Improving the Production of Salt-Tolerant Glutaminase by Integrating Multiple Copies of Mglu into the Protease and 16S rDNA Genes of Bacillus subtilis 168

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
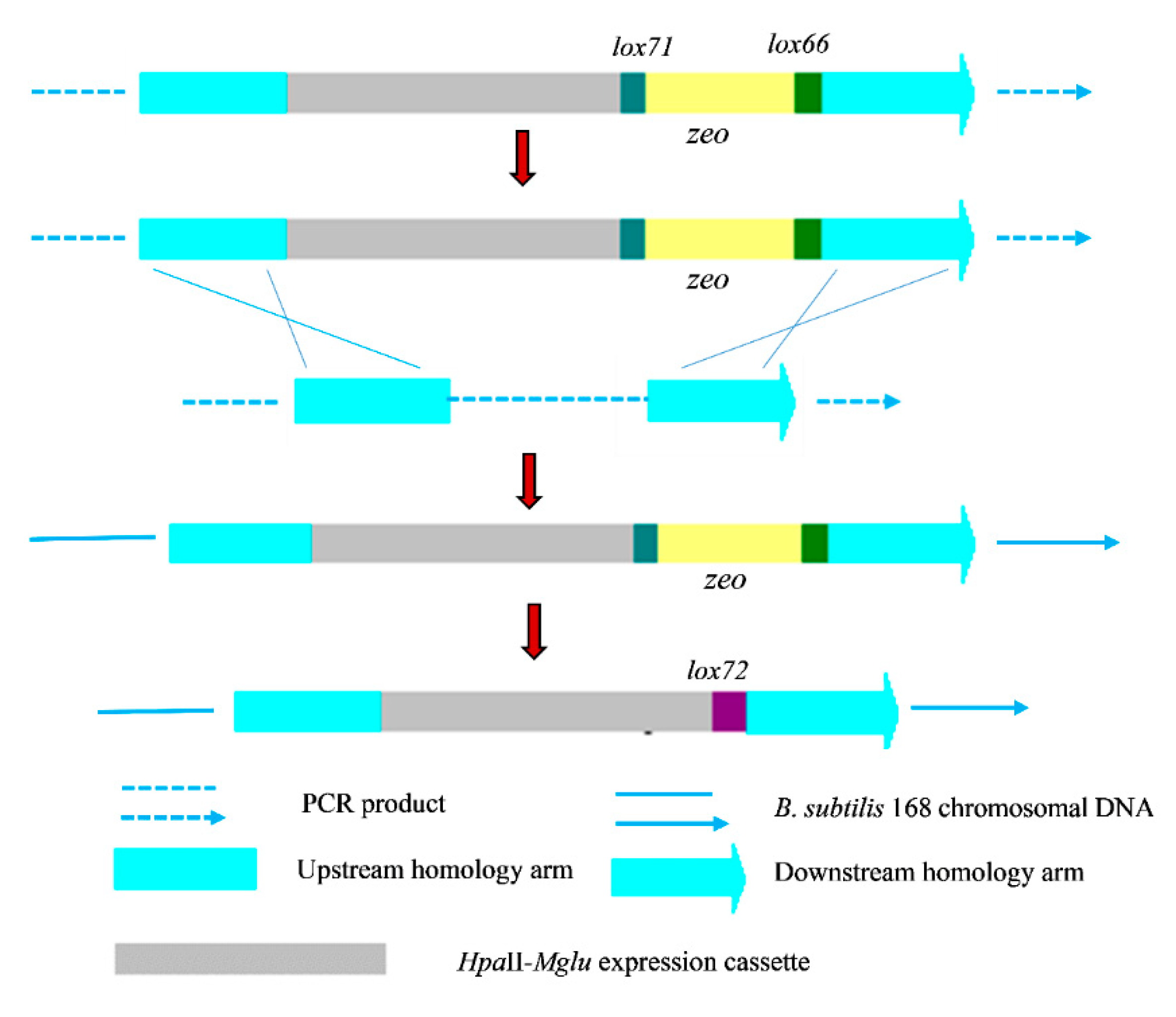
2.1. Construction of a Recombinant B. subtilis 168 Strain with Multiple Copies of Integrated Mglu
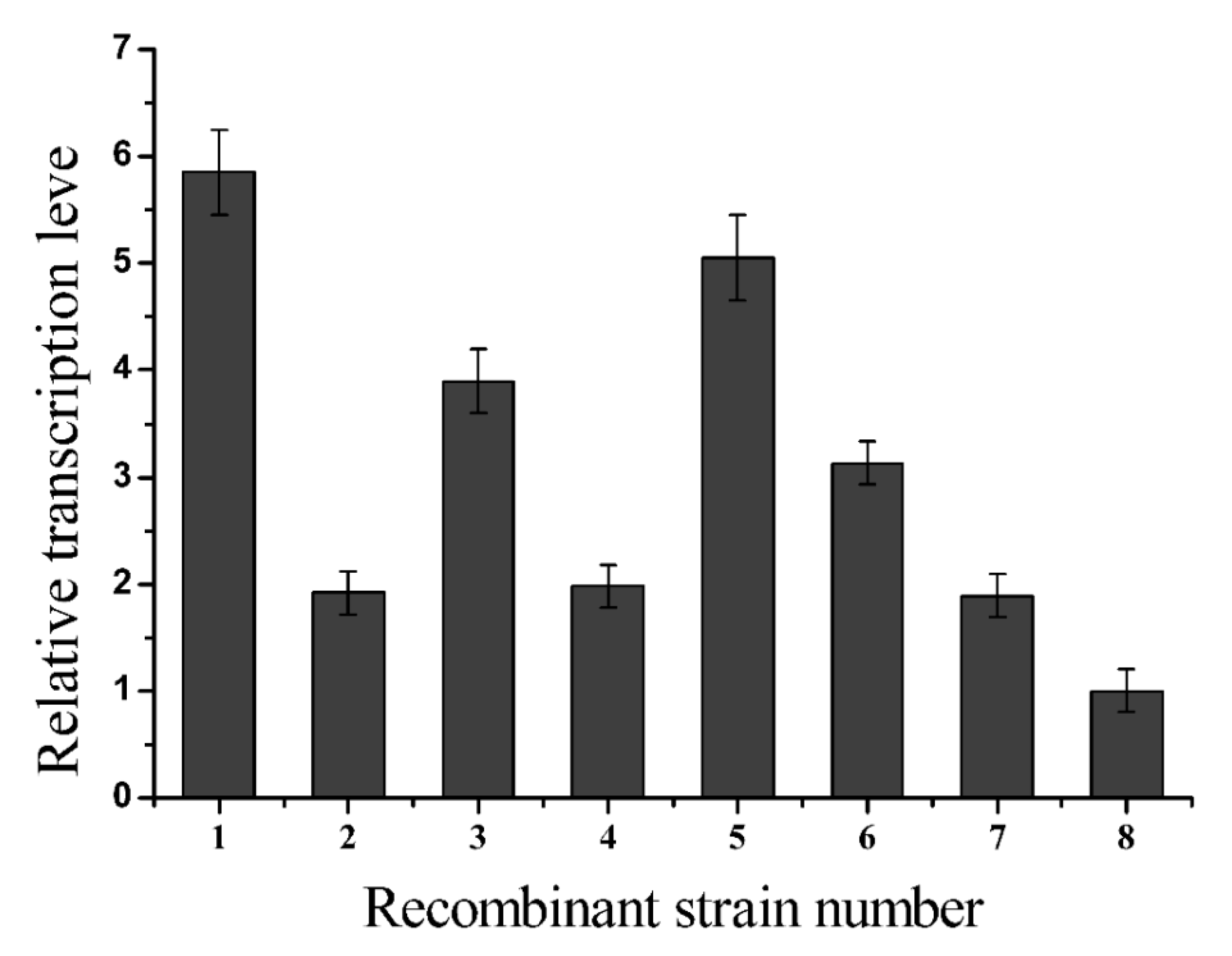
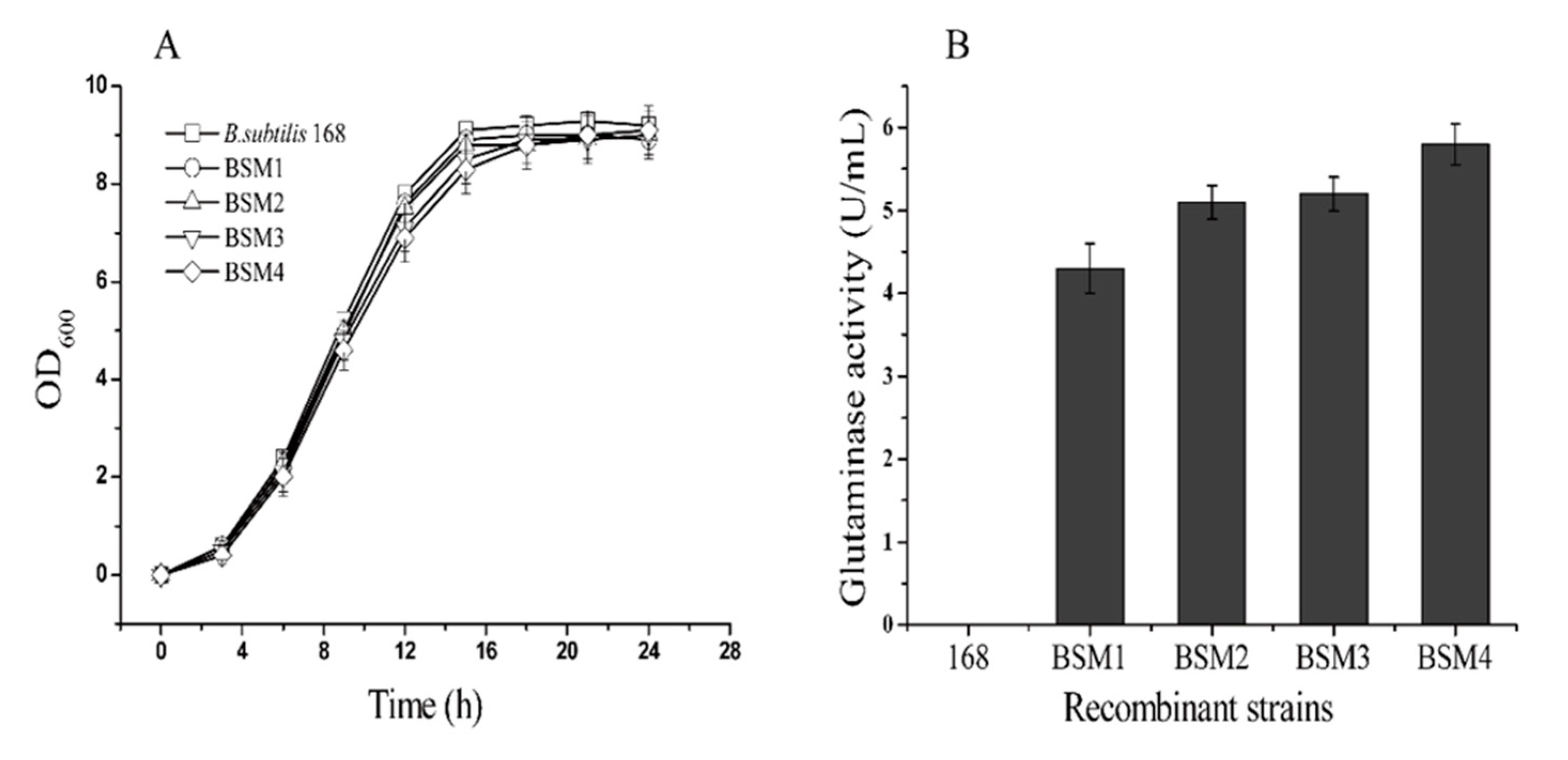
2.2. L-glutamate Enzyme Activity in the Recombinant Strains
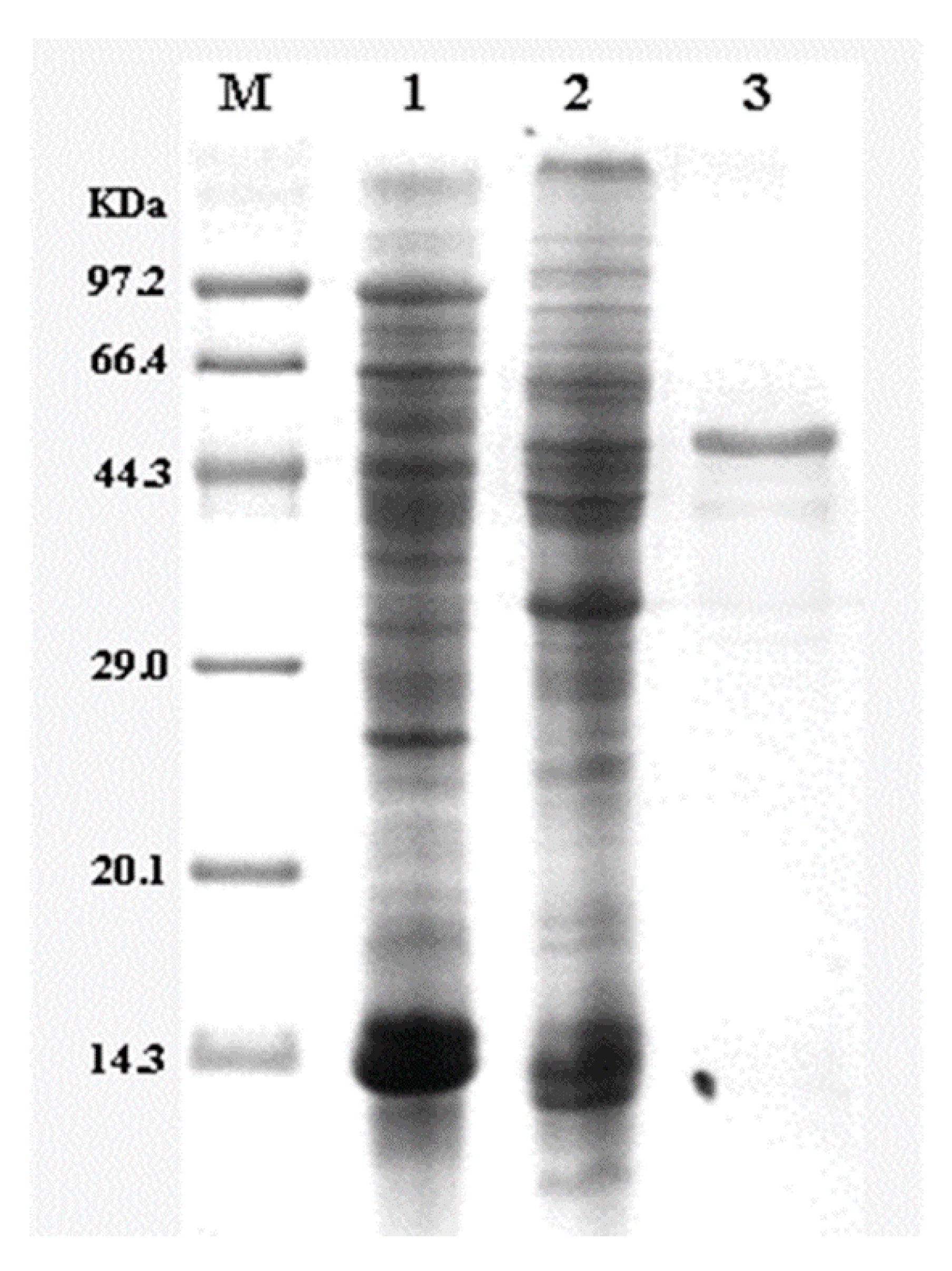
2.3. Purification of Glutaminase
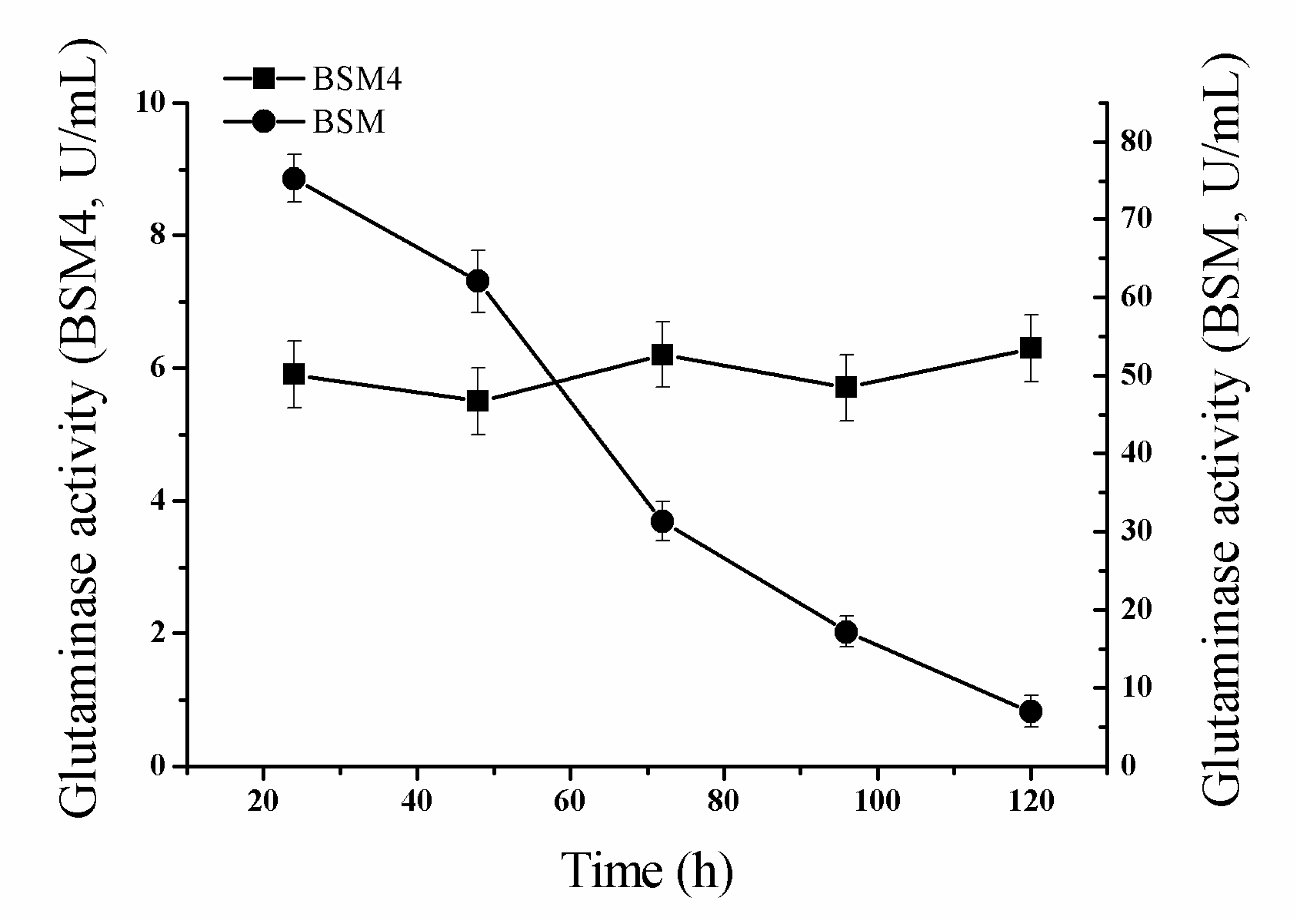
2.4. Comparison of the Genetic Stability of the Recombinant Strains
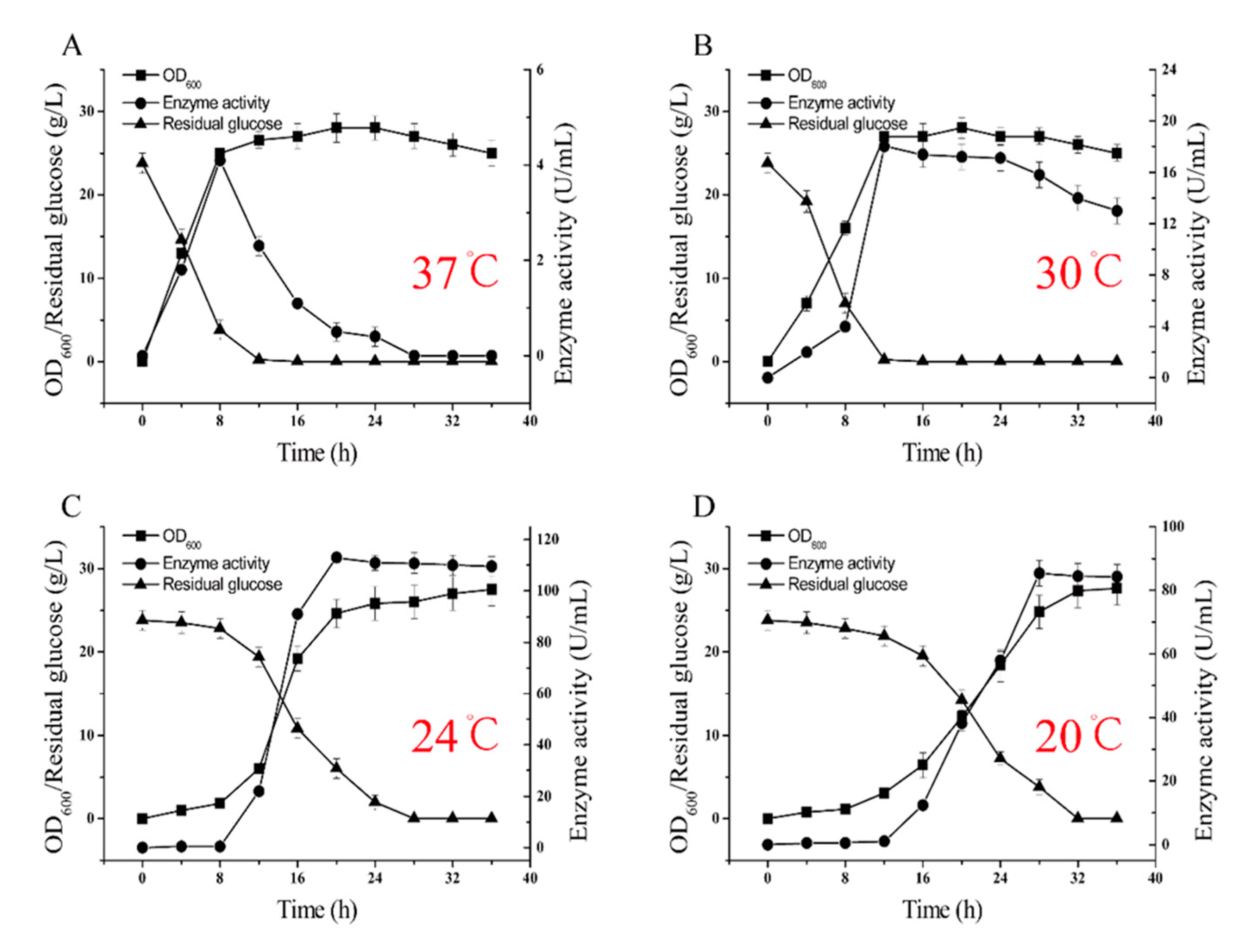
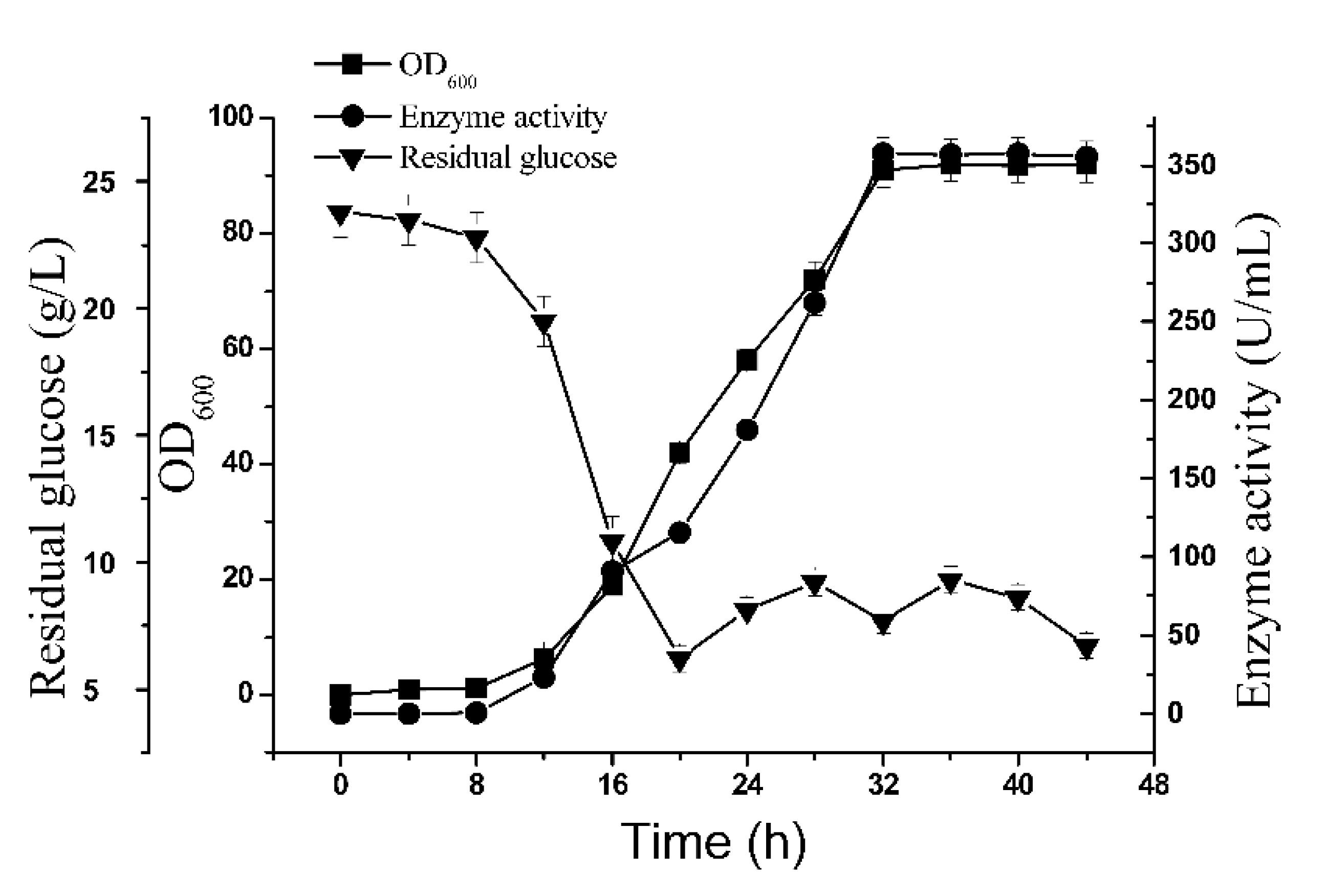
2.5. 5-L Bioreactor Fermentation Analysis of BSM4
3. Materials and Methods
3.1. Gene Source and Materials
3.2. Strains, Plasmids, and Primers
3.3. PCR Amplification of DNA Fragments
3.4. Construction of the Recombinant Plasmid and Integrated Fragments
3.5. Preparation of RNA and cDNA Synthesis
3.6. Analysis of the Relative Transcription Levels of Recombinant Glutaminase
3.7. Transformation and Selection of the Recombinant Strains
3.8. Enzyme Activity Assay
3.9. Preliminary Determination of Enzyme Activity
3.10. Purification and SDS-PAGE Analysis of Glutaminase
3.11. Enzyme Stability of the Recombinant Strains
