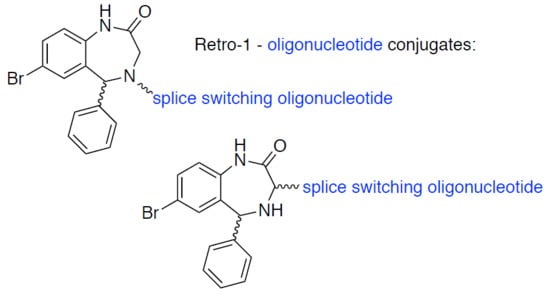
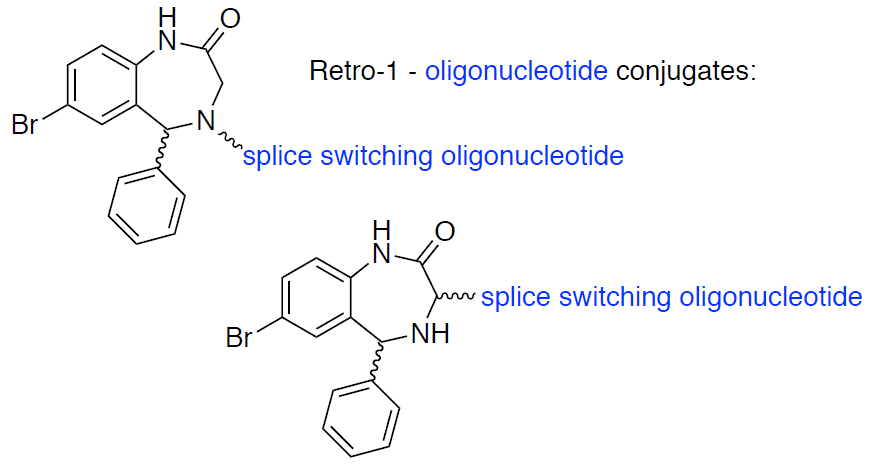
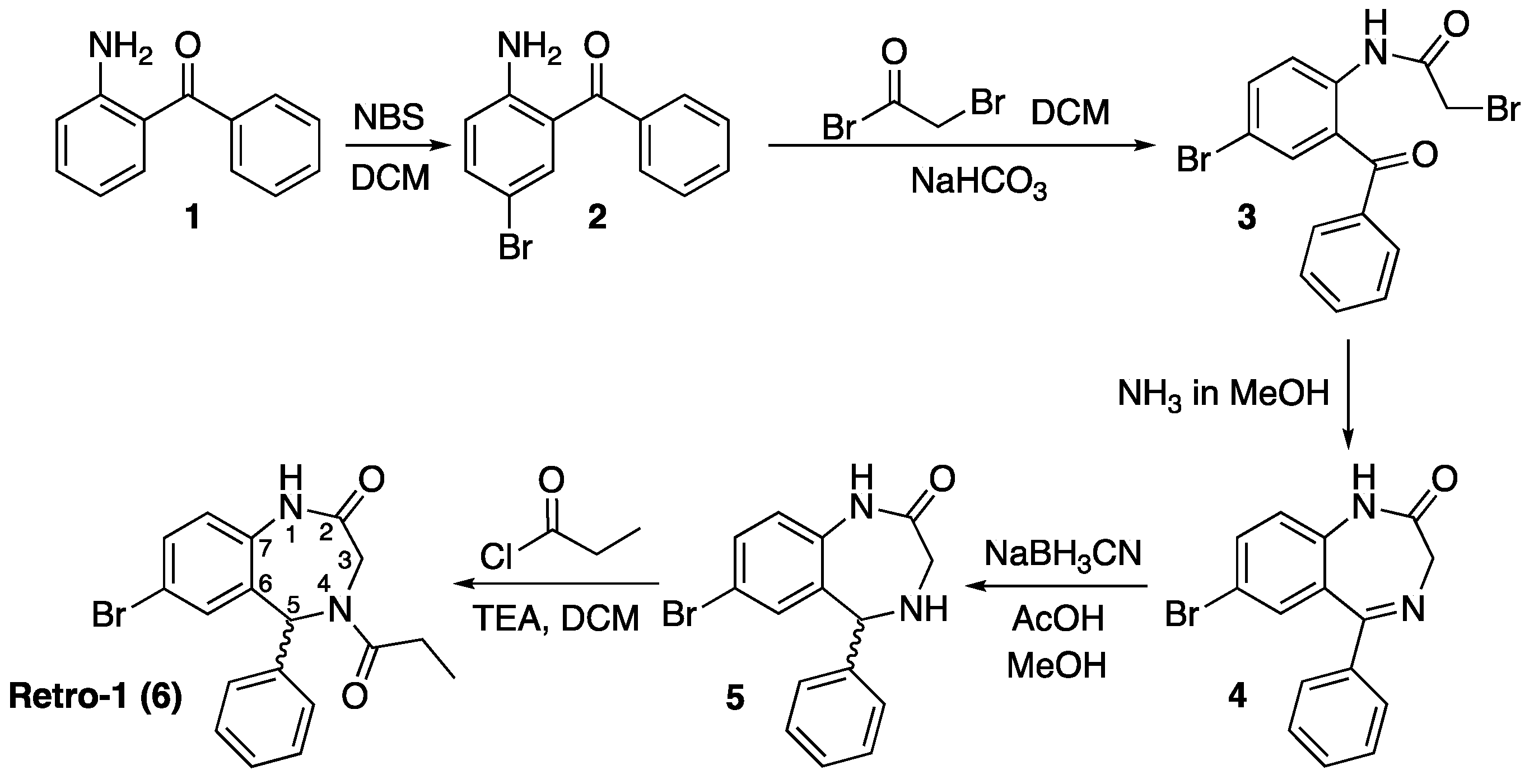
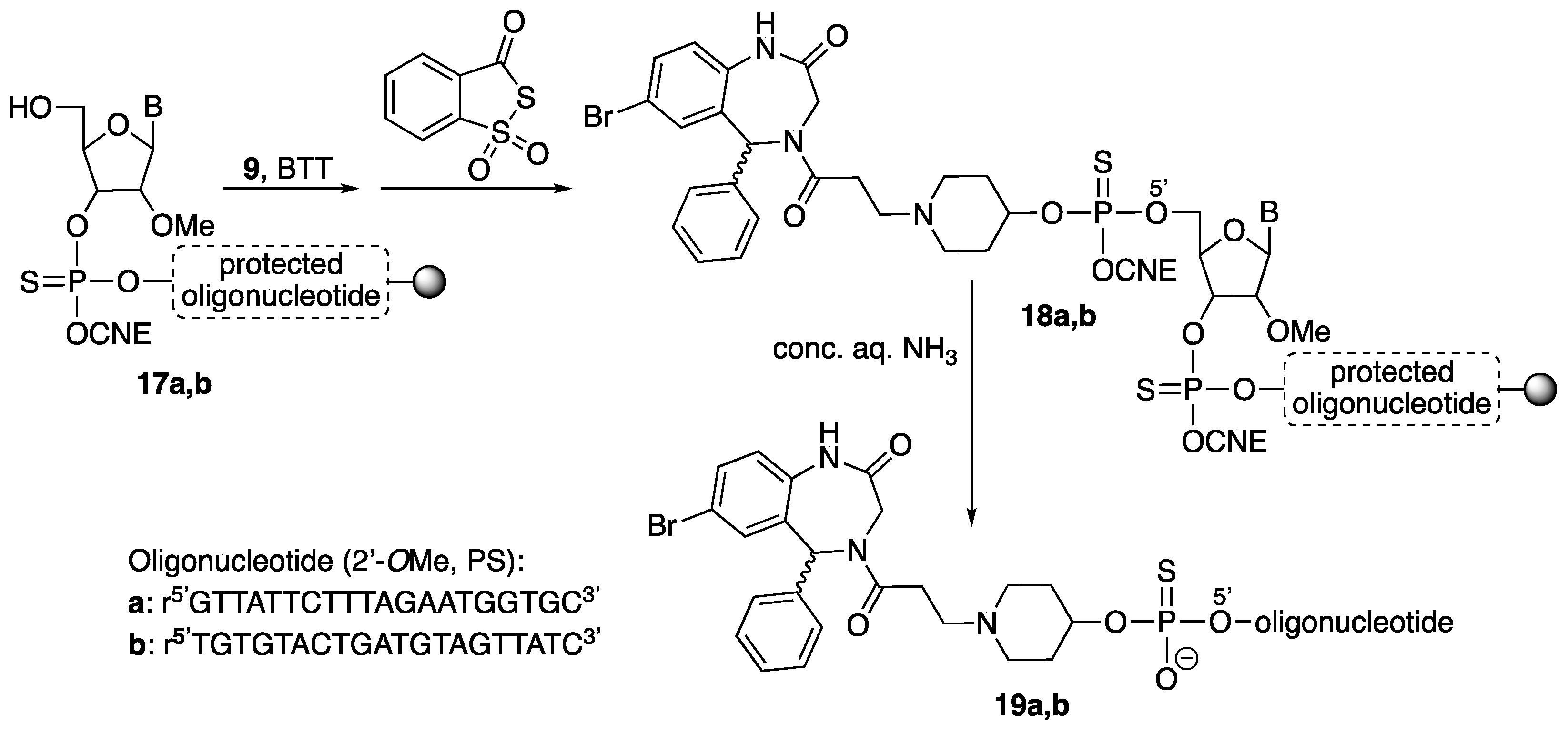
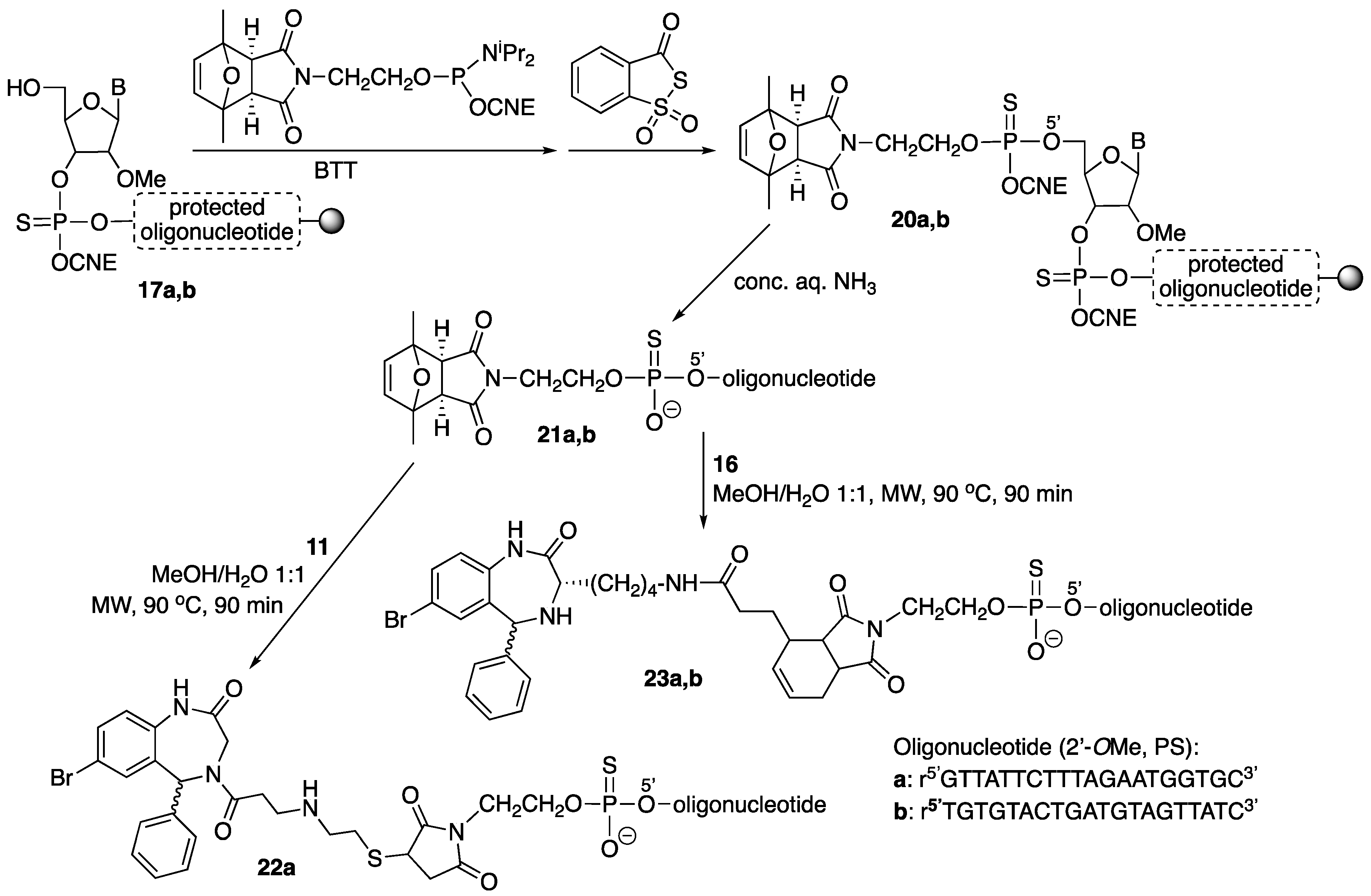
3.2. Synthesis of Small Molecules (Compounds 8–16)
Compounds
1–
6 were Synthesized as Previously Described [
17].
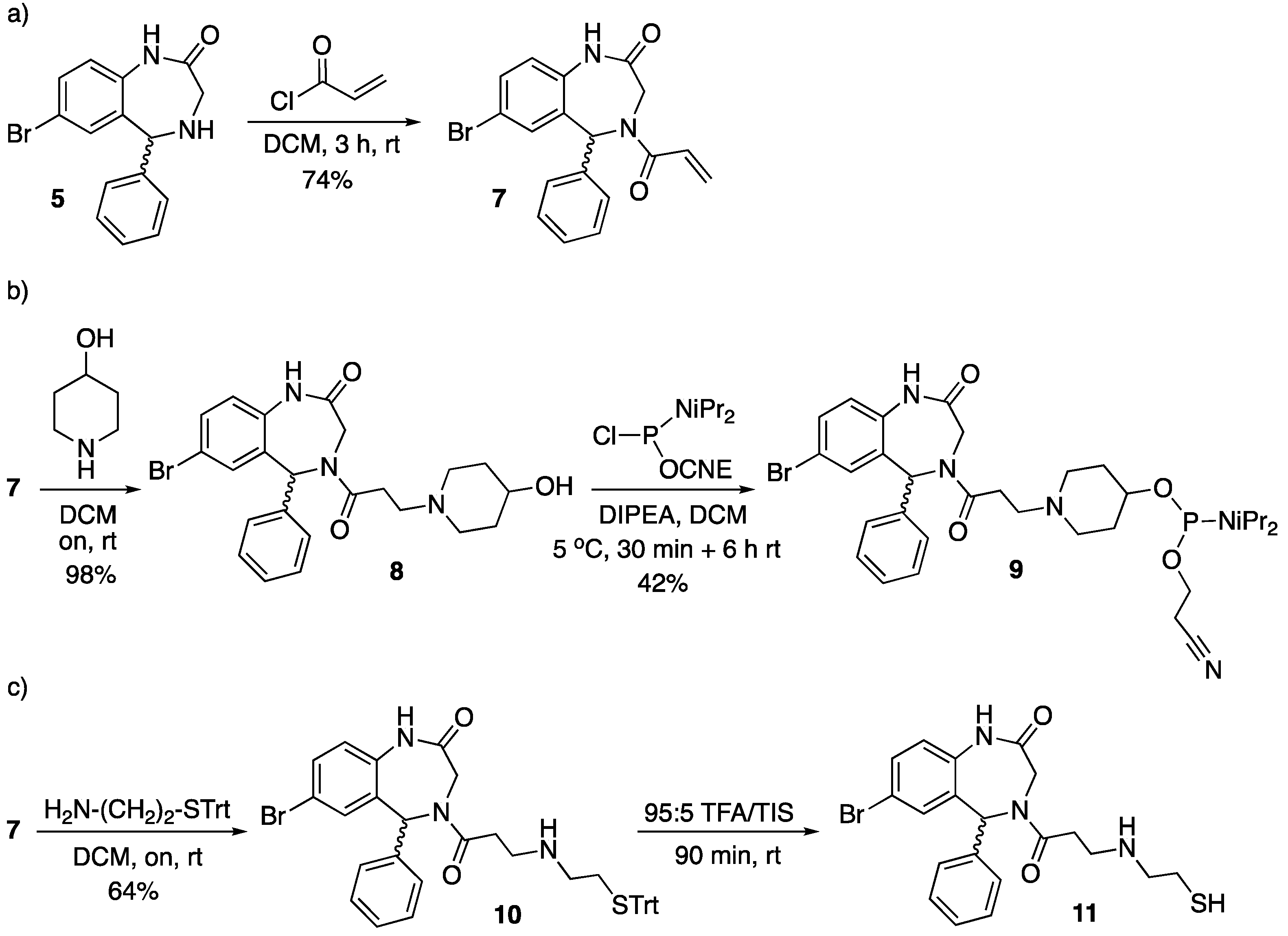
7-Bromo-1,3,4,5-tetrahydro-4-(acryloyl)-5-phenyl-2H-1,4-benzodiazepin-2-one (7). To a suspension of 5 (150 mg, 0.47 mmol) in DCM (5 mL) acryloyl chloride (50 µL, 0.62 mmol) was added, and the mixture stirred at room temperature for 3 h. Afterwards, additional DCM (30 mL) was added, and the mixture was transferred to a separatory funnel and washed with H2O (3 × 20 mL). The organic layer was dried over anh. MgSO4, filtered, and the solvent removed under low pressure. The resulting crude was purified by silica gel flash column chromatography eluting with DCM/EtOAc mixtures from 100:0 to 75:25. The title compound (7) was obtained as a white foam (130 mg, 74%).
