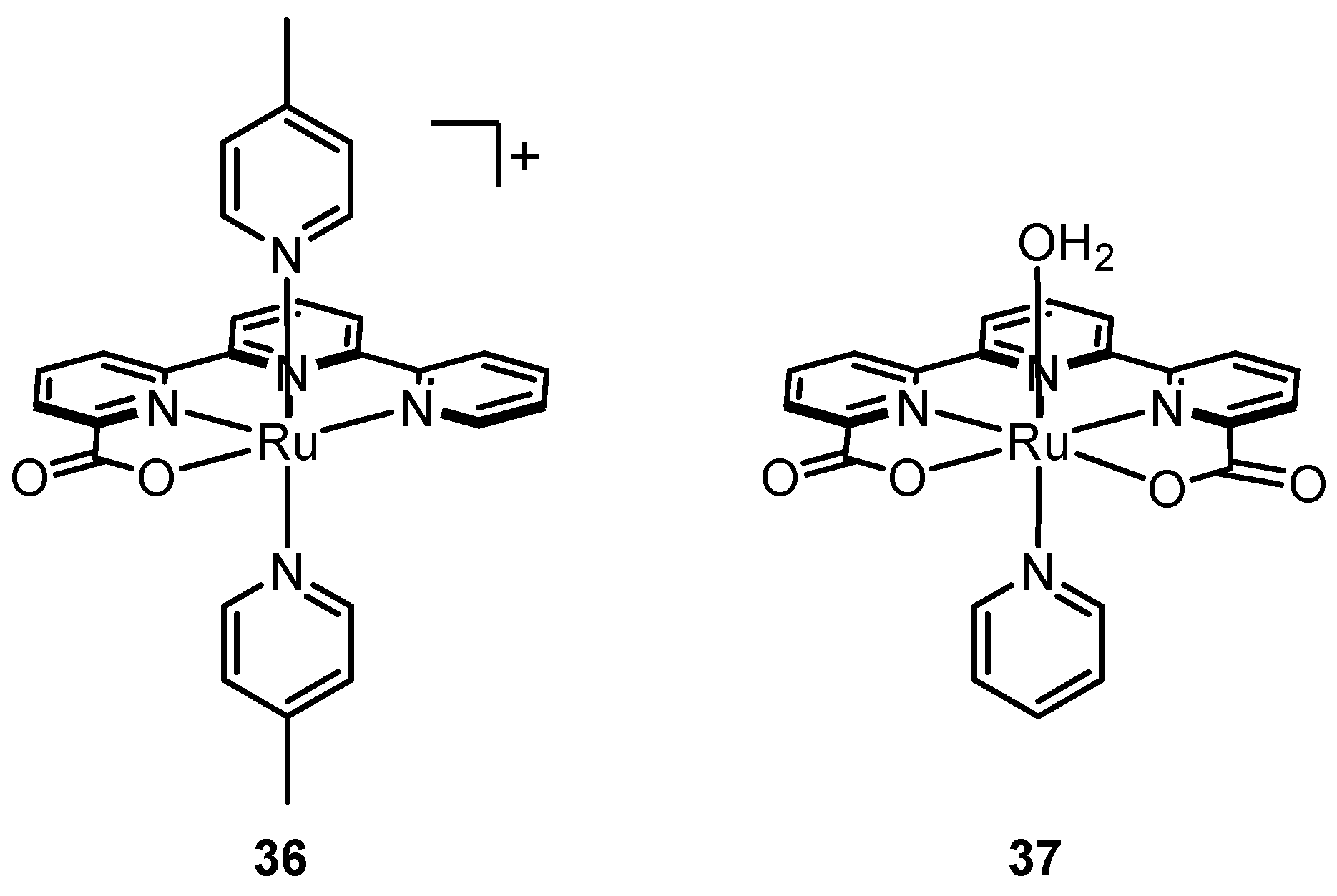
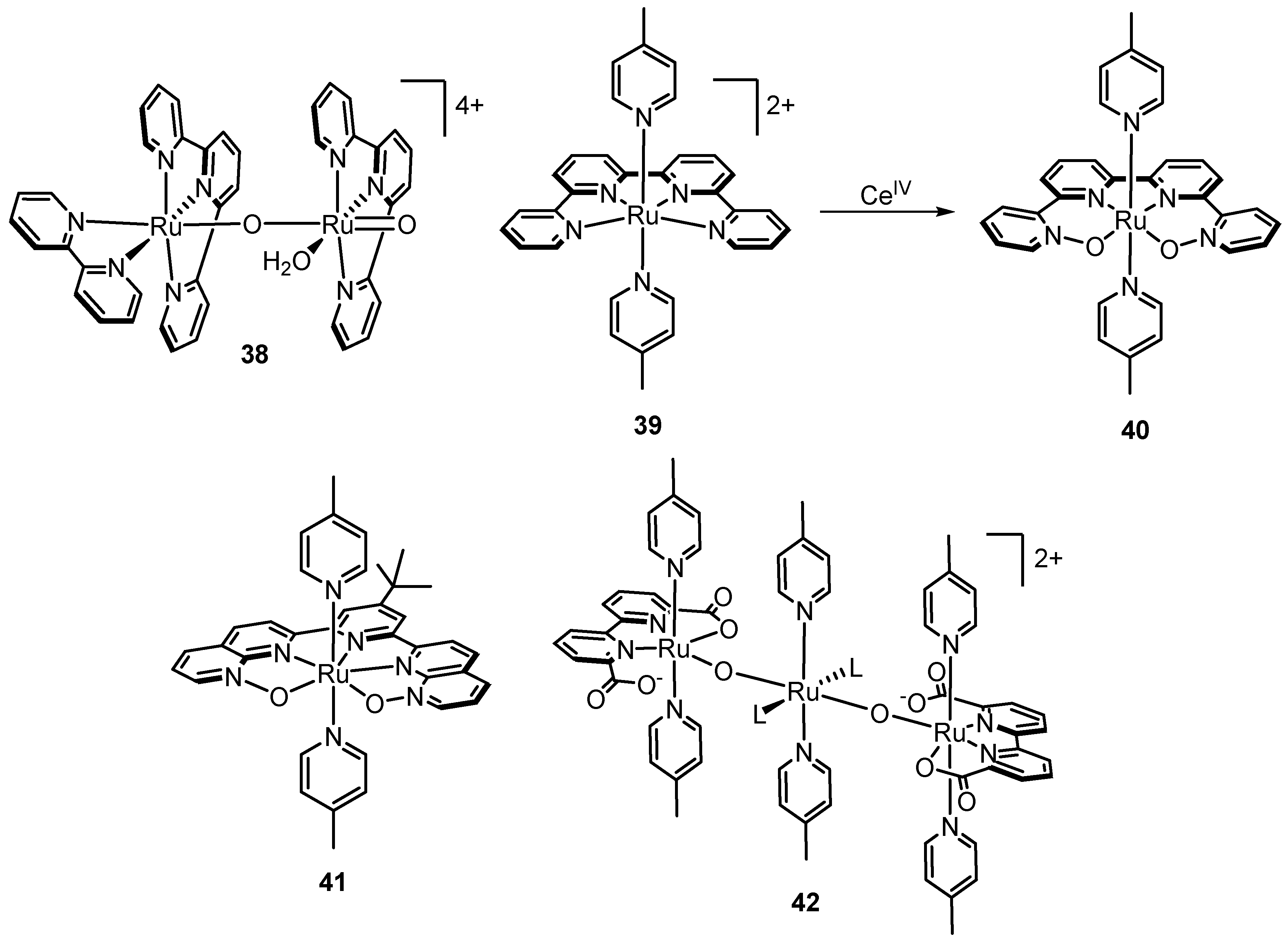
2.12. Catalyst Modification Pathways
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hammond, A.L. Solar Energy: The Largest Resource. Science 1972, 177, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.K. Prospects for hydrogen as an energy resource. Nature 1974, 249, 724–726. [Google Scholar] [CrossRef]
- Balzani, V.; Moggi, L.; Manfrin, M.F.; Bolletta, F.; Gleria, M. Solar Energy Conversion by Water Photodissociation. Science 1975, 189, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N.; Balzani, V. The Hydrogen Issue. ChemSusChem 2011, 4, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Bockris, J.O.M. The hydrogen economy: Its history. Int. J. Hydrog. Energy 2013, 38, 2579–2588. [Google Scholar] [CrossRef]
- Heidt, L.J.; McMillan, A.F. Conversion of Sunlight into Chemical Energy Available in Storage for Man’s Use. Science 1953, 117, 75–76. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Efstathiou, A.M. Hydrogen Production Technologies: Current State and Future Developments. Energy 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- da Silva Veras, T.; Mozer, T.S.; da Costa Rubim Messeder dos Santos, D.; da Silva César, A. Hydrogen: Trends, production and characterization of the main process worldwide. Int. J. Hydrog. Energy 2017, 42, 2018–2033. [Google Scholar] [CrossRef]
- Appel, A.M.; Helm, M.L. Determining the Overpotential for a Molecular Electrocatalyst. ACS Catal. 2014, 4, 630–633. [Google Scholar] [CrossRef]
- Betley, T.A.; Wu, Q.; Van Voorhis, T.; Nocera, D.G. Electronic Design Criteria for O−O Bond Formation via Metal−Oxo Complexes. Inorg. Chem. 2008, 47, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Najafpour, M.M.; Renger, G.; Hołyńska, M.; Moghaddam, A.N.; Aro, E.M.; Carpentier, R.; Nishihara, H.; Eaton-Rye, J.J.; Shen, J.R.; Allakhverdiev, S.I. Manganese Compounds as Water-Oxidizing Catalysts: From the Natural Water-Oxidizing Complex to Nanosized Manganese Oxide Structures. Chem. Rev. 2016, 116, 2886–2936. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.M.; Huang, D.L.; Crabtree, R.H.; Brudvig, G.W. Iridium-based complexes for water oxidation. Dalton Trans. 2015, 44, 12452–12472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enthaler, S.; Junge, K.; Beller, M. Sustainable metal catalysis with iron: From rust to a rising star? Angew. Chem. Int. Ed. 2008, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Ellis, W.C.; McDaniel, N.D.; Bernhard, S.; Collins, T.J. Fast Water Oxidation Using Iron. J. Am. Chem. Soc. 2010, 132, 10990–10991. [Google Scholar] [CrossRef] [PubMed]
- Fillol, J.L.; Codolà, Z.; Garcia-Bosch, I.; Gàmez, L.; Pla, J.J.; Costas, M. Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat. Chem. 2011, 3, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.M.; Waidmann, C.R.; Scheuermann, M.L.; Nesvet, J.C.; Goldberg, K.; Mayer, J.M. Developing Molecular Copper Complexes for Water Oxidation. In Molecular Water Oxidation Catalysis: A Key Topic for New Sustainable Energy Conversion Schemes; Llobet, A., Ed.; John Wiley & Sons: Chichester, West Sussex, UK, 2014; pp. 187–207. ISBN 978-1-118-41337-1. [Google Scholar]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef]
