The Cardiotoxicity Induced by Arsenic Trioxide is Alleviated by Salvianolic Acid A via Maintaining Calcium Homeostasis and Inhibiting Endoplasmic Reticulum Stress

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
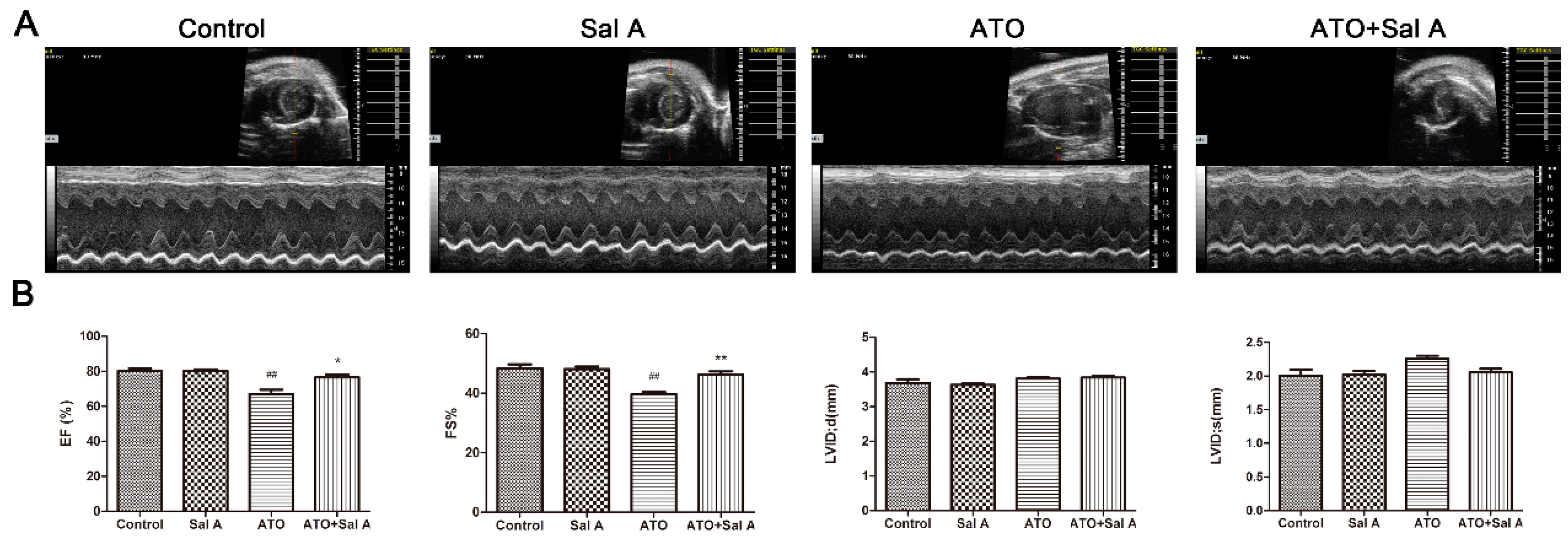
2.1. Effects of Sal A on Cardiac Function
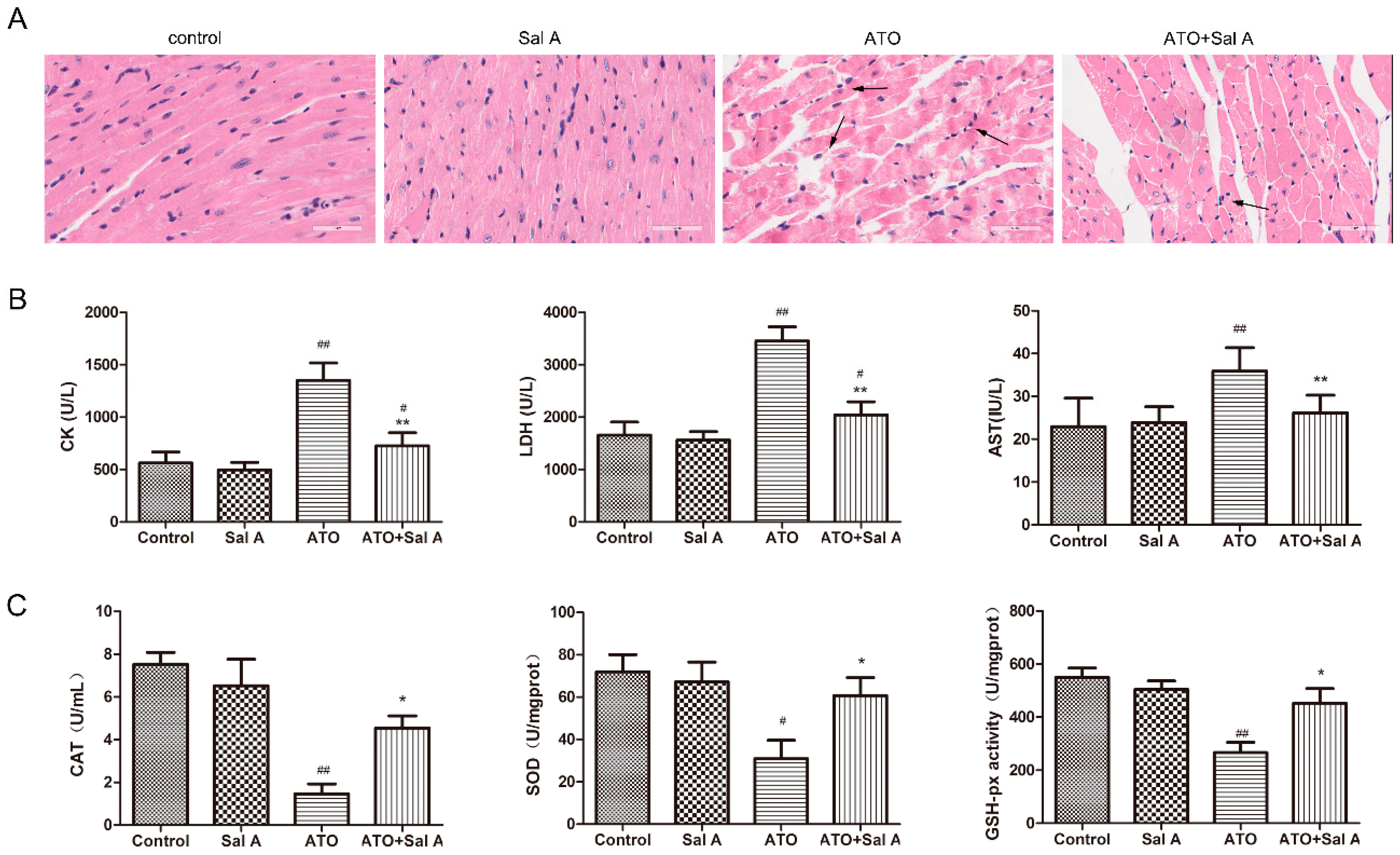
2.2. Sal A Prevents ATO-Induced Myocardial Damage
2.3. Sal A Improves Antioxidant Enzyme Activities
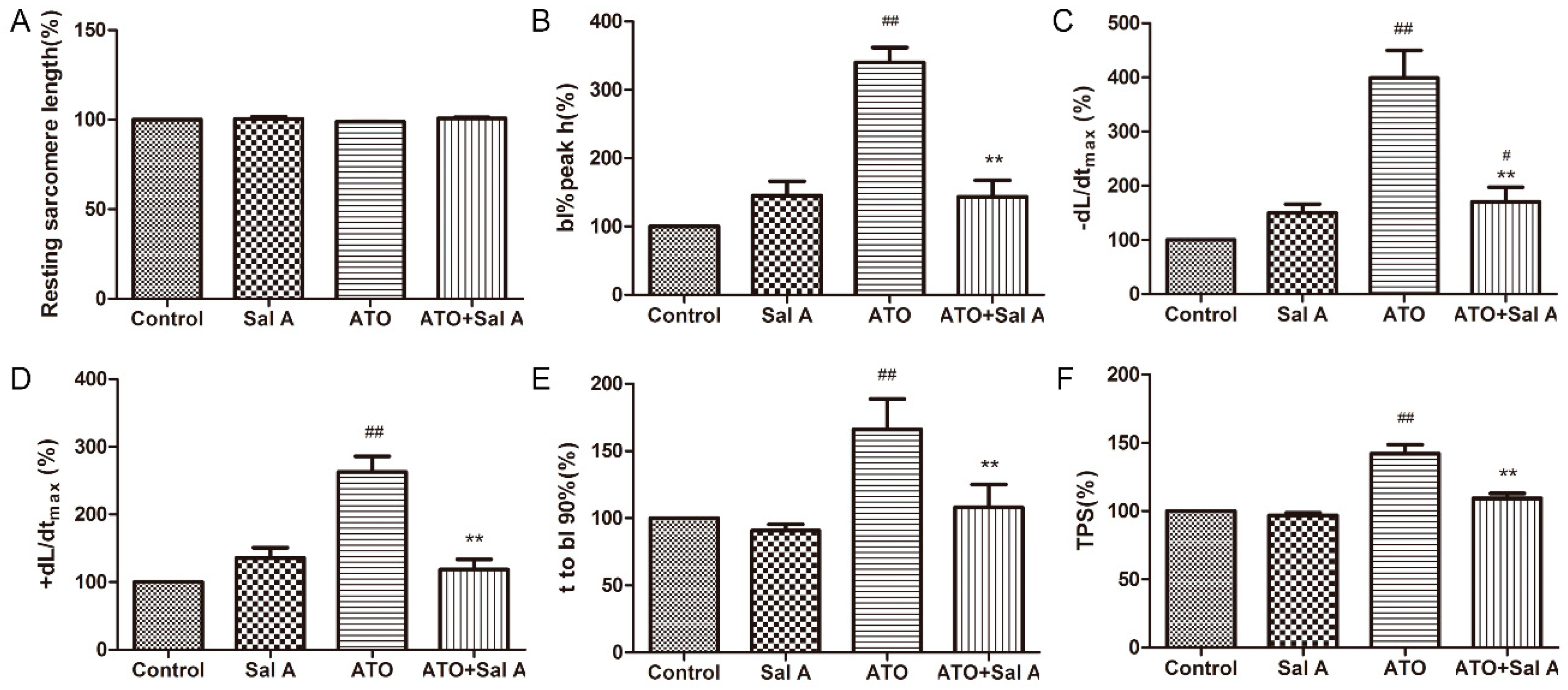
2.4. Effects of Sal A on Cardiomyocyte Contractile Function in ARVMs after ATO Treatment
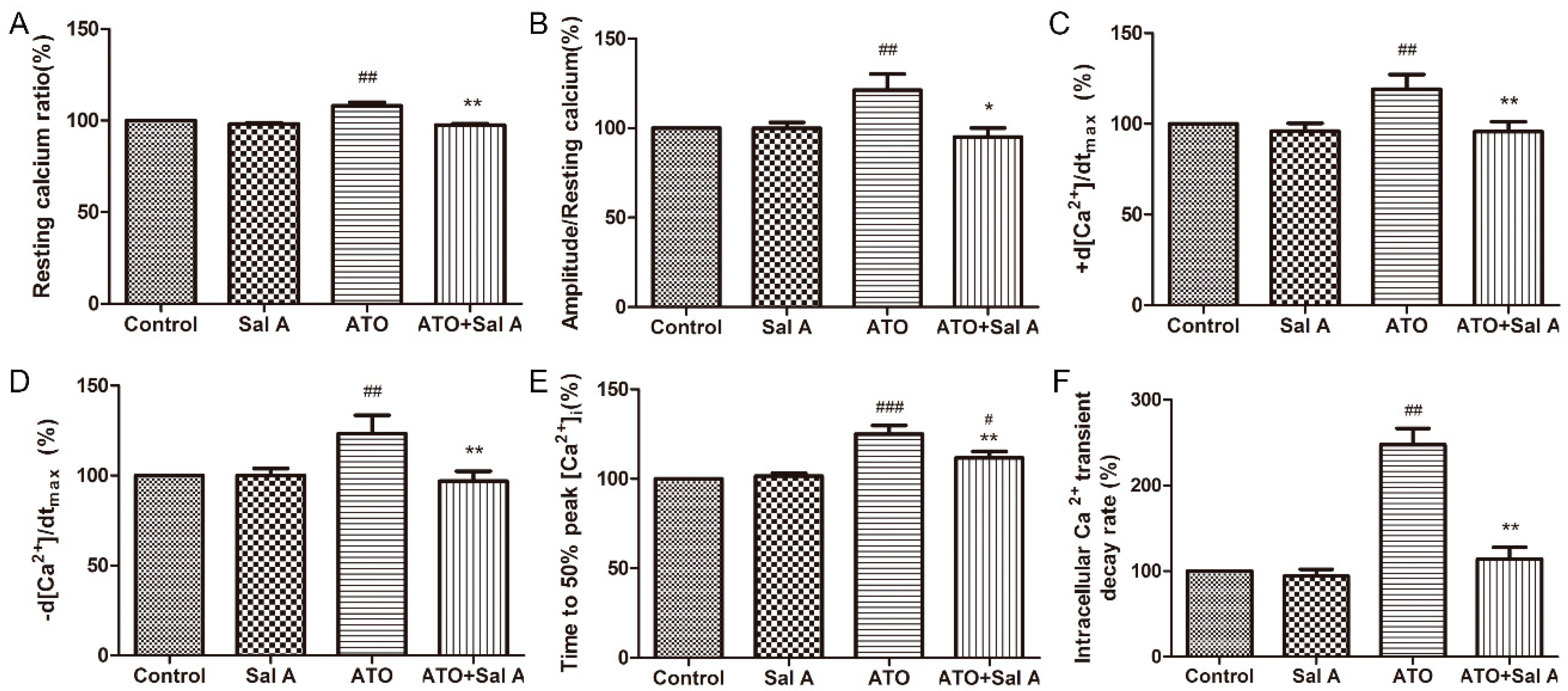
2.5. Effects of Sal A on Intracellular Ca2+ Transients in ARVMs after ATO Treatment
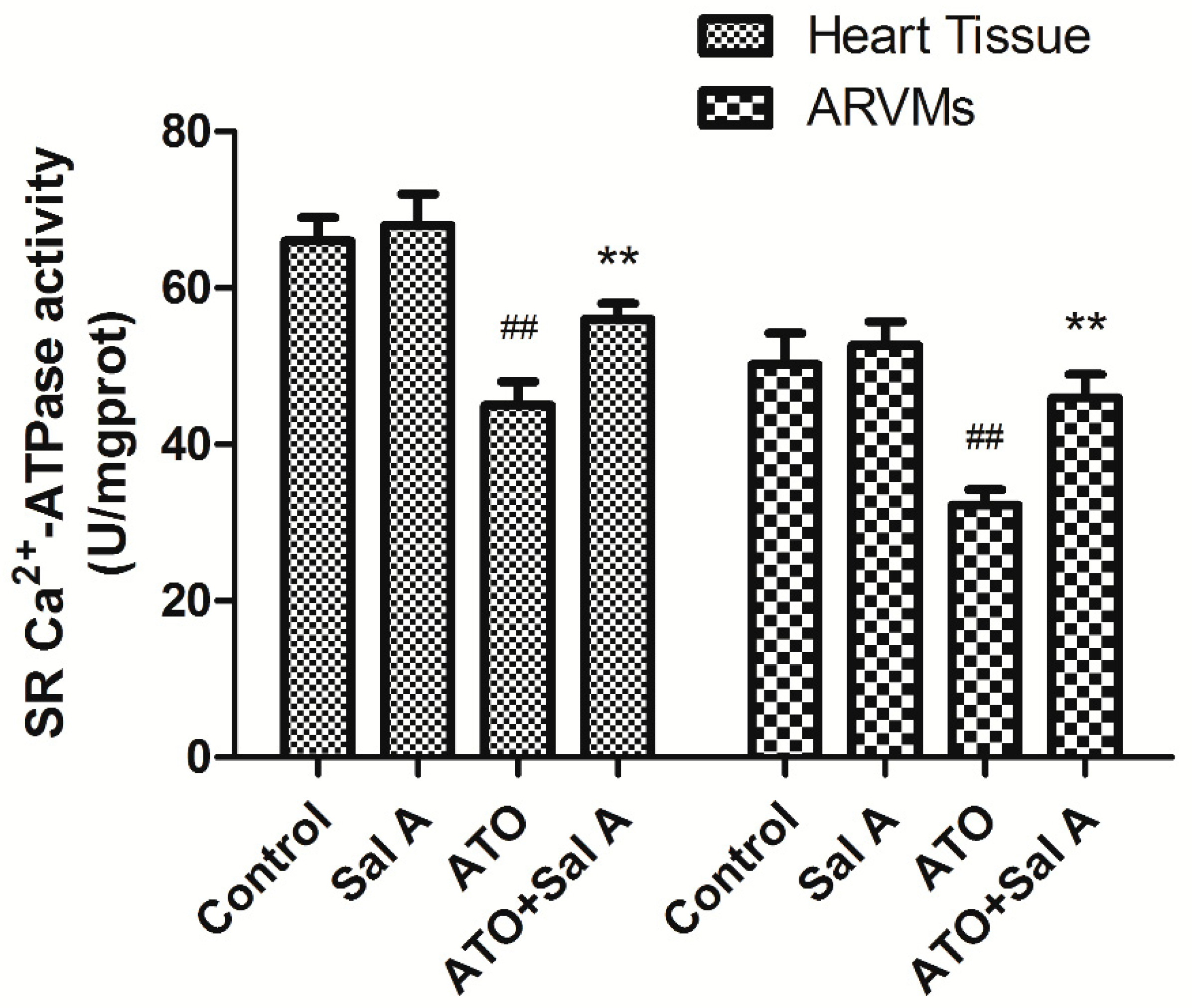
2.6. Effects of Sal A on sarco endoplasmic reticulum Ca2+-ATPase (SERCA) Activity
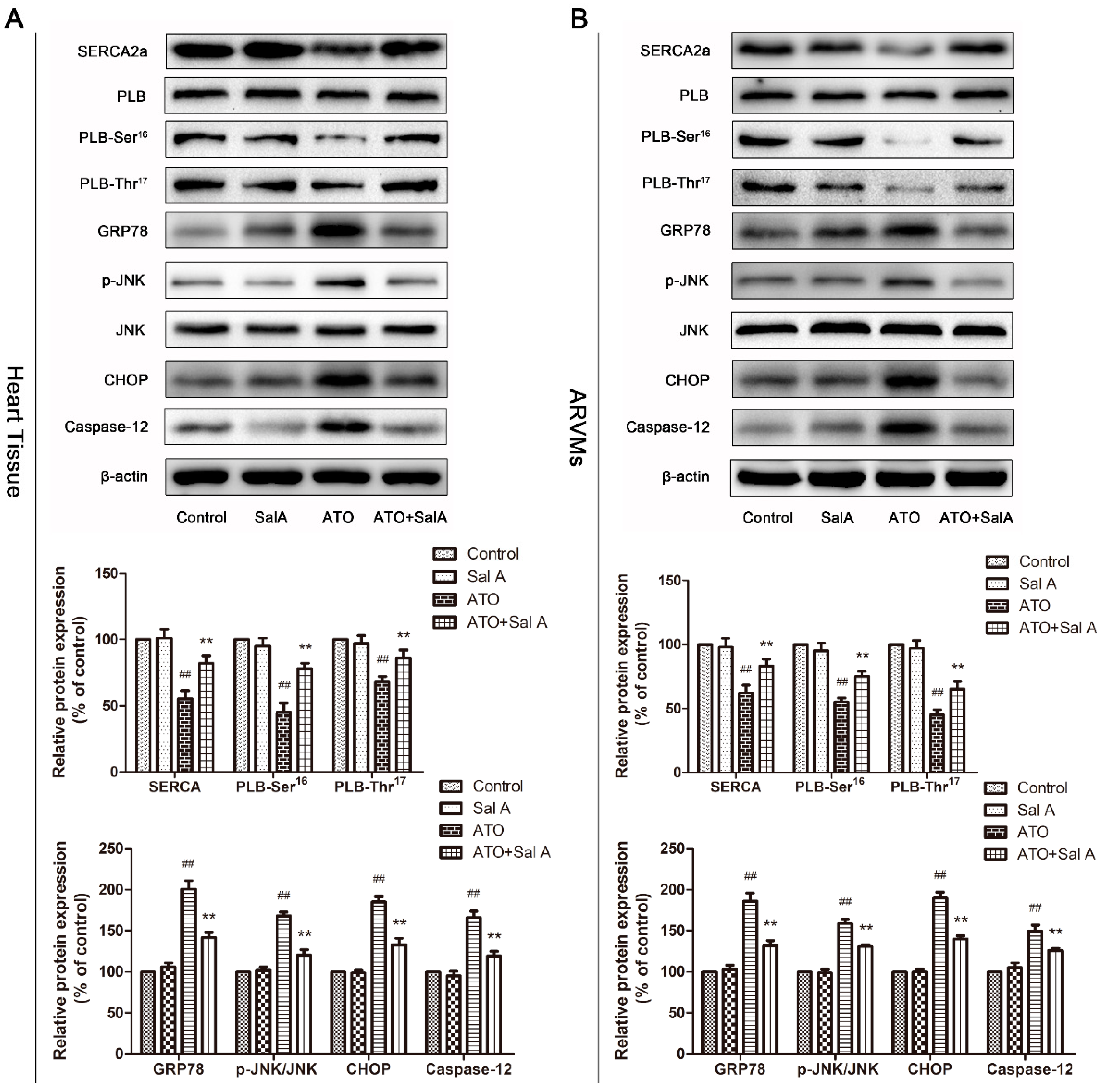
2.7. Effects of Sal A on Ca2+ and ER Stress-Related Protein Expression Levels after ATO Treatment
3. Discussion
4. Materials and Methods
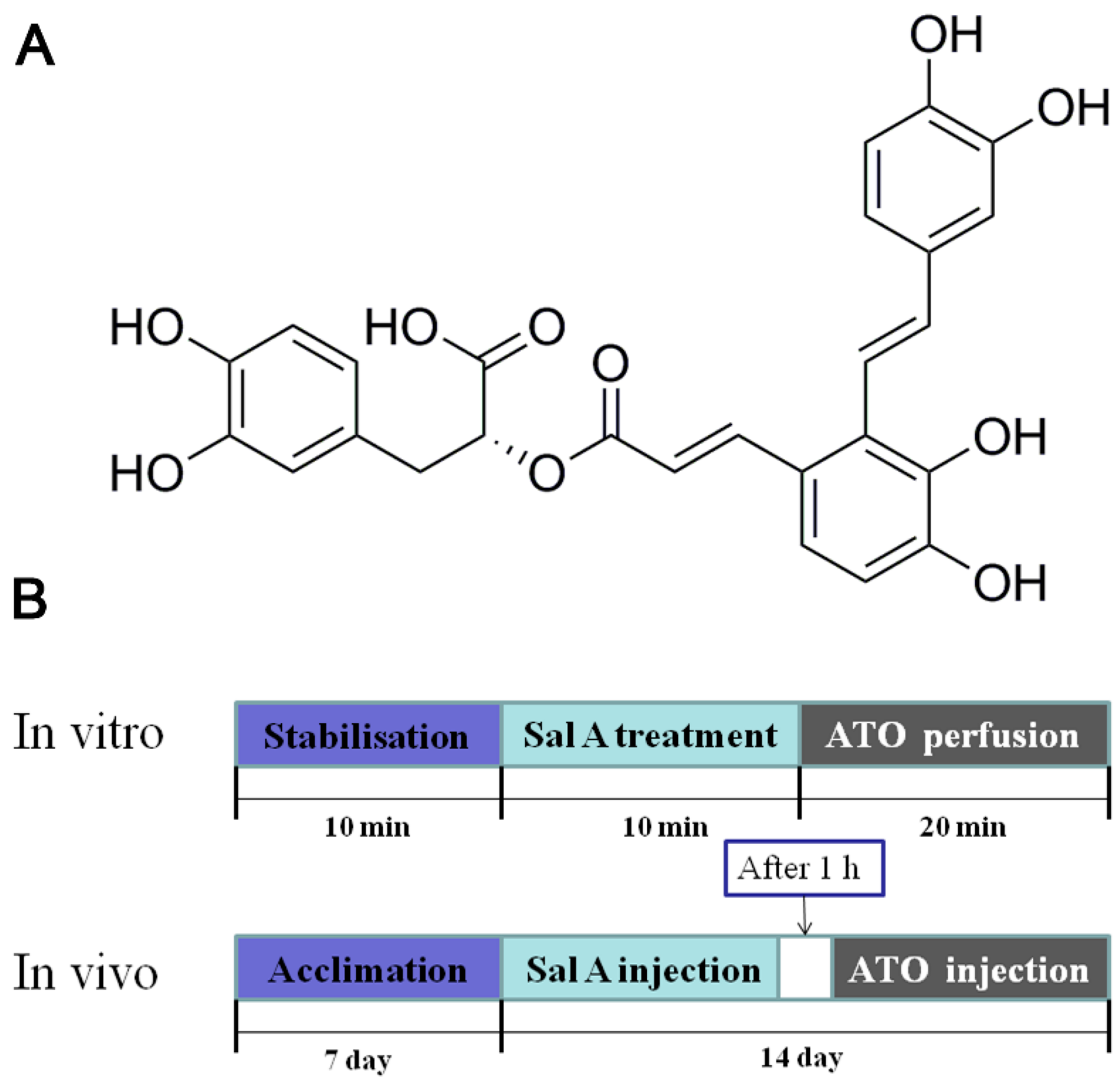
4.1. Materials and Animal Treatment