3.12. Glutaminase Fermentation in a 5-L Bioreactor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wu, G.Y.; Thompson, J.R.; Baracos, V.E. Glutamine metabolism in skeletal muscles from the broiler chick (Gallus domesticus) and the laboratory rat (Rattus norvegicus). Biochem. J. 1991, 274, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Curthoys, N.P.; Kuhlenschmidt, T.; Godfrey, S.S. Regulation of renal ammoniagenesis. Arch. Biochem. Biophys. 1976, 174, 82–89. [Google Scholar] [CrossRef]
- Glew, R.H.; Balis, J.U.; Shelley, S.; Kuhlenschmidt, T.; Jaffe, R. Isolation, characterization and localization of a 45,000 molecular weight, soluble glycoprotein from the lung in pulmonary alveolar proteinosis. Biochim. Biophys. Acta 1981, 670, 101–109. [Google Scholar] [CrossRef]
- Wakayama, M.; Nagano, Y.; Renu, N.; Kawamura, T.; Sakai, K.; Moriguchi, M. Molecular cloning and determination of the nucleotide sequence of a gene encoding salt-tolerant glutaminase from Micrococcus luteus K-3. J. Ferment. Bioeng. 1996, 82, 592–597. [Google Scholar] [CrossRef]
- Yoshimune, K.; Shirakihara, Y.; Wakayama, M.; Yumoto, I. Crystal structure of salt-tolerant glutaminase from Micrococcus luteus K-3 in the presence and absence of its product L-glutamate and its activator Tris. FEBS J. 2010, 277, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Masuo, N.; Ito, K.; Yoshimune, K.; Hoshino, M.; Matsushima, K.; Koyama, Y.; Moriguchi, M. Molecular cloning, overexpression, and purification of Micrococcus luteus K-3-type glutaminase from Aspergillus oryzae RIB40. Protein Expr. Purif. 