- Yin, Q.; Tan, J.M.; Besson, C.; Geletii, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A Fast Soluble Carbon-Free Molecular Water Oxidation Catalyst Based on Abundant Metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef]
- Rigsby, M.L.; Mandal, S.; Nam, W.; Spencer, L.C.; Llobet, A.; Stahl, S.S. Cobalt analogs of Ru-based water oxidation catalysts: Overcoming thermodynamic instability and kinetic lability to achieve electrocatalytic O2 evolution. Chem. Sci. 2012, 3, 3058–3062. [Google Scholar] [CrossRef]
- Chen, B.T.; Morlanés, N.; Adogla, E.; Takanabe, K.; Rodionov, V.O. An Efficient and Stable Hydrophobic Molecular Cobalt Catalyst for Water Electro-oxidation at Neutral pH. ACS Catal. 2016, 6, 4647–4652. [Google Scholar] [CrossRef]
- Dogutan, D.K.; McGuire, R.; Nocera, D.G. Electocatalytic Water Oxidation by Cobalt(III) Hangman β-Octafluoro Corroles. J. Am. Chem. Soc. 2011, 133, 9178–9180. [Google Scholar] [CrossRef] [PubMed]
- Gortsema, F.P.; Cobble, J.W. The Induced Oxidation of Bound Water by Ruthenium(IV). J. Am. Chem. Soc. 1959, 81, 5516. [Google Scholar] [CrossRef]
- Moyer, B.; Meyer, T. Oxobis(2,2′-bipyridine)pyridineruthenium(IV) Ion, [(bpy)2(py)Ru=O]2+. J. Am. Chem. Soc. 1978, 2, 3601–3603. [Google Scholar] [CrossRef]
- Moyer, B.A.; Meyer, T.J. Properties of the oxo/aqua system (bpy)2(py)RuO2+/(bpy)2(py)Ru(OH2)2+. Inorg. Chem. 1981, 20, 436–444. [Google Scholar] [CrossRef]
- Binstead, R.A.; Moyer, B.A.; Samuels, G.J.; Meyer, T.J. Proton-coupled electron transfer between [Ru(bpy)2(py)OH2]2+ and [Ru(bpy)2(py)O]2+. A solvent isotope effect (kH2O/kD2O) of 16.1. J. Am. Chem. Soc. 1981, 103, 2897–2899. [Google Scholar] [CrossRef]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Miller, R.; Brandt, W.; Puke, M. Metal-Amine Coördination Compounds. V. The Ruthenium-2,2′-Bipyridine System. J. Am. Chem. Soc. 1955, 77, 3178–3180. [Google Scholar] [CrossRef]
- Dwyer, F.P.; Goodwin, H.A.; Gyarfas, E.C. Mono- and Bis-(2,2’-bipyridine) and -(1,10-phenanthroline) Chelates of Ruthenium and Osmium. II. Bischelates of Bivalent and Tervalent Ruthenium. Aust. J. Chem. 1963, 16, 544–548. [Google Scholar] [CrossRef]
- Dwyer, F.P.; Gyarfas, E.C. Persistence of Optical Activity in an Oxidation-Reduction Reaction. Nature 1949, 163, 918. [Google Scholar] [CrossRef]
- Keene, F.R.; Salmon, D.J.; Meyer, T.J. Oxidation of primary amines bound to bis(2,2′-bipyridine)ruthenium(II). J. Am. Chem. Soc. 1976, 98, 1884–1889. [Google Scholar] [CrossRef]
- Gagliardi, C.J.; Vannucci, A.K.; Concepcion, J.J.; Chen, Z.C.; Meyer, T.J. The Role of Proton Coupled Electron Transfer in Water Oxidation. Energy Environ. Sci. 2012, 5, 7704–7717. [Google Scholar] [CrossRef]
- Meyer, T.J.; Huynh, M.H.V. The Remarkable Reactivity of High Oxidation State Ruthenium and Osmium Polypyridyl Complexes. Inorg. Chem. 2003, 42, 8140–8160. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Tsuge, K.; Tanaka, K. Electrochemical Oxidation of Water to Dioxygen Catalyzed by the Oxidized Form of the Bis(ruthenium – hydroxo) Complex in H2O. Angew. Chem. Int. Ed. 2000, 39, 1479–1482. [Google Scholar] [CrossRef]