TLC (DCM/EtOAc 80:20): Rf = 0.50; IR (ATR, solid): 3221, 2920, 2359, 2340, 1682, 1647, 1515, 1416, 665 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.88 and 8.85 (s, 1H), 7.45 (s, 1H), 7.46 and 7.40 (d, J = 8.5 Hz, 1H), 7.32–7.24 (m, 3H), 7.05 (s, 1H), 7.04 and 6.16 (s, 1H), 6.93 and 6.89 (d, J = 8.5 Hz, 1H), 6.61 and 6.54 (dd, J = 10.4, 16.4 Hz, 1H), 6.45 and 6.41 (s, 1H), 5.84–5.78 (m, 1H), 4.34–4.02 (m, 2H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 170.39, 166.04, 138.44, 134.30, 132.09, 130.61, 129.02, 127.93, 126.67, 122.91, 117.43, 59.46, 48.72 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 170.39, 166.63, 137.52, 135.52, 134.71, 133.15, 132.73, 130.86, 130.00, 129.06, 128.39, 128.21, 127.08, 126.79, 123.67, 117.55, 63.49, 46.30 ppm; ESI-HRMS (positive mode): m/z 371.0391/373.0368 (81Br) [M + H]+, M calcd for C18H16BrN2O2 371.0390.
7-Bromo-1,3,4,5-tetrahydro-4-[1-oxo-3-(4-hydroxypiperidin-1-yl)propyl]-5-phenyl-2H-1,4-benzodiazep-in-2-one (8). 7 (100 mg, 0.27 mmol) and 4-hydroxypiperidine (273.1 mg, 2.70 mmol) were dissolved in DCM (5 mL) and reacted overnight at room temperature. Afterwards, the solvent was removed under low pressure, the resulting crude dissolved in EtOAc (40 mL) and washed with H2O (3 × 20 mL). The organic phase was dried over anh. MgSO4, filtered, and the solvent removed under vacuum. The title compound (8) was obtained as a white solid (125 mg, 98%).
TLC (DCM:EtOAc 50:50): Rf = 0.20; IR (ATR, solid): 3214, 3119, 2983, 2353, 2334, 1865, 1650, 1558, 1553, 1508, 1239, 783, 666 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.70 and 8.68 (s, 1H), 7.46–7.38 (m, 2H), 7.34–7.27 (m, 3H), 7.06–7.00 (m, 2H), 6.95 and 6.20 (s, 1H), 6.92–6.87 (m, 1H), 4.45–3.98 (m, 2H), 3.65 (p, J = 4.5 Hz, 1H), 2.80–2.54 (m, 6H), 2.20–2.12 (m, 2H), 1.87–1.79 (m, 4H), 1.57–1.47 (m, 1H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 171.64, 170.40, 138.33, 134.86, 134.25, 132.05, 130.34, 129.14, 128.96, 127.74, 122.87, 117.39, 67.49, 63.07, 59.29, 53.75, 51.19, 34.18, 31.60 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 171.96, 170.94, 137.60, 135.50, 133.17, 132.60, 130.37, 128.44, 128.10, 127.05, 123.45, 117.19, 67.44, 60.42, 53.82, 51.30, 46.10, 34.19, 31.30 ppm; ESI-HRMS (positive mode): m/z 472.1222/474.1205 (81Br) [M + H]+, M calcd for C23H27BrN3O3 472.1230.
7-Bromo-1,3,4,5-tetrahydro-4-[1-oxo-3-(4-hydroxypiperidin-1-yl)propyl]-5-phenyl-2H-1,4-benz-odiazepin-2-one, 2-cyanoethyl N,N-diisopropylphosphoramidite (9). 8 (453.5 mg, 0.96 mmol) was dissolved in anh. DCM (15 mL). Subsequently, anh. N,N-diisopropylethylamine (310 µL, 2.41 mmol) was added and the mixture was stirred in an ice bath for 5 min under an argon atmosphere. Afterwards, a solution of chloro(2-cyanoethoxy)diisopropylaminophosphine (250 mg, 1.06 mmol) in anh. DCM (1 mL) was added, and the mixture was reacted in an ice bath for 30 min. Then, it was allowed to warm up and left stirring for 6 h at room temperature until complete phosphitylation as shown by TLC. The solvent was removed under low pressure; the crude was dissolved in EtOAc (50 mL) and washed with aq. NaHCO3(sat.) (3 × 30 mL). The organic phase was dried over anh. MgSO4, filtered, and the solvent removed under low pressure. The resulting crude was further purified by silica gel flash column chromatography eluting with DCM/EtOAc/NEt3 mixtures from 98:0:2 to 48:50:2. The title compound (9) was obtained as a white foam (272.4 mg, 42%).
TLC (DCM/EtOAc/NEt3 48:50:2): Rf = 0.50; 1H NMR (CDCl3, 400 MHz): δ 7.90 and 7.86 (s, 1H), 7.48–7.40 (m, 2H), 7.33–7.27 (m, 3H), 7.05–7.01 (m, 2H), 6.95 and 6.22 (s, 1H), 6.88–6.80 (m, 1H), 4.44–4.00 (m, 2H), 3.90–3.71 (m, 3H), 3.65–3.45 (m, 2H), 2.80–2.56 (m, 8H), 2.38–2.23 (m, 2H), 1.93–1.79 (m, 2H), 1.76–1.60 (m, 2H), 1.27 and 1.17 (dd, J = 6.8, 5.5 Hz, 12H) ppm; 31P NMR (CDCl3, 162 MHz): δ 145.85 ppm; ESI-HRMS (positive mode): m/z 672.2298/674.2281 (81Br) [M + H]+, M calcd for C32H44BrN5O4P 672.2309.
7-Bromo-5-phenyl-4-{3-[(2-(tritylthio)ethyl)amino]propanoyl}-1,3,4,5-tetrahydro-2H-benzo[e][1,4]di-azepin-2-one (10). 7 (370 mg, 0.99 mmol) and S-tritylcysteamine (1.50 g, 4.49 mmol) were dissolved in DCM (5 mL) and reacted overnight at room temperature. Afterwards, the solvent was removed under low pressure, and the resulting crude was purified by silica gel flash column chromatography eluting with hexanes/EtOAc/MeOH mixtures from 30:70:0 to 0:100:3. The title compound (10) was obtained as a white solid (441 mg, 64%).
TLC (EtOAc/MeOH 97:3): Rf = 0.35; IR (ATR, solid): 3211, 2926, 1729, 1666, 1664, 1482, 1448, 1368, 1242, 1216, 1188, 1058, 821, 745, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.44–7.40 (m, 9H), 7.30–7.18 (m, 14H), 7.03–6.99 (m, 1H), 6.90 and 6.11 (s, 1H), 4.42–3.96 (m, 2H), 2.80–2.66 (m, 2H), 2.60–2.45 (m, 3H), 2.41–2.31 (m, 3H), 1.87 (br s, 1H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 171.49, 169.75, 145.01, 138.34, 134.80, 134.45, 133.43, 132.28, 130.38, 129.15, 128.00, 127.85, 127.13, 126.80, 122.89, 117.61, 66.71, 63.16, 48.73, 44.67, 41.11, 33.67, 31.86 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 171.86, 170.35, 144.90, 137.43, 135.47, 133.43, 132.83, 130.41, 129.33, 128.62, 128.30, 128.04, 127.13, 126.80, 123.40, 117.61, 66.80, 63.16, 48.40, 44.89, 41.11, 33.31, 31.81 ppm; ESI-HRMS (positive mode): m/z 690.1776/692.1765 (81Br) [M + H]+, M calcd for C39H37BrN3O2S 690.1784.