4.2. Echocardiographic Measurements
4.3. Heart Histopathological Measurements
4.4. Serum Analysis of LDH, CK, AST, GSH-PX, CAT, and SOD
4.5. Isolation of Adult Rat Ventricular Myocytes (ARVMs)
4.6. Measurement of Sarcomere Shortening and Ca2+ Transients in ARVMs
4.7. SERCA Activity Detection
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; de The, H. How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer 2002, 2, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.X.; Chen, G.Q.; Ni, J.H.; Li, X.S.; Xiong, S.M.; Qiu, Q.Y.; Zhu, J.; Tang, W.; Sun, G.L.; Yang, K.Q.; et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997, 89, 3354–3360. [Google Scholar] [PubMed]
- Fox, E.; Razzouk, B.I.; Widemann, B.C.; Xiao, S.; O’Brien, M.; Goodspeed, W.; Reaman, G.H.; Blaney, S.M.; Murgo, A.J.; Balis, F.M.; et al. Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood 2008, 111, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westervelt, P.; Brown, R.A.; Adkins, D.R.; Khoury, H.; Curtin, P.; Hurd, D.; Luger, S.M.; Ma, M.K.; Ley, T.J.; DiPersio, J.F. Sudden death among patients with acute promyelocytic leukemia treated with arsenic trioxide. Blood 2001, 98, 266–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.Y.; Chang, C.S.; Tang, J.L.; Tien, H.F.; Kuo, T.L.; Huang, S.F.; Yao, Y.T.; Chou, W.C.; Chung, C.Y.; Wang, C.H.; et al. Acute and chronic arsenic poisoning associated with treatment of acute promyelocytic leukaemia. Br. J. Haematol. 1998, 103, 1092–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Feng, T.; Chen, H.; Shan, H.; Zhang, Y.; Lu, Y.; Yang, B. Arsenic trioxide-induced apoptosis in H9c2 cardiomyocytes: Implications in cardiotoxicity. Basic Clin. Pharmacol. Toxicol. 2008, 102, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Girija, S.; Soumya, R.S.; Raghu, K.G. Polyphenol-rich apple (Malus domestica L.) peel extract attenuates arsenic trioxide induced cardiotoxicity in H9c2 cells via its antioxidant activity. Food Funct. 