- Honda, K.; Frank, A.J. Catalytic oxidation of water by an oxo-bridged ruthenium complex in homogeneous media and in colloidal SiO2 suspensions. J. Chem. Soc. Chem. Commun. 1984, 1635–1636. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Eggleston, D.S.; Murphy, W.R.; Geselowitz, D.A.; Gersten, S.W.; Hodgson, D.J.; Meyer, T.J. Structure and redox properties of the water-oxidation catalyst [(bpy)2(OH2)RuORu(OH2)(bpy)2]4+. J. Am. Chem. Soc. 1985, 107, 3855–3864. [Google Scholar] [CrossRef]
- Vining, W.J.; Meyer, T.J. Redox properties of the water oxidation catalyst (bpy)2(H2O)RuORu(H2O)(bpy)24+ in thin polymeric films. Electrocatalytic oxidation of Cl− to Cl2. Inorg. Chem. 1986, 25, 2023–2033. [Google Scholar] [CrossRef]
- Ramaraj, R.; Kira, A.; Kaneko, M. Electrochemistry and stability studies of oxo-bridged dinuclear ruthenium(III) complexes for water oxidation. J. Chem. Soc. Faraday Trans. 1986, 82, 3515–3524. [Google Scholar] [CrossRef]
- Doppelt, P.; Meyer, T.J. Synthesis and redox properties of the unsymmetrical, oxo-bridged dimer [(bpy)2(py)RuIIIORuIII(H2O)(bpy)2]4+. Inorg. Chem. 1987, 26, 2027–2034. [Google Scholar] [CrossRef]
- Rotzinger, F.P.; Munavalli, S.; Comte, P.; Hurst, J.K.; Graetzel, M.; Pern, F.J.; Frank, A.J. A molecular water-oxidation catalyst derived from ruthenium diaqua bis(2,2′-bipyridyl-5,5′-dicarboxylic acid). J. Am. Chem. Soc. 1987, 109, 6619–6626. [Google Scholar] [CrossRef]
- Ramaraj, R.; Kira, A.; Kaneko, M. Oxygen Evolution by Water Oxidation with Polynuclear Ruthenium Complexes. J. Chem. Soc. Chem. Commun. 1987, 83, 1539–1551. [Google Scholar] [CrossRef]
- Raven, S.J.; Meyer, T.J. Reactivity of the Oxo-Bridged Ion [(bpy)2(O)RuIVORuV(O)(bpy)2]3+. J. Am. Chem. Soc. 1988, 27, 4478–4483. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Rotzinger, F.P.; Comte, P.; Grätzel, M. Spontaneous Oxidation of Water to Oxygen by the Mixed-valence µ-oxo Ruthenium Dimer L2(H2O)RuIII-O-RuIV(OH)L2 (L = 2,2′-bipyridyl-5,5′-dicarboxylic acid). J. Chem. Soc. Chem. Commun. 1988, 872–874. [Google Scholar] [CrossRef]
- Comte, P.; Nazeeruddin, M.K.; Rotzinger, F.P.; Frank, A.J.; Grätzel, M. Artificial analogues of the oxygen-evolving complex in photosynthesis: The oxo-bridged ruthenium dimer L2(H2O)RuIII-O-RuIII(H2O)L2, L = 2,2′-bipyridyl-4,4′-dicarboxylate. J. Mol. Catal. 1989, 52, 63–84. [Google Scholar] [CrossRef]
- Zhou, J.; Xi, W.; Hurst, J. Structure and Water-Oxidizing Capabilities of Dimeric Ruthenium EDTA Complex Ions. Inorg. Chem. 1990, 1, 160–166. [Google Scholar] [CrossRef]
- Geselowitz, D.; Meyer, T.J. Water oxidation by [(bpy)2(O)RuVORuV(O)(bpy)2]4+. An oxygen-labeling study. Inorg. Chem. 1990, 29, 3894–3896. [Google Scholar] [CrossRef]
- Hurst, J.K.; Zhou, J.; Lei, Y. Pathways for water oxidation catalyzed by the [(bpy)2Ru(OH2)]2O4+ Ion. Inorg. Chem. 1992, 31, 1010–1017. [Google Scholar] [CrossRef]
- Petach, H.H.; Elliott, C.M. Characterization and Catalytic Activity of Covalently Linked Bipyridyl Ruthenium Oxo Dimers. J. Electrochem. Soc. 1992, 139, 2217–2221. [Google Scholar] [CrossRef]
- Lei, Y.; Hurst, J.K. Structural Investigations of the Catalytic Mechanisms of Water Oxidation by the [(bpy)2Ru(OH2)]2O4+ Ion. Inorg. Chem. 1994, 33, 4460–4467. [Google Scholar] [CrossRef]