2004, 38, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Masuo, N.; Yoshimune, K.; Ito, K.; Matsushima, K.; Koyama, Y.; Moriguchi, M. Micrococcus luteus K-3-type glutaminase from Aspergillus oryzae RIB40 is salt-tolerant. J. Biosci. Bioeng. 2005, 100, 576–578. [Google Scholar] [CrossRef]
- Kumar, L.; Singh, B.; Adhikari, D.K.; Mukherjee, J.; Ghosh, D. A temperature and salt-tolerant L-glutaminase from gangotri region of uttarakhand himalaya: Enzyme purification and characterization. Appl. Biochem. Biotechnol. 2012, 166, 1723–1735. [Google Scholar] [CrossRef]
- Jeon, J.M.; Lee, H.I.; Han, S.H.; Chang, C.S.; So, J.S. Partial purification and characterization of glutaminase from Lactobacillus reuteri KCTC3594. Appl. Biochem. Biotechnol. 2010, 162, 146–154. [Google Scholar] [CrossRef]
- Sato, I.; Kobayashi, H.; Hanya, Y.; Abe, K.; Murakami, S.; Scorzetti, G.; Fell, J.W. Cryptococcus nodaensis sp nov, a yeast isolated from soil in Japan that produces a salt-tolerant and thermostable glutaminase. J. Indust. Microbiol. Biotechnol. 1999, 22, 127–132. [Google Scholar] [CrossRef]
- TOMITA, K.; YANO, T.; KITAGATA, T. Action of Glutaminase in a Model System of a Soy Sauce Fermentation. J. Agric. Chem. Soc. Japn. 1989, 53, 1873–1878. [Google Scholar]