2014, 5, 502–511. [Google Scholar] [CrossRef]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, N.; Luan, R.; Li, Y.; Wang, D.; Zou, W.; Xing, Y.; Tao, L.; Cao, F.; Wang, H. Apelin protects sarcoplasmic reticulum function and cardiac performance in ischaemia-reperfusion by attenuating oxidation of sarcoplasmic reticulum Ca2+-ATPase and ryanodine receptor. Cardiovasc. Res. 2013, 100, 114–124. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Sun, G.B.; Wang, M.; Liao, P.; Du, Y.Y.; Yang, K.; Sun, X.B. Arsenic trioxide triggered calcium homeostasis imbalance and induced endoplasmic reticulum stress-mediated apoptosis in adult rat ventricular myocytes. Toxicol. Res. 2016, 5, 682–688. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Sun, G.B.; Luo, Y.; Wang, M.; Wang, W.; Du, Y.Y.; Yu, Y.L.; Sun, X.B. Salvianolic acid a protects H9c2 cells from arsenic trioxide-induced injury via inhibition of the mapk signaling pathway. Cell. Physiol. Biochem. 2017, 41, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, J.X.; Meng, H.X.; Lin, C.R. Blocking effect of salvianolic acid A on calcium channels in isolated rat ventricular myocytes. Chin. J. Integr. Med. 