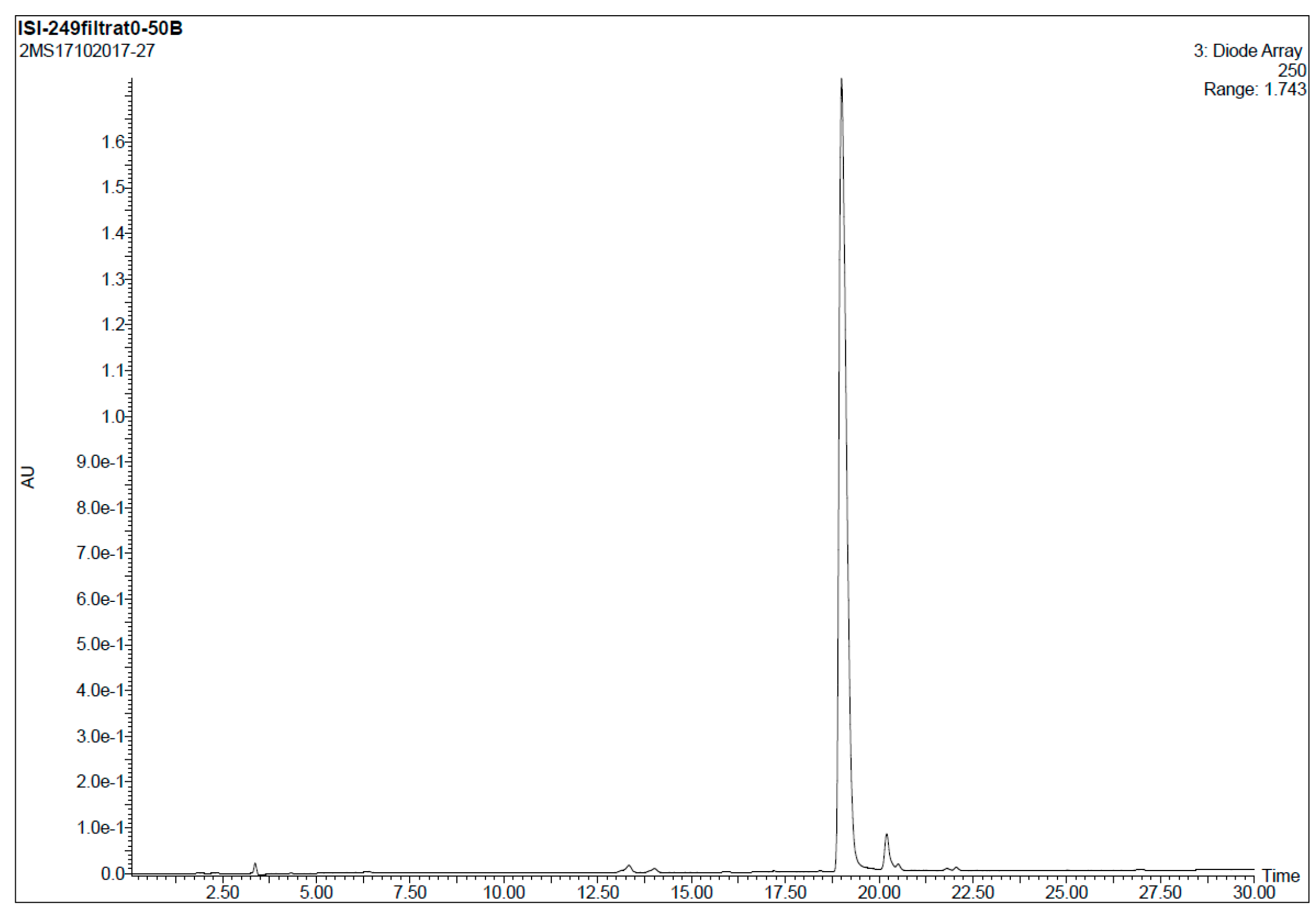
7-Bromo-4-{3-[(2-mercaptoethyl)amino]propanoyl}-5-phenyl-1,3,4,5-tetrahydro-2H-benzo[e][1,4]diaze-pin-2-one (
11).
10 (20 mg, 0.03 mmol) was dissolved in a mixture of TFA/TIS (95:5) and reacted at room temperature for 90 min. Afterwards, the solvent was removed under a N
2 stream and the resulting crude dissolved in a mixture MeOH/H
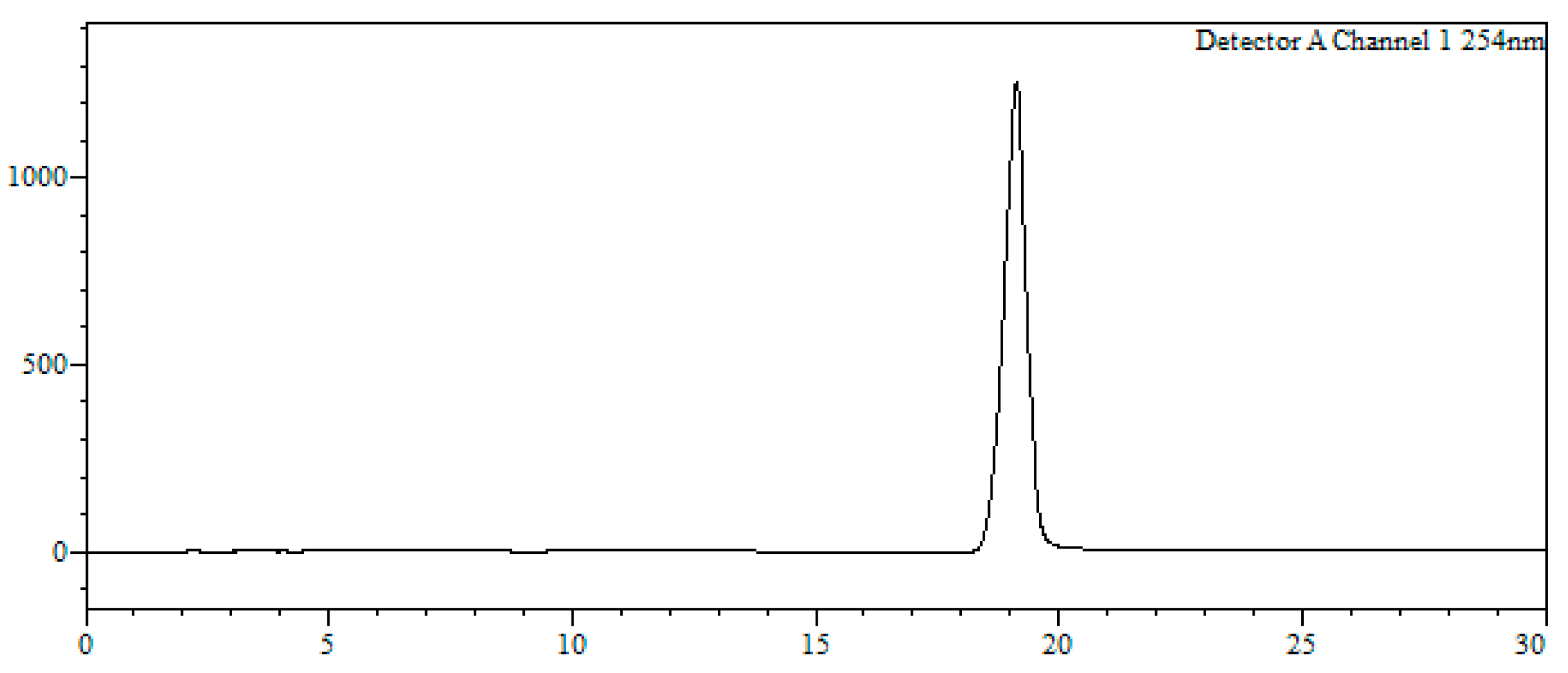
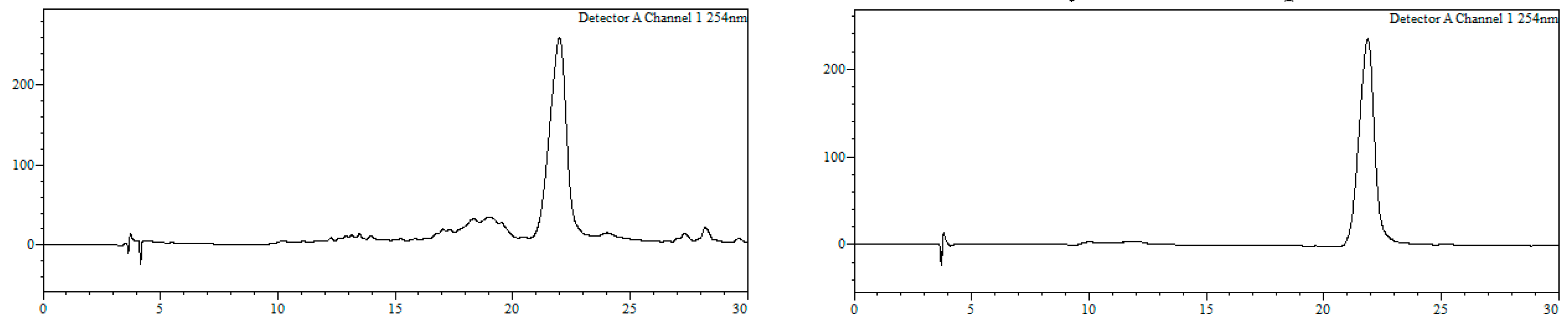
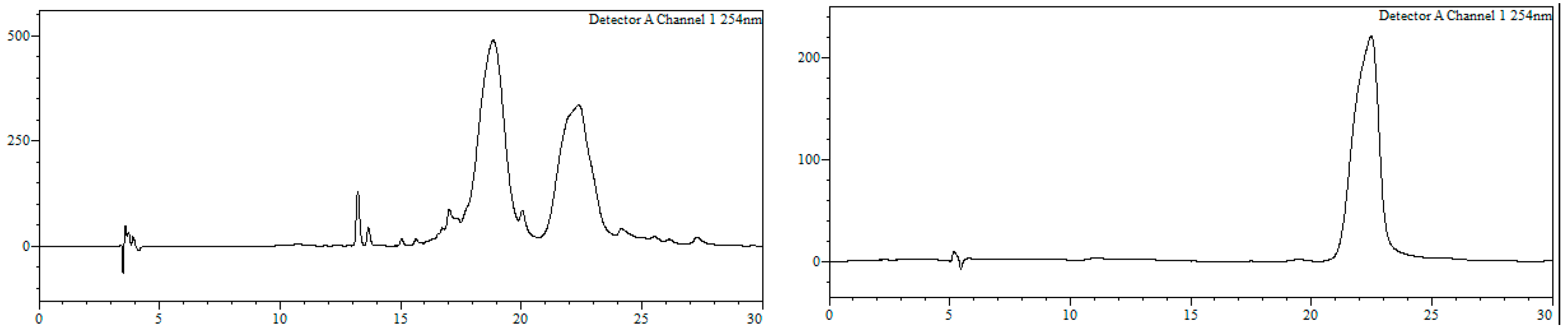
2O (1:1, 2 mL) and filtered through a hydrophilic PTFE (polytetrafluoroethylene) syringe filter (0.22 µm), lyophilized and used without further purification (quantitative thiol deprotection as assessed by HPLC,
Figure 2).