- Lei, Y.; Hurst, J.K. Dynamical investigations of the catalytic mechanisms of water oxidation by the [(bpy)2Ru(OH2)]2O4+ ion. Inorg. Chim. Acta 1994, 226, 179–185. [Google Scholar] [CrossRef]
- Lai, Y.K.; Wong, K.Y. Electrochemistry of oxo-bridged ruthenium dimers with 4,4′-dichloro- and 5,5′-dichloro-2,2′-bipyridine and their catalytic properties towards water oxidation. J. Electroanal. Chem. 1995, 380, 193–200. [Google Scholar] [CrossRef]
- Chronister, C.W.; Binstead, R.A.; Ni, J.; Meyer, T.J. Mechanism of Water Oxidation Catalyzed by the μ-Oxo Dimer [(bpy)2(OH2)RuIIIORuIII(OH2)(bpy)2]4+. Inorg. Chem. 1997, 36, 3814–3815. [Google Scholar] [CrossRef]
- Yagi, M.; Takahashi, Y.; Oginoa, I.; Kanekoa, M. Dioxygen evolution induced by visible light atlayered sensitizer/water oxidationcatalyst. J. Chem. Soc. Faraday Trans. 1997, 93, 3125–3127. [Google Scholar] [CrossRef]
- Lebeau, E.L.; Adeyemi, S.A.; Meyer, T.J. Water Oxidation by [(tpy)(H2O)2RuIIIORuIII(H2O)2(tpy)]4+. Inorg. Chem. 1998, 37, 6476–6484. [Google Scholar] [CrossRef] [PubMed]
- Nagoshi, K.; Yamashita, S.; Yagi, M. Catalytic activity of [(bpy)2(H2O)Ru-O-Ru(H2O)(bpy)2]4+ for four-electron water oxidation. J. Mol. Catal. A Chem. 1999, 144, 71–76. [Google Scholar] [CrossRef]
- Wada, T.; Tsuge, K.; Tanaka, K. Syntheses and Redox Properties of Bis(hydroxoruthenium) Complexes with Quinone and Bipyridine Ligands. Water-Oxidation Catalysis. Inorg. Chem. 2001, 40, 329–337. [Google Scholar] [CrossRef]
- Sens, C.; Romero, I.; Rodríguez, M.; Llobet, A.; Parella, T.; Benet-Buchholz, J. A New Ru Complex Capable of Catalytically Oxidizing Water to Molecular Dioxygen. J. Am. Chem. Soc. 2004, 126, 7798–7799. [Google Scholar] [CrossRef]
- Romain, S.; Bozoglian, F.; Sala, X.; Llobet, A. Oxygen−Oxygen Bond Formation by the Ru-Hbpp Water Oxidation Catalyst Occurs Solely via an Intramolecular Reaction Pathway. J. Am. Chem. Soc. 2009, 131, 2768–2769. [Google Scholar] [CrossRef]
- Zong, R.; Thummel, R.P. A New Family of Ru Complexes for Water Oxidation. J. Am. Chem. Soc. 2005, 127, 12802–12803. [Google Scholar] [CrossRef]
- Goswami, S.; Chakravarty, A.R.; Chaktravorty, A. The RuII(OH2)–RuIV(O) couple in a ruthenium complex of 2-(phenylazo)pyridine: Homogeneous catalysis of the oxidation of water to dioxygen. J. Chem. Soc. Chem. Commun. 1982, 1288–1289. [Google Scholar] [CrossRef]
- Takeuchi, K.J.; Samuels, G.J.; Gersten, S.W.; Gilbert, J.A.; Meyer, T.J. Multiple oxidation states of ruthenium and osmium based on dioxo/diaqua couples. Inorg. Chem. 1983, 22, 1407–1409. [Google Scholar] [CrossRef]
- Collin, J.P.; Sauvage, J.P. Synthesis and study of mononuclear ruthenium(II) complexes of sterically hindering diimine chelates. Implications for the catalytic oxidation of water to molecular oxygen. Inorg. Chem. 1986, 25, 135–141. [Google Scholar] [CrossRef]
- Ramaraj, R.; Kira, A.; Kaneko, M. Monoculear ruthenium–ammine complexes as catalysts for water oxidation. J. Chem. Soc. Chem. Commun. 1987, 227–228. [Google Scholar] [CrossRef]
- Dobson, J.C.; Meyer, T.J. Redox properties and ligand loss chemistry in aqua/hydroxo/oxo complexes derived from cis- and trans-[(bpy)2RuII(OH2)2]2+. Inorg. Chem. 1988, 27, 3283–3291. [Google Scholar] [CrossRef]