- Sietske, A.D.B.; Diderichsen, B. On the safety of Bacillus subtilis and B. amyloliquefaciens: A review. Appl. Microbiol. Biotechnol. 1991, 36, 1–4. [Google Scholar] [CrossRef]
- Negri, A.; Potocki, W.; Iwanicki, A.; Obuchowski, M.; Hinc, K. Expression and display of Clostridium difficile protein FliD on the surface of Bacillus subtilis spores. J. Med. Microbiol. 2013, 62, 1379–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.; Zhang, W.; Feng, Y.; Jiang, B.; Zhou, L. Recent advances on applications and biotechnological production of D-psicose. Appl. Microbiol. Biotechnol. 2012, 94, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.R.; Wipat, A. Sequencing and functional analysis of the genome of Bacillus subtilis strain 168. FEBS Lett. 1996, 389, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Zhao, Y.; Zhang, H.; Liu, Z.; Lu, Z. Improving methyl parathion hydrolase to enhance its chlorpyrifos-hydrolysing efficiency. Lett. Appl. Microbiol. 2014, 58, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, D. 5-Genetic transformation in Bacillus subtilis. Bacillus Subtilis 1982, 147–178. [Google Scholar]
- Young, M. Gene amplification in Bacillus subtilis. J. Gen. Microbiol. 1984, 130, 1613–1621. [Google Scholar] [CrossRef]