HPLC-MS: Analysis conditions: 0 → 50 % B in 30 min, tR = 19.0 min. ESI-MS (positive mode) of the main peak: m/z 448.2/450.2 (81Br), M calcd for C20H23BrN3O2S 448.07.
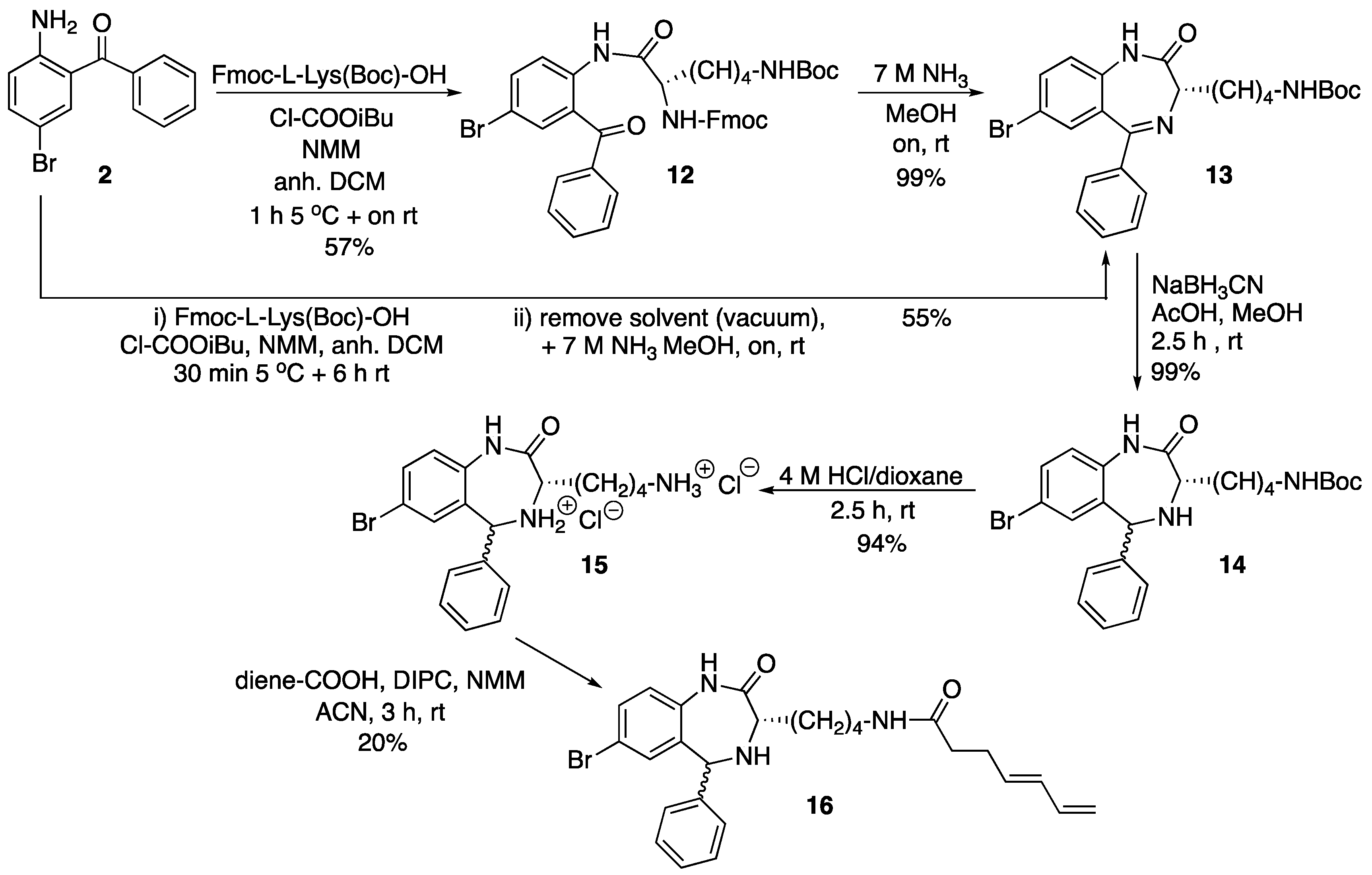
N-(Nα-Fmoc-Nε-l-lysinyl)-2-amino-5-bromobenzophenone (12). N-Methylmorpholine (200 µL, 1.81 mmol) and isobutylchloroformate (94 µL, 0.73 mmol) were added to a solution of Fmoc-l-Lys(Boc)-OH (340.5 mg, 0.73 mmol) in anh. DCM (5 mL), and the mixture cooled in an ice bath. After 15 min, a solution of 2 (200.0 mg, 0.73 mmol) in anh. DCM (1 mL) was added and the mixture was stirred for 1 h at 5 °C and overnight at room temperature. Afterwards, further DCM was added (15 mL) and the mixture was washed with aq. HCl 10% (2 × 10 mL). The organic phase was dried over anh. MgSO4, filtered and the solvent evaporated under low vacuum. The resulting crude was purified by silica gel flash column chromatography eluting with DCM/MeOH mixtures from 100:0 to 97:3. The title compound (12) was obtained as a pale white foam (301 mg, 57%).
TLC (DCM/MeOH 95:5): Rf = 0.40; IR (ATR, solid): 2980, 2905, 2355, 1730, 1658, 1599, 1571, 1497, 1425, 1437, 1391, 1254, 1248, 1173, 1158, 1071, 1043, 757, 744, 694, 533 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.57 (d, J = 8.8 Hz, 1H), 7.85–7.52 (m, 9H), 7.46–7.23 (m, 7H), 5.95 (br. s, 1H), 4.78–4.63 (m, 1H), 4.55–4.34 (m, 2H), 4.32–4.20 (m, 2H), 3.13 (t, J = 8.0 Hz, 2H), 2.02 (m, 2H), 1.93–1.78 (m, 2H), 1.77–1.50 (m, 2H), 1.45 (s, 9H) ppm; 13C NMR (CDCl3, 101 MHz): δ 197.84, 171.21, 156.47, 156.31, 143.69, 141.23, 138.78, 137.60, 136.75, 135.51, 132.91, 129.94, 128.46, 127.73, 127.66, 127.09, 127.05, 125.47, 125.14, 120.01, 119.89, 115.13, 79.21, 77.48, 77.16, 76.84, 67.44, 56.46, 47.20, 39.77, 31.73, 29.81, 28.47, 22.42 ppm. ESI-HRMS (positive mode): m/z 726.2173/728.2161 (81Br) [M + H]+; M calcd for C39H40BrN3O6 726.2173.
Tert-Butyl (S)-[4-(7-bromo-2-oxo-5-phenyl-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)butyl]carbamate (13). 12 (150 mg, 0.21 mmol) was dissolved in a 7 M ammonia solution (in MeOH, 3 mL) and reacted at room temperature overnight. Afterwards, the reaction mixture was taken to dryness and the crude purified by silica gel flash column chromatography eluting with a 70:30 hexanes/EtOAc mixture. The title compound (13) was obtained as a pale yellow solid (104 mg, 99%). For characterization see below.
One-pot synthesis of compound13from2. A solution of Fmoc-l-Lys(Boc)-OH (1.87 g, 4.00 mmol) in anh. DCM (15 mL) was cooled in an ice bath. N-Methylmorpholine (1.37 mL, 9.09 mmol) and isobutyl chloroformate (0.81 mL, 3.64 mmol) were added. After 10 min, a solution of 2 (1.00 g, 3.64 mmol) in anh. DCM (2 mL) was added, and the mixture was stirred for 30 min at 5 °C and 5.5 h at room temperature. Afterwards, the solvent was removed under vacuum and the crude dissolved in 7 M NH3 (in MeOH, 40 mL), and the solution was left to react overnight at room temperature. Subsequently, the solvent was removed under vacuum and the resulting crude dissolved in EtOAc (100 mL) and washed with 10% aq. HCl (3 × 30 mL). The organic phase was dried over anh. MgSO4, filtered and the solvent evaporated under vacuum. The crude material was further purified by silica gel flash column chromatography eluting with a 70:30 hexanes/EtOAc mixture. The title compound (13) was obtained as a pale yellow solid (970 mg, 55%).