- Khan, M.M.; Ramachandraiah, G.; Mehta, S.H.; Abdi, S.H.R.; Kumar, S. Mononuclear bis(dimethylglyoximato)ruthenium(III) complexes with different appended axial groups: Highly efficient catalysts for water oxidation. J. Mol. Catal. 1990, 58, 199–203. [Google Scholar] [CrossRef]
- Pramanik, N.C.; Bhattacharya, S. Chemical oxidation of water to dioxygen. Homogeneous catalysis by a ruthenium aquo-complex. Transit. Met. Chem. 1997, 22, 524–526. [Google Scholar] [CrossRef]
- Pramanik, N.C.; Pramanik, K.; Ghosh, P.; Bhattacharya, S. Chemistry of [Ru(tpy)(pap)(L′)n+ (tpy = 2,2′,6′,2′′-terpyridine; pap = 2-(phenylazo)pyridine; L′ = Cl−, H2O, CH3CN, 4-picoline, N3−; n = 1, 2). X-ray crystal structure of [Ru(tpy)(pap)(CH3CN)](CIO4)2 and catalytic oxidation of water to dioxygen by [Ru(tpy) (pap) (H2O)]2+. Polyhedron 1998, 17, 1525–1534. [Google Scholar] [CrossRef]
- Tseng, H.W.; Zong, R.; Muckerman, J.T.; Thummel, R.P. Mononuclear Ruthenium(II) Complexes That Catalyze Water Oxidation. Inorg. Chem. 2008, 47, 11763–11773. [Google Scholar] [CrossRef]
- Masaoka, S.; Sakai, K. Clear Evidence Showing the Robustness of a Highly Active Oxygen-evolving Mononuclear Ruthenium Complex with an Aqua Ligand. Chem. Lett. 2009, 38, 182–183. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Koivisto, B.D.; Henderson, M.A.; Berlinguette, C.P. Insight into Water Oxidation by Mononuclear Polypyridyl Ru Catalysts. Inorg. Chem. 2010, 49, 2202–2209. [Google Scholar] [CrossRef]
- Kaveevivitchai, N.; Zong, R.; Tseng, H.; Chitta, R.; Thummel, R.P. Further Observations on Water Oxidation Catalyzed by Mononuclear Ru(II) Complexes. Inorg. Chem. 2012, 51, 2930–2939. [Google Scholar] [CrossRef]
- Yan, L.; Zong, R.; Pushkar, Y. Unexpected Ligand Lability in Condition of Water Oxidation Catalysis. J. Catal. 2015, 330, 255–260. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Templeton, J.L.; Meyer, T.J. One Site is Enough. Catalytic Water Oxidation by [Ru(tpy)(bpm)(OH2)]2+ and [Ru(tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc. 2008, 130, 16462–16463. [Google Scholar] [CrossRef] [PubMed]
- Pushkar, Y.; Moonshiram, D.; Purohit, V.; Yan, L.; Alperovich, I. Spectroscopic Analysis of Catalytic Water Oxidation by [RuII(bpy)(tpy)H2O]2+ Suggests That RuV═O Is Not a Rate-Limiting Intermediate. J. Am. Chem. Soc. 2014, 136, 11938–11945. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, D.J.; Ganesamoorthy, C.; Henderson, M.A.; Koivisto, B.D.; Osthoff, H.D.; Berlinguette, C.P. Electronic Modification of the [RuII(tpy)(bpy)(OH2)]2+ Scaffold: Effects on Catalytic Water Oxidation. J. Am. Chem. Soc. 2010, 132, 16094–16106. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Tajima, S.; Komi, M.; Yamazaki, H. Highly active and tunable catalysts for O2 evolution from water based on mononuclear ruthenium(II) monoaquo complexes. Dalton Trans. 2011, 40, 3802–3804. [Google Scholar] [CrossRef] [PubMed]
- Durham, B.; Wilson, S.R.; Hodgson, D.J.; Meyer, T.J. Cis-trans photoisomerization in Ru(bpy)2(OH2)22+. Crystal structure of trans-[Ru(bpy)2(OH2)(OH)](ClO4)2. J. Am. Chem. Soc. 1980, 102, 600–607. [Google Scholar] [CrossRef]