- Albertini, A.M.; Galizzi, A. Amplification of a chromosomal region in Bacillus subtilis. J. Bacteriol. 1985, 162, 1203–1211. [Google Scholar]
- He, W.; Mu, W.; Jiang, B.; Yan, X.; Zhang, T. Food-grade expression of d-Psicose 3-epimerase with tandem repeat genes in Bacillus subtilis. J. Agric. Food Chem. 2016, 64, 5701–5707. [Google Scholar] [CrossRef]
- Sargent, M.G.; Bennett, M.F. Amplification of a major membrane-bound DNA sequence of Bacillus subtilis. J. Bacteriol. 1985, 161, 589–595. [Google Scholar] [PubMed]
- Niaudet, B.; Jannière, L.; Ehrlich, S.D. Recombination between repeated DNA sequences occurs more often in plasmids than in the chromosome of Bacillus subtilis. Mol. Gen. Genet. Mgg 1984, 197, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, S.; Hranueli, D.; Young, M. Evidence that recombination between reiterated sequences in the Bacillus subtilis chromosome does not occur via unequal crossing over. Biochimie 1992, 74, 713–721. [Google Scholar] [CrossRef]
- Petit, M.A.; Mesas, J.M.; Noirot, P.; Morel-Deville, F.; Ehrlich, S.D. Induction of DNA amplification in the Bacillus subtilis chromosome. EMBO J. 1992, 11, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Garber, R.C.; Turgeon, B.G.; Selker, E.U.; Yoder, O.C. Organization of ribosomal RNA genes in the fungus Cochliobolus heterostrophus. Curr. Genet. 1988, 14, 573–582. [Google Scholar] [CrossRef]
