
Novel Therapeutics for Epstein–Barr Virus



Abstract
:1. Introduction
2. Why Is There No Antiviral Drug Approved for the Treatment of EBV Infections?
3. Medical Need for Anti-EBV Therapeutics Targeting Lytic Replication
4. Antivirals Against EBV Evaluated in The Clinic
4.1. Nucleoside Analogues (Acyclovir, Valacyclovir, Ganciclovir, and Valganciclovir)
4.2. Nucleotide Analogues
4.3. Pyrophosphate Analogues
5. Anti-EBV Compounds Under Investigation
5.1. Inhibitors of EBV Protein Kinase BGLF4
5.2. Inhibitors of EBV DNA Polymerase
5.3. Inhibitors of EBV Nuclear Antigen 1 (EBNA1)
6. Cellular Targets
7. Medicinal Plants
8. Use of Antivirals in Lytic Induction Therapy
9. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Katz, B.Z.; Shiraishi, Y.; Mears, C.J.; Binns, H.J.; Taylor, R. Chronic fatigue syndrome after infectious mononucleosis in adolescents. Pediatrics 2009, 124, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Ryan, J.L.; Morgan, D.R.; Dominguez, R.L.; Thorne, L.B.; Elmore, S.H.; Mino-Kenudson, M.; Lauwers, G.Y.; Booker, J.K.; Gulley, M.L. High levels of Epstein–Barr virus DNA in latently infected gastric adenocarcinoma. Lab. Invest. 2009, 89, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; El Guindy, A.; Countryman, J.; Ye, J.; Gradoville, L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 2007, 97, 81–109. [Google Scholar] [PubMed]
- Heslop, H.E. How I treat EBV lymphoproliferation. Blood 2009, 114, 4002–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein–Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein–Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef]
- Keith, K.A.; Hartline, C.B.; Bowlin, T.L.; Prichard, M.N. A standardized approach to the evaluation of antivirals against DNA viruses: Polyomaviruses and lymphotropic herpesviruses. Antiviral Res. 2018, 159, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Duraffour, S.; Naesens, L.; Krecmerova, M.; Van den, O.J.; Snoeck, R.; Andrei, G. Evaluation of novel acyclic nucleoside phosphonates against human and animal gammaherpesviruses revealed an altered metabolism of cyclic prodrugs upon Epstein–Barr virus reactivation in P3HR-1 cells. J. Virol. 2013, 87, 12422–12432. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Duraffour, S.; Topalis, D.; Snoeck, R.; Andrei, G. Spectrum of activity and mechanisms of resistance of various nucleoside derivatives against gammaherpesviruses. Antimicrob. Agents Chemother. 2014, 58, 7312–7323. [Google Scholar] [CrossRef] [PubMed]
- Gershburg, E.; Pagano, J.S. Epstein–Barr virus infections: Prospects for treatment. J. Antimicrob. Chemother. 2005, 56, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Ernberg, I.; Andersson, J. Acyclovir efficiently inhibits oropharyngeal excretion of Epstein–Barr virus in patients with acute infectious mononucleosis. J. Gen. Virol. 1986, 67, 2267–2272. [Google Scholar] [CrossRef] [PubMed]
- Tynell, E.; Aurelius, E.; Brandell, A.; Julander, I.; Wood, M.; Yao, Q.Y.; Rickinson, A.; Akerlund, B.; Andersson, J. Acyclovir and prednisolone treatment of acute infectious mononucleosis: A multicenter, double-blind, placebo-controlled study. J. Infect. Dis. 1996, 174, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Negro, F. The paradox of Epstein–Barr virus-associated hepatitis. J. Hepatol. 2006, 44, 839–841. [Google Scholar] [CrossRef]
- Drebber, U.; Kasper, H.U.; Krupacz, J.; Haferkamp, K.; Kern, M.A.; Steffen, H.M.; Quasdorff, M.; Zur Hausen, A.; Odenthal, M.; Dienes, H.P. The role of Epstein–Barr virus in acute and chronic hepatitis. J. Hepatol. 2006, 44, 879–885. [Google Scholar] [CrossRef]
- Kimura, H.; Cohen, J.I. Chronic Active Epstein–Barr Virus Disease. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein–Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef]