TLC (hexanes/EtOAc 1:1): Rf = 0.42; IR (ATR, solid): 3414, 3309, 2929, 2359, 2337, 1682, 1653, 1508, 1232, 1156, 669 cm−1; 1H NMR (CDCl3, 400 MHz): δ 9.45 (s, 1H), 7.60 (dd, J = 8.6, 2.3 Hz, 1H), 7.52–7.34 (m, 6H), 7.08 (d, J = 8.6 Hz, 1H), 4.63 (s br, 1H), 3.50 (dd, J = 8.2, 5.7 Hz, 1H), 3.19 (d, J = 7.6 Hz, 2H), 2.32–2.14 (m, 2H), 1.69–1.56 (m, 2H), 1.44 (s, 11H) ppm; 13C NMR (CDCl3, 101 MHz): δ 172.13, 168.09, 156.03, 138.64, 137.65, 134.57, 133.30, 130.51, 129.67, 129.10, 128.34, 123.04, 116.00, 79.01, 63.22, 40.47, 30.66, 30.00, 28.44, 23.32 ppm; ESI-HRMS (positive mode): m/z 486.1386/488.1370 (81Br) [M + H]+, M calcd for C24H29BrN3O3 486.1387.
7-Bromo-1,3,4,5-tetrahydro-4-[4-(N-Boc-amino)butyl]-5-phenyl-2H-1,4-benzodiazepin-2-one (14). To a solution of 13 (800 mg, 1.65 mmol) and NaBH3CN (155.4 mg, 2.48 mmol) in MeOH (10 mL), AcOH (500 µL, 8.24 mmol) was added, and the mixture was stirred at room temperature for 2.5 h until complete reduction of the imine as assessed by TLC. Afterwards, the solvent was removed under low pressure, the crude was dissolved in EtOAc (50 mL) and washed with aq. NaHCO3(sat) (2 × 20 mL). The organic layer was dried over anh. MgSO4, filtered and the solvent removed under low pressure. The title compound (14) was obtained as a pale yellow solid (799 mg, 99%).
TLC (hexanes/EtOAc 1:1): Rf = 0.52; IR (ATR, solid): 3290, 2967, 2929, 2866, 1663, 1479, 1365, 1245, 1159, 817, 700 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.99 and 7.67 (s, 1H), 7.45–7.27 (m, 5H), 7.13 and 6.76 (d, J = 2.3 Hz, 1H), 6.89 and 6.80 (d, 8.4 Hz, 1H), 5.34 and 5.24 (s, 1H), 4.56 (br s, 1H), 3.57 and 3.31 (t, J = 7.2 Hz, 1H), 3.14–3.05 (m, 2H), 1.94–1.44 (m, 6H), 1.42 (s, 9H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 175.02, 156.00, 142.23, 139.97, 136.56, 132.23, 128.64, 128.45, 127.19, 122.69, 117.53, 79.06, 63.98, 59.94, 40.29, 31.65, 29.93, 28.41, 23.50 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 174.27, 156.00, 142.33, 139.97, 135.88, 131.35, 128.57, 128.20, 127.54, 123.32, 118.88, 79.06, 58.97, 56.14, 40.29 31.65, 30.04, 28.42, 23.21 ppm; ESI-HRMS (positive mode): m/z 488.1559/490.1555 (81Br) [M + H]+, M calcd for C24H31BrN3O3 488.1543.