- Udem, S.A.; Warner, J.R. Ribosomal RNA synthesis in Saccharomyces cerevisiae. J. Mol. Biol. 1972, 65, 227–242. [Google Scholar] [CrossRef]
- Tran, L.; Wu, X.C.; Wong, S.L. Cloning and expression of a novel protease gene encoding an extracellular neutral protease from Bacillus subtilis. J. Bacteriol. 1991, 173, 6364–6372. [Google Scholar] [CrossRef]
- Yang, M.Y.; Ferrari, E.; Henner, D.J. Cloning of the neutral protease gene of Bacillus subtilis and the use of the clone to create an in vitro-derived deletion mutant. J. Bacteriol. 1984, 160, 15–21. [Google Scholar]
- Aoki, T.; Noguchi, N.; Sasatsu, M.; Kono, M. Complete nucleotide sequence of pTZ12, a chloramphenicol-resistance plasmid of Bacillus subtilis. Gene 1987, 51, 107–111. [Google Scholar] [CrossRef]
- Moriguchi, M.; Sakai, K.; Tateyama, R.; Furuta, Y.; Wakayama, M. Isolation and characterization of salt-tolerant glutaminases from marine Micrococcus luteus K-3. J. Ferment. Bioeng. 1994, 77, 621–625. [Google Scholar] [CrossRef]
- Jiang, W.; Fang, B. Construction of a tunable multi-enzyme-coordinate expression system for biosynthesis of chiral drug intermediates. Sci. Rep. 2016, 6, 30462. [Google Scholar] [CrossRef] [Green Version]
- Sathish, T.; Prakasham, R.S. Enrichment of glutaminase production byBacillus subtilisRSP-GLU in submerged cultivation based on neural networkâ genetic algorithm approach. J. Chem. Technol. Biotechnol. 2010, 85, 50–58. [Google Scholar] [CrossRef]