- Walsh, J.L.; Durham, B. Trans isomers of ruthenium(II) complexes containing two bipyridine ligands. Inorg. Chem. 1982, 21, 329–332. [Google Scholar] [CrossRef]
- Coe, B.J.; Meyer, T.J.; White, P.S. Control of axial ligand substitution in trans-bis(2,2’-bipyridine)ruthenium(II) complexes. Crystal and molecular structure of trans-(4-ethylpyridine)(dimethyl sulfoxide)bis(2,2′-bipyridine)ruthenium(II) hexafluorophosphate, trans-[Ru(bpy)2(4-Etpy)(DMSO)](PF6)2. Inorg. Chem. 1993, 32, 4012–4020. [Google Scholar] [CrossRef]
- Concepción, J.; Loeb, B.; Simón-Manso, Y.; Zuloaga, F. Influence of L-type ligands on the relative stability and interconversion of cis–trans-[Ru(phen)2L2]n+ type complexes. A theoretical study. Polyhedron 2000, 19, 2297–2302. [Google Scholar] [CrossRef]
- Chan, C.W.; Lai, T.F.; Che, C.M. Electrochemical oxidation of diaquaruthenium(II) complexes of quaterpyridines and crystal structure of [RuL1(PPh3)2][ClO4]2(L1= 3″,5,5′,5‴-tetramethyl-2,2′:6′,2″: 6″,2‴-quaterpyridine). J. Chem. Soc. Dalton. Trans. 1994, 895–899. [Google Scholar] [CrossRef]
- Zong, R.; Thummel, R.P. 2,9-Di-(2′-pyridyl)-1,10-phenanthroline: A Tetradentate Ligand for Ru(II). J. Am. Chem. Soc. 2004, 126, 10800–10801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zong, R.; Tseng, H.W.; Thummel, R.P. Ru(II) Complexes of Tetradentate Ligands Related to 2,9-Di(pyrid-2′-yl)-1,10-phenanthroline. Inorg. Chem. 2008, 47, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Tseng, H.W.; Zong, R.; Wang, D.; Thummel, R. Preparation and Study of a Family of Dinuclear Ru(II) Complexes That Catalyze the Decomposition of Water. Inorg. Chem. 2008, 47, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Lomoth, R.; Huang, P.; Zheng, J.T.; Sun, L.C.; Hammarström, L.; Åkermark, B.; Styring, S. Synthesis and Characterization of a Dinuclear Manganese(III,III) Complex with Three Phenolate Ligands. Eur. J. Inorg. Chem. 2002, 2965–2974. [Google Scholar] [CrossRef]
- Xu, Y.; Åkermark, T.; Gyollai, V.; Zou, D.; Eriksson, L.; Duan, L.; Zhang, R.; Åkermark, B.; Sun, L. A New Dinuclear Ruthenium Complex as an Efficient Water Oxidation Catalyst. Inorg. Chem. 2009, 48, 2717–2719. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fischer, A.; Duan, L.; Tong, L.; Gabrielsson, E.; Åkermark, B.; Sun, L. Chemical and Light-Driven Oxidation of Water Catalyzed by an Efficient Dinuclear Ruthenium Complex. Angew. Chem. Int. Ed. 2010, 49, 8934–8937. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Fischer, A.; Xu, Y.; Sun, L. Isolated Seven-Coordinate Ru(IV) Dimer Complex with [HOHOH]– Bridging Ligand as an Intermediate for Catalytic Water Oxidation. J. Am. Chem. Soc. 2009, 131, 10397–10399. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Kowalczyk, M.; Badiei, Y.M.; Polyansky, D.E.; Concepcion, J.J.; Zong, R.; Thummel, R.P.; Fujita, E. New Water Oxidation Chemistry of a Seven-Coordinate Ruthenium Complex with a Tetradentate Polypyridyl Ligand. Inorg. Chem. 2014, 53, 6904–6913. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Richmond, C.J.; Matheu, R.; Poater, A.; Falivene, L.; Benet-Buchholz, J.; Sala, X.; Cavallo, L.; Llobet, A. Supramolecular Water Oxidation with Ru–bda-Based Catalysts. Chem. Eur. J. 2014, 20, 17282–17286. [Google Scholar] [CrossRef]