7-Bromo-1,3,4,5-tetrahydro-4-(4-aminobutyl)-5-phenyl-2H-1,4-benzodiazepin-2-one dihydrochloride (15). In a 10 mL round-bottom flask, 14 (100 mg, 0.21 mmol) was dissolved in 4 M HCl (in dioxane, 5 mL) and the reaction mixture was left stirring for 2.5 h at room temperature. Afterwards the solvent was removed under low pressure to afford the title product (15) as a pale yellow solid (94 mg, 99%).
TLC (DCM/MeOH 9:1): Rf = 0.20; IR (ATR, solid): 3474, 3199, 2911, 2711, 2524, 1704, 1590, 1568, 1479, 1448, 1375, 1315, 1270, 1245, 1131, 998, 916, 827, 697 cm−1; 1H NMR (CD3OD, 400 MHz): δ 7.75 and 7.73 (s, 1H), 7.69–7.55 (m, 4H), 7.44–7.38 (m, 1H), 7.34–7.29 (m, 2H), 7.19 (d, J = 8.5 Hz) and 7.09 (d, J = 9.2 Hz, 1H), 6.02 and 5.71 (s, 1H), 4.12 (dd, J = 9.0, 4.6 Hz) and 3.86 (dd, J = 10.5, 3.1 Hz, 1H), 2.99–2.89 (m, 2H), 2.32–2.10 (m, 1H), 2.00–1.80 (m, 1H), 1.71 (h, J = 7.3 Hz, 2H), 1.58–1.34 (m, 2H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 166.45, 138.28, 135.43, 135.17, 133.98, 130.67, 130.20, 129.58, 130.20, 129.58, 128.34, 125.45, 120.24, 60.83, 57.09, 40.24, 28.13, 27.96, 23.88 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 167.82, 136.80, 135.87, 134.92, 133.34, 131.13, 130.35, 129.58, 128.34, 126.16, 119.84, 63.60, 57.38, 40.24, 29.17, 28.05, 23.66 ppm; ESI-HRMS (positive mode): m/z 388.1016/390.1000 (81Br) [M + H]+, M calcd for C19H23BrN3O 388.1019.
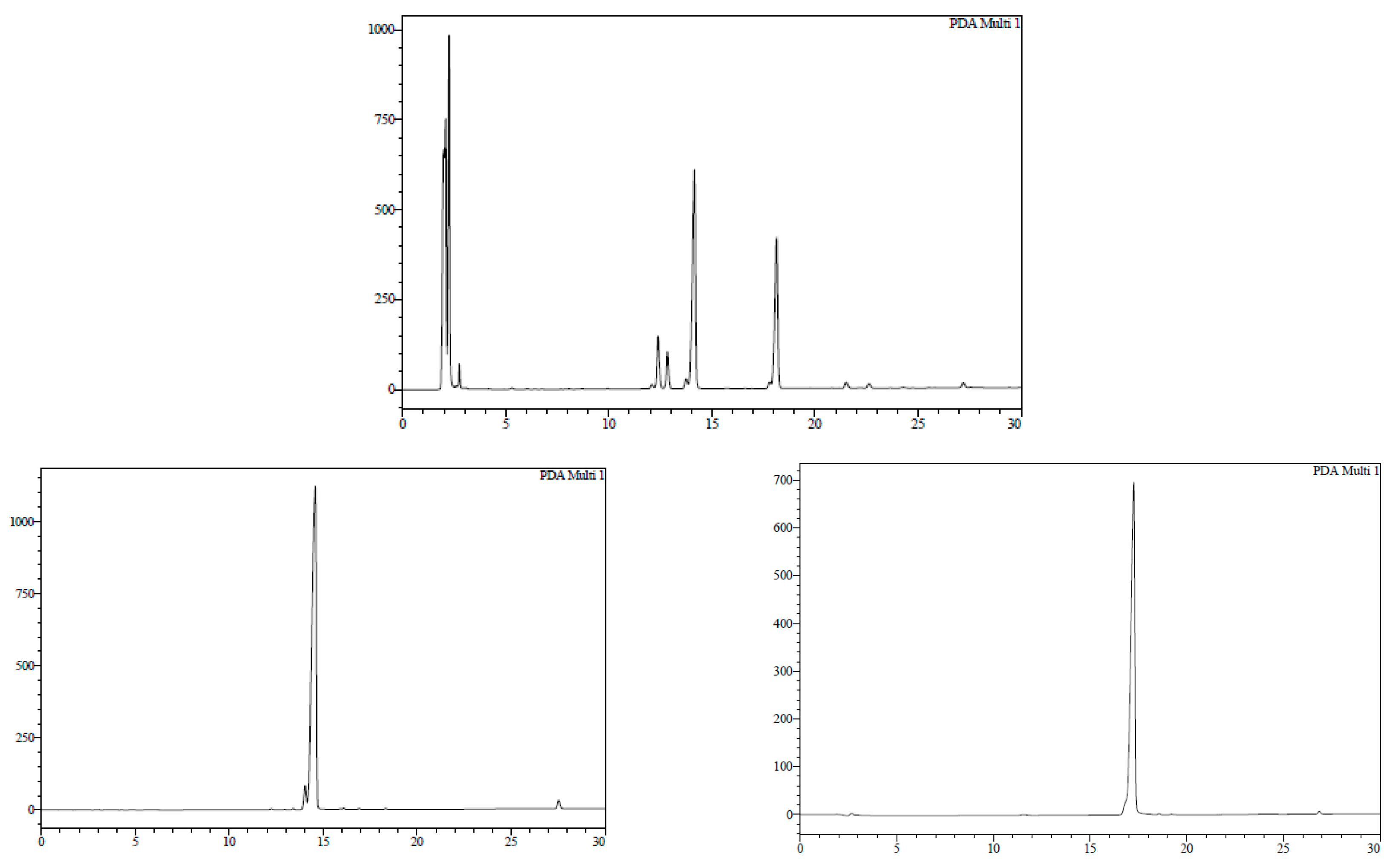
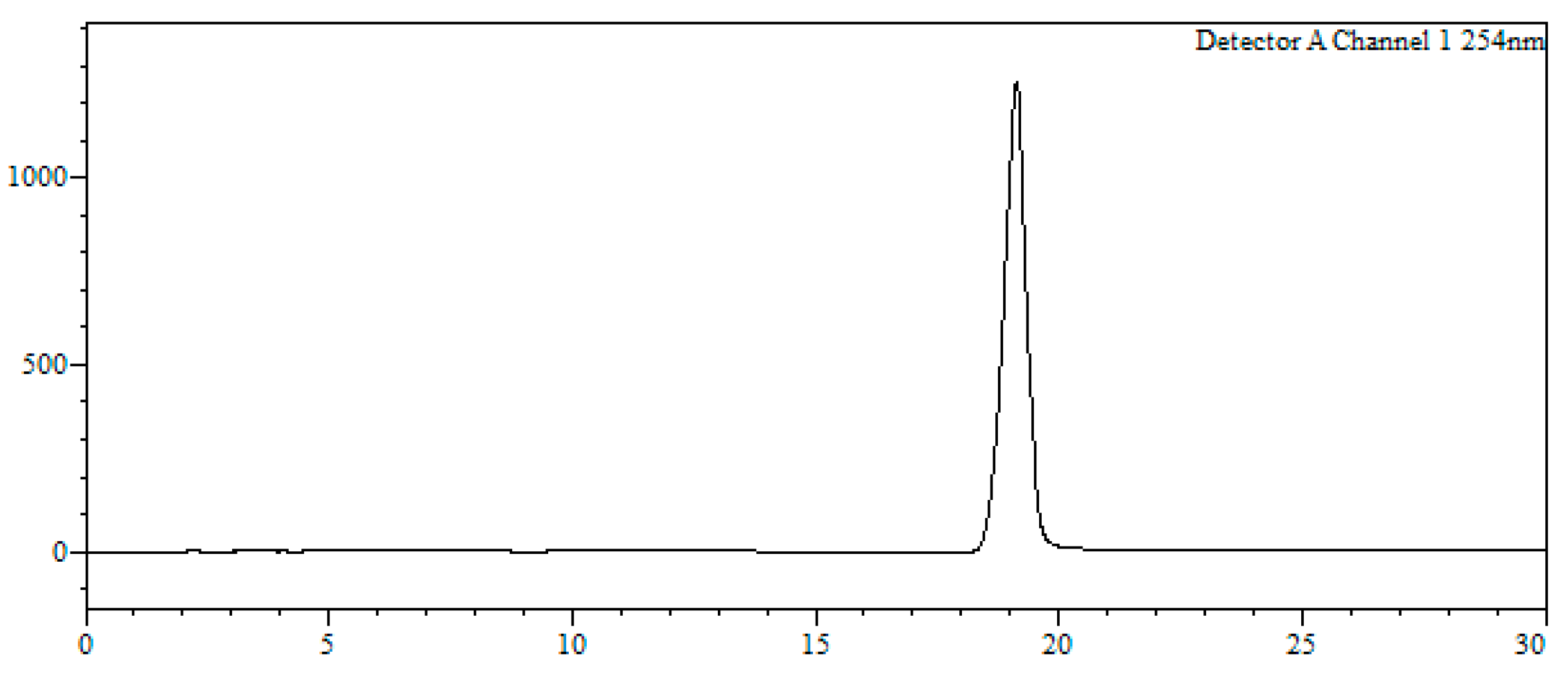
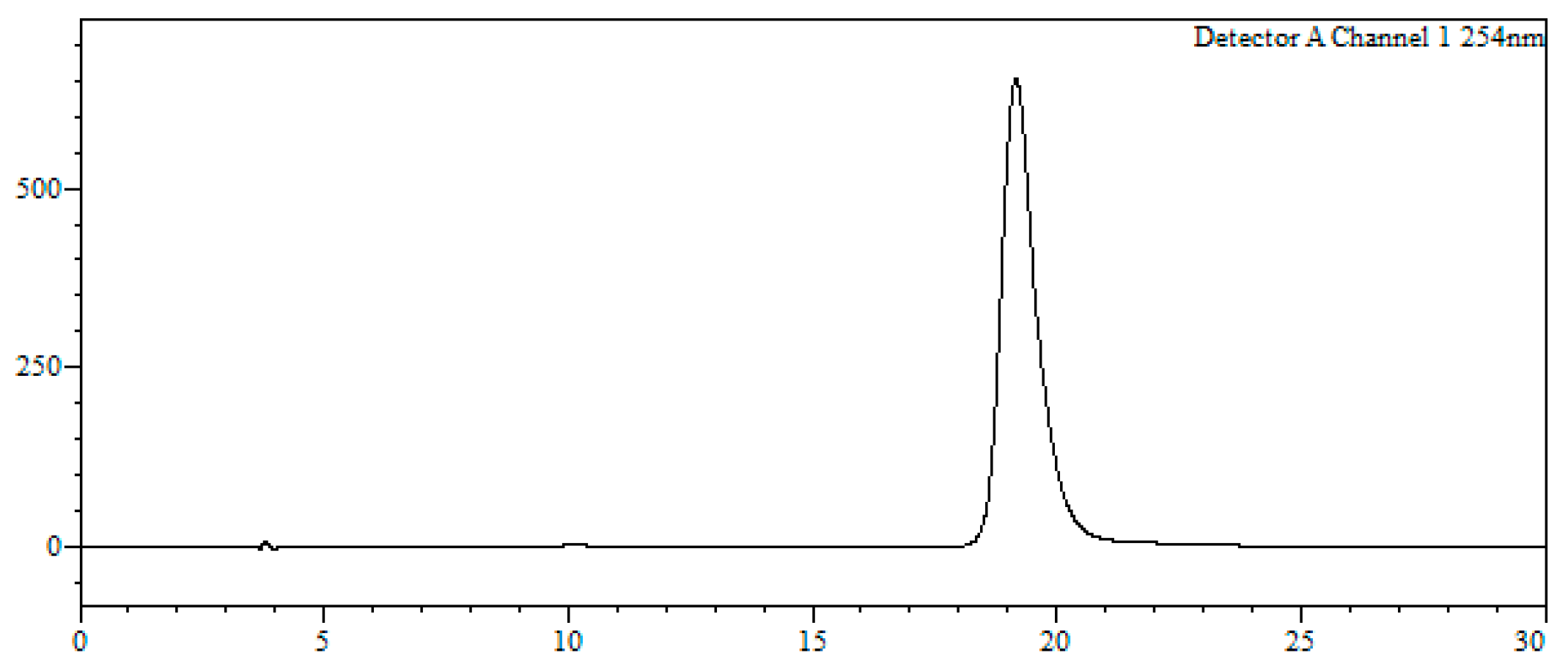
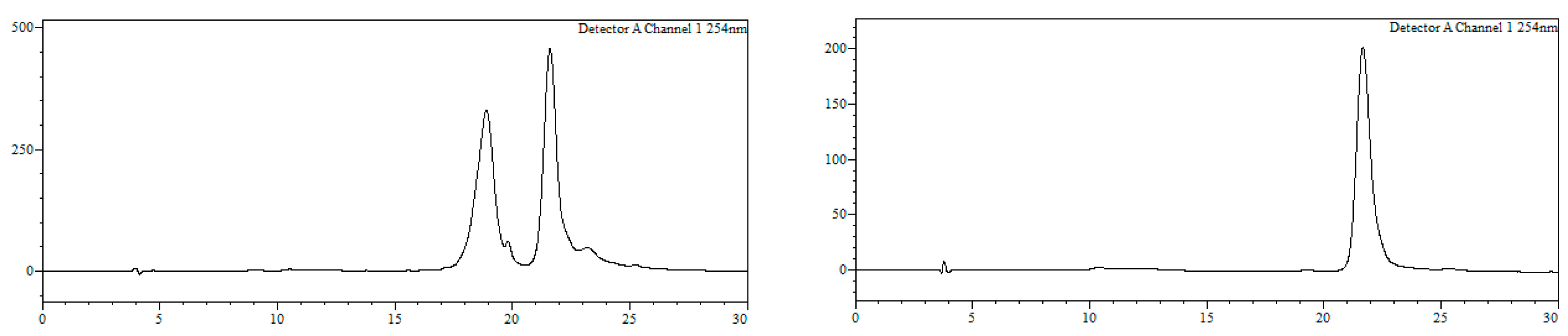
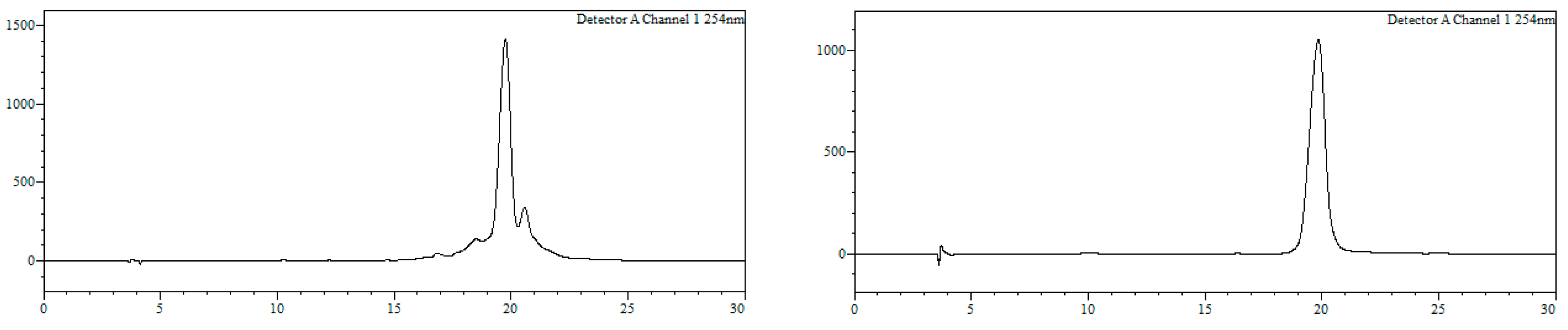
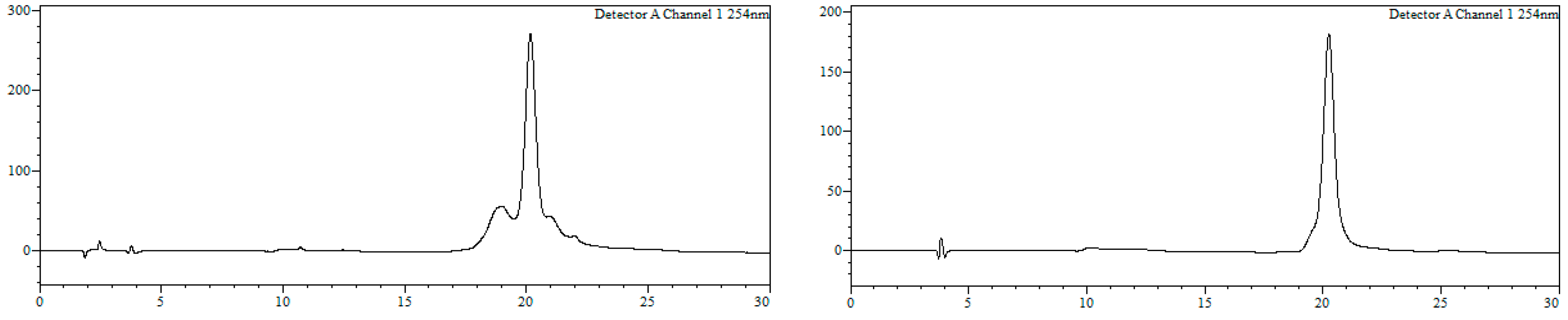
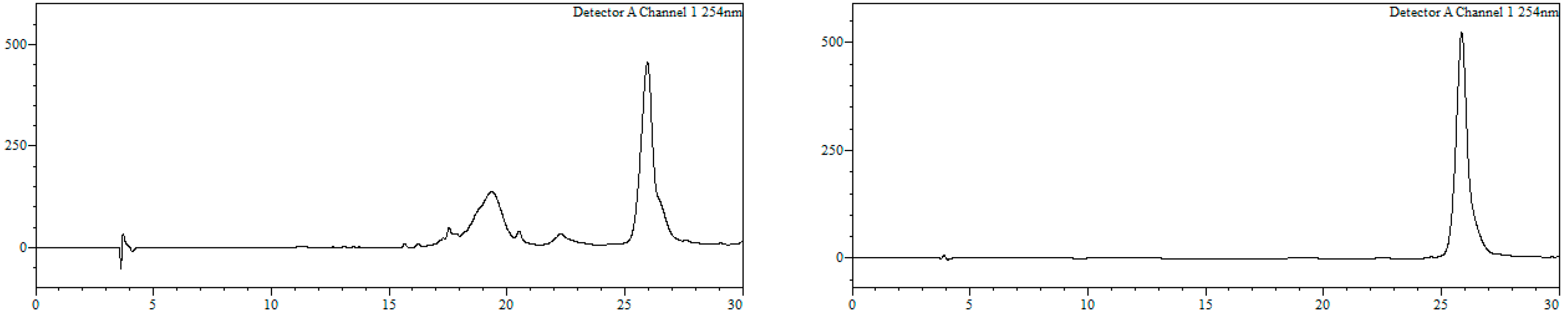
7-Bromo-1,3,4,5-tetrahydro-4-{4-[N-[(4E)-4,6-heptadienoyl]-amino]butyl}-5-phenyl-2H-1,4-benzodiazepin-2-one (16). N,N’-Diisopropylcarbodiimide (61 µL, 0.39 mmol) and N-methylmorpholine (2.6 µL, 0.03 mmol) were added to a solution of (4E)-4,6-heptadienoic acid (49 mg, 0.39 mmol) in HPLC-quality acetonitrile (1 mL), and the mixture was left