- Sathish, T.; Lakshmi, G.S.; Rao Ch, S.; Brahmaiah, P.; Prakasham, R.S. Mixture design as first step for improved glutaminase production in solid-state fermentation by isolated Bacillus. Lett. Appl. Microbiol. 2008, 47, 256–262. [Google Scholar] [CrossRef]
- Wakayama, M.; Yamagata, T.; Kamemura, A.; Bootim, N.; Yano, S.; Tachiki, T.; Yoshimune, K.; Moriguchi, M. Characterization of salt-tolerant glutaminase from Stenotrophomonas maltophilia NYW-81 and its application in Japanese soy sauce fermentation. J. Ind. Microbiol. Biotechnol. 2005, 32, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Shevchuk, N.A.; Bryksin, A.V.; Nusinovich, Y.A.; Cabello, F.C.; Sutherland, M.; Ladisch, S. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucl. Acids Res. 2004, 32, e19. [Google Scholar] [CrossRef]
- Teramoto, H.; Inui, M.; Yukawa, H. OxyR acts as a transcriptional repressor of hydrogen peroxide-inducible antioxidant genes in Corynebacterium glutamicum R. FEBS J. 2013, 280, 3298–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostopoulos, C.; Spizizen, J. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 1961, 81, 741–746. [Google Scholar]
- Hastie, A.R.; Pruitt, S.C. Yeast two-hybrid interaction partner Screening throughin vivoCre-mediated Binary Interaction Tag generation. Nucl Acids Res. 2007, 35, e141. [Google Scholar] [CrossRef]