2012, 18, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Jiang, Z.; Bi, L.; Yang, Y.; Chen, W. Salvianolic acid A attenuates TNF-alpha- and D-GalN-induced ER stress-mediated and mitochondrial-dependent apoptosis by modulating Bax/Bcl-2 ratio and calcium release in hepatocyte LO2 cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Rajadurai, M.; Stanely, M.P.P. Preventive effect of naringin on cardiac markers, electrocardiographic patterns and lysosomal hydrolases in normal and isoproterenol-induced myocardial infarction in Wistar rats. Toxicology 2007, 230, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, D.; Xiao, R.P. Dual site phospholamban phosphorylation and its physiological relevance in the heart. Trends Cardiovasc. Med. 2002, 12, 51–56. [Google Scholar] [CrossRef]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sun, G.B.; Sun, X.; Wang, H.W.; Meng, X.B.; Qin, M.; Sun, J.; Luo, Y.; Sun, X.B. Cardioprotective effect of salvianolic acid B against arsenic trioxide-induced injury in cardiac H9c2 cells via the PI3K/Akt signal pathway. Toxicol. Lett. 2013, 216, 100–107. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhang, B.; Wang, M.; Wang, W.; Liao, P.; Sun, G.B.; Sun, X.B. Calcium homeostasis and endoplasmic reticulum stress are involved in Salvianolic acid B-offered protection against cardiac toxicity of arsenic trioxide. Oncotarget 2017, 8, 97384–97393. [Google Scholar] [CrossRef]
- Feng, S.; Geng, J.; Sun, R.; Huang, J.; Tan, Z.; Yan, C.X.; Wang, G. Protective effect of salvianolic acid a on brain endothelial cells after treatment with deprivation and reperfusion of oxygen-glucose. Chin. Herb. Med. 2017, 9, 335–343. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Subcell Biochem. 2007, 45, 481–506. [Google Scholar] [PubMed]
- Bers, D.M. Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 2000, 87, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lipskaia, L.; Keuylian, Z.; Blirando, K.; Mougenot, N.; Jacquet, A.; Rouxel, C.; Sghairi, H.; Elaib, Z.; Blaise, R.; Adnot, S.; et al. Expression of sarco (endo) plasmic reticulum calcium ATPase (SERCA) system