stirring for 10 min. Afterwards, 15 (50 mg, 0.13 mmol) and an additional amount of N-methylmorpholine (2.6 µL, 0.03 mmol) were poured onto the mixture. Additional amounts of N-methylmorpholine (2.6 µL, 0.03 mmol) were added after 20 and 40 min, respectively, and the solution was stirred for up to 2 h at room temperature. Subsequently, acetonitrile was added (4 mL), and the crude was filtered using a hydrophilic PTFE syringe filter (0.22 µm), purified by RP-HPLC (Jupiter Proteo C18 (10 μm, 250 nm, 250 × 10 mm) from Phenomenex, solvent A: H2O 0.1% formic acid; solvent B: ACN 0.1% formic acid, linear gradient 40 → 80% B, 3 mL/min, detection wavelength 254 nm) and lyophilized. The title compound (16) was obtained as a white solid (10.7 mg, 20%).
TLC (EtOAc/MeOH 9:1): Rf = 0.10; IR (ATR, solid): 2980, 2905, 2355, 1730, 1658, 1599, 1571, 1497, 1425, 1437, 1391, 1254, 1248, 1173, 1158, 1071, 1043, 757, 744, 694, 533 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.65–7.53 (m, 4H), 7.42–7.28 (m, 4H), 7.16 and 7.04 (d, J = 8.5 Hz, 1H), 7.47 and 6.97 (d, J = 2.2 Hz, 1H), 6.35–6.21 (m, 1H), 6.13–6.02 (m, 1H), 5.80 and 5.68 (s, 1H), 5.70–5.61 (m, 1H), 5.06 (dd, J = 16.9, 1.9 Hz, 1H), 4.92 (dt, J = 10.2, 2.2 Hz, 1H), 4.17–3.77 (m, 1H), 3.72 (dd, J = 10.1, 3.7 Hz, 1H), 3.23–3.07 (m, 2H), 2.39–2.31 (m, 2H), 2.27–2.12 (m, 3H), 1.81–1.63 (m, 1H), 1.55–1.31 (m, 4H) ppm; 13C NMR (CDCl3, 101 MHz): δ 173.99, 173.93, 136.91, 135.65, 133.59, 132.93, 132.50, 131.86, 129.62, 129.28, 128.67, 127.77, 126.88, 123.98, 118.74, 114.45, 59.34, 55.99, 38.24, 35.25, 29.37, 28.76, 28.34, 22.80 ppm; ESI-HRMS (positive mode): m/z 496.1584/498.1573 (81Br) [M + H]+, M calcd. for C26H30BrN3O3 496.1594.
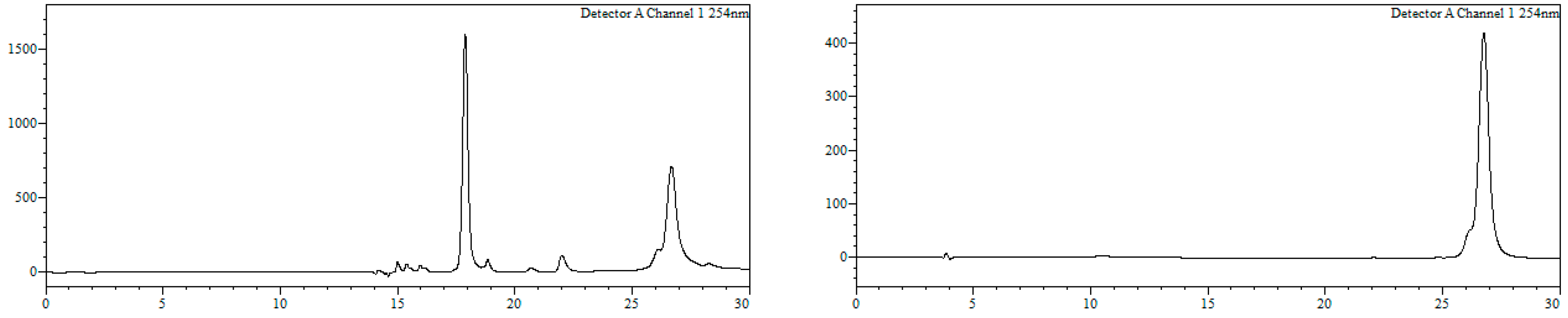
RP-HPLC analysis conditions (
Figure 3): Jupiter Proteo C
18 (4 μm, 254 nm, 250 × 4.6 mm) from Phenomenex, 30 → 70% B in 30 min, 1 mL/min, t
R = 14.3 min and 18.1 min; purification conditions: Jupiter Proteo C
18 (10 μm, 254 nm, 250 × 10 mm) from Phenomenex, 40 → 80% B in 30 min, 3 mL/min (t
R = 7.5 min and 8.9 min).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}