Sample Availability: Samples of the plasmids and strains are available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Xu, Z.; Liu, S.; Qian, K.; Xu, M.; Yang, T.; Xu, J.; Rao, Z. Improving the Production of Salt-Tolerant Glutaminase by Integrating Multiple Copies of Mglu into the Protease and 16S rDNA Genes of Bacillus subtilis 168. Molecules 2019, 24, 592. https://doi.org/10.3390/molecules24030592
Zhang X, Xu Z, Liu S, Qian K, Xu M, Yang T, Xu J, Rao Z. Improving the Production of Salt-Tolerant Glutaminase by Integrating Multiple Copies of Mglu into the Protease and 16S rDNA Genes of Bacillus subtilis 168. Molecules. 2019; 24(3):592. https://doi.org/10.3390/molecules24030592
Chicago/Turabian StyleZhang, Xian, Zhaoyang Xu, Song Liu, Kai Qian, Meijuan Xu, Taowei Yang, Jianzhong Xu, and Zhiming Rao. 2019. "Improving the Production of Salt-Tolerant Glutaminase by Integrating Multiple Copies of Mglu into the Protease and 16S rDNA Genes of Bacillus subtilis 168" Molecules 24, no. 3: 592. https://doi.org/10.3390/molecules24030592
APA StyleZhang, X., Xu, Z., Liu, S., Qian, K., Xu, M., Yang, T., Xu, J., & Rao, Z. (2019). Improving the Production of Salt-Tolerant Glutaminase by Integrating Multiple Copies of Mglu into the Protease and 16S rDNA Genes of Bacillus subtilis 168. Molecules, 24(3), 592. https://doi.org/10.3390/molecules24030592