- Duan, L.; Moyses, C.; Ahlquist, M.S.G.; Sun, L. Highly efficient and robust molecular ruthenium catalysts for water oxidation. Proc. Natl. Acad. Sci. USA 2012, 109, 15584–15588. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Concepcion, J.J.; Binstead, R.A.; Rudd, J.A.; Vannucci, A.K.; Dares, C.J.; Coggins, M.K.; Meyer, T.J. Base-enhanced catalytic water oxidation by a carboxylate–bipyridine Ru(II) complex. Proc. Natl. Acad. Sci. USA 2015, 112, 4935–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, Q.; Huang, P.; Fan, T.; Wang, Y.; Duan, L.; Wang, L.; Li, F.; Rinkevicius, Z.; Mamedov, F.; Ahlquist, M.S.G.; Styring, S.; Sun, L. Rearranging from 6- to 7-coordination initiates the catalytic activity: An EPR study on a Ru-bda water oxidation catalyst. Coord. Chem. Rev. 2017, 346, 206–215. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Zhong, D.K.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Mechanism of Water Oxidation by [Ru(bda)(L)2]: The Return of the “Blue Dimer”. Chem. Commun. 2015, 51, 4105–4108. [Google Scholar] [CrossRef]
- Lebedev, D.; Pineda-Galvan, Y.; Tokimaru, Y.; Federov, A.; Kaeffer, N.; Copéret, C.; Pushkar, Y. The Key RuV=O Intermediate of Site-Isolated Mononuclear Water Oxidation Catalyst Detected by in situ X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2018, 140, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Concepcion, J.J.; Hu, X.; Yang, W.; Hoertz, P.G.; Meyer, T.J. Concerted O atom–proton transfer in the O—O bond forming step in water oxidation. Proc. Natl. Acad. Sci. USA 2010, 107, 7225–7229. [Google Scholar] [CrossRef] [PubMed]
- Grotjahn, D.B.; Lev, D.A. A General Bifunctional Catalyst for the Anti-Markovnikov Hydration of Terminal Alkynes to Aldehydes Gives Enzyme-Like Rate and Selectivity Enhancements. J. Am. Chem. Soc. 2004, 126, 12232. [Google Scholar] [CrossRef] [PubMed]
- Grotjahn, D.B.; Kragulj, E.J.; Zeinalipour-Yazdi, C.D.; Miranda-Soto, V.; Lev, D.A.; Cooksy, A.L. Finding the Proton in a Key Intermediate of anti-Markovnikov Alkyne Hydration by a Bifunctional Catalyst. J. Am. Chem. Soc. 2008, 130, 10860. [Google Scholar] [CrossRef] [PubMed]
- Arita, A.J.; Cantada, J.; Grotjahn, D.B.; Cooksy, A.L. Computational Study of the Extensive Role of Heterocyclic Ligands in Acetylene Hydration by a Bifunctional Organometallic Catalyst. Organometallics 2013, 32, 6867–6870. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Larsen, C.R.; Gustafson, J.L.; Nair, R.; Sharma, A. Extensive Isomerization of Alkenes Using a Bifunctional Catalyst: An Alkene Zipper. J. Am. Chem. Soc. 2007, 129, 9592–9593. [Google Scholar] [CrossRef]
- Matheu, R.; Ertem, M.Z.; Benet-Buchholz, J.; Coronado, E.; Batista, V.S.; Sala, X.; Llobet, A. Intramolecular Proton Transfer Boosts Water Oxidation Catalyzed by a Ru Complex. J. Am. Chem. Soc. 2015, 137, 10786–10795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheu, R.; Neudeck, S.; Meyer, F.; Sala, X.; Llobet, A. Foot of the Wave Analysis for Mechanistic Elucidation and Benchmarking Applications in Molecular Water Oxidation Catalysis. ChemSusChem 2016, 9, 3361–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.; Duan, L.; Huang, P.; Chen, H.; Daniel, Q.; Ahlquist, M.S.G.; Sun, L. The Ru-tpc Water Oxidation Catalyst and Beyond: Water Nucleophilic Attack Pathway versus Radical Coupling Pathway. ACS Catal. 2017, 7, 2956–2966. [Google Scholar] [CrossRef]