in normal mouse cardiovascular tissues, heart failure and atherosclerosis. Biochim. Biophys. Acta 2014, 1843, 2705–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Teng, Z.; Lu, F.H.; Zhao, Y.J.; Li, H.; Ren, H.; Chen, H.; Pan, Z.W.; Lv, Y.J.; Yang, B.F.; et al. Post-conditioning protects cardiomyocytes from apoptosis via PKC(epsilon)-interacting with calcium-sensing receptors to inhibit endo(sarco)plasmic reticulum-mitochondria crosstalk. Mol. Cell. Biochem. 2010, 341, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Healy, S.J.; Jager, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Ichijo, H. Survival and apoptosis signals in ER stress: The role of protein kinases. J. Chem. Neuroanat. 2004, 28, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Zhang, J.; Wang, S.; Wang, M.; Ye, T.; Du, Y.; Xie, X.; Ye, J.; Sun, G.; Sun, X. The Cardiotoxicity Induced by Arsenic Trioxide is Alleviated by Salvianolic Acid A via Maintaining Calcium Homeostasis and Inhibiting Endoplasmic Reticulum Stress. Molecules 2019, 24, 543. https://doi.org/10.3390/molecules24030543
Wang R, Zhang J, Wang S, Wang M, Ye T, Du Y, Xie X, Ye J, Sun G, Sun X. The Cardiotoxicity Induced by Arsenic Trioxide is Alleviated by Salvianolic Acid A via Maintaining Calcium Homeostasis and Inhibiting Endoplasmic Reticulum Stress. Molecules. 2019; 24(3):543. https://doi.org/10.3390/molecules24030543
Chicago/Turabian StyleWang, Ruiying, Jingyi Zhang, Shan Wang, Min Wang, Tianyuan Ye, Yuyang Du, Xueheng Xie, Jingxue Ye, Guibo Sun, and Xiaobo Sun. 2019. "The Cardiotoxicity Induced by Arsenic Trioxide is Alleviated by Salvianolic Acid A via Maintaining Calcium Homeostasis and Inhibiting Endoplasmic Reticulum Stress" Molecules 24, no. 3: 543. https://doi.org/10.3390/molecules24030543
APA StyleWang, R., Zhang, J., Wang, S., Wang, M., Ye, T., Du, Y., Xie, X., Ye, J., Sun, G., & Sun, X. (2019). The Cardiotoxicity Induced by Arsenic Trioxide is Alleviated by Salvianolic Acid A via Maintaining Calcium Homeostasis and Inhibiting Endoplasmic Reticulum Stress. Molecules, 24(3), 543. https://doi.org/10.3390/molecules24030543