- Matheu, R.; Ertem, M.Z.; Gimbert-Suriñach, C.; Benet-Buchholz, J.; Sala, X.; Llobet, A. Hydrogen Bonding Rescues Overpotential in Seven-Coordinated Ru Water Oxidation Catalysts. ACS Catal. 2017, 7, 6525–6532. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, D.W.; Xie, Y.; Szalda, D.J.; Concepcion, J.J. Lability and Basicity of Bipyridine-Carboxylate-Phosphonate Ligand Accelerate Single-Site Water Oxidation by Ruthenium-Based Molecular Catalysts. J. Am. Chem. Soc. 2017, 139, 15347–15355. [Google Scholar] [CrossRef] [PubMed]
- Wrzolek, P.; Wahl, S.; Schwalbe, M. Electrocatalytic investigation on the water oxidation ability of a hangman complex based on the [Ru(tpy)(bpy)(OH2)]2+ motif. Catal. Today 2017, 290, 28–32. [Google Scholar] [CrossRef]
- Wang, Y.; Rinkevicus, Z.; Ahlquist, M.S.G. Formation of N-oxide in the third oxidation of [RuII(tpy)(L)(OH2)]2+. Chem. Commun. 2017, 53, 5622–5624. [Google Scholar] [CrossRef] [PubMed]
- López, I.; Ertem, M.Z.; Maji, S.; Benet-Buchholz, J.; Keidel, A.; Kuhlman, U.; Hildebrandt, P.; Cramer, C.J.; Batista, V.S.; Llobet, A. A Self-Improved Water-Oxidation Catalyst: Is One Site Really Enough? Angew. Chem. Int. Ed. 2014, 53, 205–209. [Google Scholar] [CrossRef]
- Liu, Y.; Ng, S.; Yiu, S.; Lam, W.W.Y.; Wei, X.; Lau, K.; Lau, T. Catalytic Water Oxidation by Ruthenium(II) Quaterpyridine (qpy) Complexes: Evidence for Ruthenium(III) qpy-N,N’’’-dioxide as the Real Catalysts. Angew. Chem. Int. Ed. 2014, 53, 14468–14471. [Google Scholar] [CrossRef]
- Moonshiram, D.; Pineda-Galvan, Y.; Erman, D.; Palenik, M.; Zong, R.; Thummel, R.; Pushkar, Y. Uncovering the Role of Oxygen Atom Transfer in Ru-Based Catalytic Water Oxidation. J. Am. Chem. Soc. 2016, 138, 15605–15616. [Google Scholar] [CrossRef]
- Kagalwala, H.N.; Tong, L.; Zong, R.; Kohler, L.; Ahlquist, M.S.G.; Fan, T.; Gagnon, K.J.; Thummel, R.P. Evidence for Oxidative Decay of a Ru-Bound Ligand during Catalyzed Water Oxidation. ACS Catal. 2017, 7, 2607–2615. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.; Yiu, S.; Wong, C.; Lau, T. Intermediates in the Oxidative Degradation of a Ruthenium-Bound 2,2′-Bipyridyl–Phenoxy Ligand during Catalytic Water Oxidation. ChemCatChem 2018, 10, 501–504. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamdar, J.M.; Grotjahn, D.B. An Overview of Significant Achievements in Ruthenium-Based Molecular Water Oxidation Catalysis. Molecules 2019, 24, 494. https://doi.org/10.3390/molecules24030494
Kamdar JM, Grotjahn DB. An Overview of Significant Achievements in Ruthenium-Based Molecular Water Oxidation Catalysis. Molecules. 2019; 24(3):494. https://doi.org/10.3390/molecules24030494
Chicago/Turabian StyleKamdar, Jayneil M., and Douglas B. Grotjahn. 2019. "An Overview of Significant Achievements in Ruthenium-Based Molecular Water Oxidation Catalysis" Molecules 24, no. 3: 494. https://doi.org/10.3390/molecules24030494