- Wass, M.; Bauer, M.; Pfannes, R.; Lorenz, K.; Odparlik, A.; Muller, L.P.; Wickenhauser, C. Chronic active Epstein–Barr virus infection of T-cell type, systemic form in an African migrant: Case report and review of the literature on diagnostics standards and therapeutic options. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Petrara, M.R.; Giunco, S.; Serraino, D.; Dolcetti, R.; De, R.A. Post-transplant lymphoproliferative disorders: From epidemiology to pathogenesis-driven treatment. Cancer Lett. 2015, 369, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharnidharka, V.R.; Webster, A.C.; Martinez, O.M.; Preiksaitis, J.K.; Leblond, V.; Choquet, S. Post-transplant lymphoproliferative disorders. Nat. Rev. Dis. Primers 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Dierickx, D.; Habermann, T.M. Post-Transplantation Lymphoproliferative Disorders in Adults. N. Engl. J. Med. 2018, 378, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Morrow, R.; Huang, R.; Fixler, D. Persistent Epstein–Barr viral load in Epstein–Barr viral naive pediatric heart transplant recipients: Risk of late-onset post-transplant lymphoproliferative disease. World J. Transplant. 2016, 6, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Colombini, E.; Guzzo, I.; Morolli, F.; Longo, G.; Russo, C.; Lombardi, A.; Merli, P.; Barzon, L.; Murer, L.; Piga, S.; et al. Viral load of EBV DNAemia is a predictor of EBV-related post-transplant lymphoproliferative disorders in pediatric renal transplant recipients. Pediatr. Nephrol. 2017, 32, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Hocker, B.; Bohm, S.; Fickenscher, H.; Kusters, U.; Schnitzler, P.; Pohl, M.; John, U.; Kemper, M.J.; Fehrenbach, H.; Wigger, M.; et al. (Val-)Ganciclovir prophylaxis reduces Epstein–Barr virus primary infection in pediatric renal transplantation. Transpl. Int. 2012, 25, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Malouf, M.A.; Chhajed, P.N.; Hopkins, P.; Plit, M.; Turner, J.; Glanville, A.R. Anti-viral prophylaxis reduces the incidence of lymphoproliferative disease in lung transplant recipients. J. Heart Lung Transplant. 2002, 21, 547–554. [Google Scholar] [CrossRef]
- Hierro, L.; Diez-Dorado, R.; Diaz, C.; De, L.; Frauca, E.; Camarena, C.; Munoz-Bartolo, G.; Gonzalez, D.Z.; Lopez, S.M.; Jara, P. Efficacy and safety of valganciclovir in liver-transplanted children infected with Epstein–Barr virus. Liver Transpl. 2008, 14, 1185–1193. [Google Scholar] [CrossRef]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein–Barr virus lytic cycle involvement in diffuse large B cell lymphoma. Hematol. Oncol. 2018, 36, 98–103. [Google Scholar] [CrossRef]
- Jones, R.J.; Seaman, W.T.; Feng, W.H.; Barlow, E.; Dickerson, S.; Delecluse, H.J.; Kenney, S.C. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer 2007, 121, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, G.K.; Kumar, P.; Wang, L.; Damania, B.; Gulley, M.L.; Delecluse, H.J.; Polverini, P.J.; Kenney, S.C. Epstein–Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J. Virol. 2005, 79, 13984–13992. [Google Scholar] [CrossRef] [PubMed]
- Beatty, P.R.; Krams, S.M.; Martinez, O.M. Involvement of IL-10 in the autonomous growth of EBV-transformed B cell lines. J. Immunol. 1997, 158, 4045–4051. [Google Scholar] [PubMed]
- Coen, N.; Duraffour, S.; Haraguchi, K.; Balzarini, J.; van den Oord, J.J.; Snoeck, R.; Andrei, G. Antiherpesvirus activities of two novel 4’-thiothymidine derivatives, KAY-2-41 and KAH-39-149, are dependent on viral and cellular thymidine kinases. Antimicrob. Agents Chemother. 2014, 58, 4328–4340. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Singh, U.; Vuyyuru, V.; Van den Oord, J.J.; Balzarini, J.; Duraffour, S.; Snoeck, R.; Cheng, Y.C.; Chu, C.K.; Andrei, G. Activity and mechanism of action of HDVD, a novel pyrimidine nucleoside derivative with high levels of selectivity and potency against gammaherpesviruses. J. Virol. 2013, 87, 3839–3851. [Google Scholar] [CrossRef] [PubMed]
- Ville, S.; Imbert-Marcille, B.M.; Coste-Burel, M.; Garandeau, C.; Meurette, A.; Cantarovitch, D.; Giral, M.; Hourmant, M.; Blancho, G.; Dantal, J. Impact of antiviral prophylaxis in adults Epstein–Barr Virus-seronegative kidney recipients on early and late post-transplantation lymphoproliferative disorder onset: A retrospective cohort study. Transpl. Int. 2018, 31, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Østensen, A.B.; Sanengen, T.; Holter, E.; Line, P.D.; Almaas, R. No effect of treatment with intravenous ganciclovir on Epstein–Barr virus viremia demonstrated after pediatric liver transplantation. Pediatr. Transplant. 2017, 21. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Hwang, Y.Y.; Chan, T.S.; Pang, A.W.; Leung, A.Y.; Tse, E.; Kwong, Y.L. Valganciclovir suppressed Epstein Barr virus reactivation during immunosuppression with alemtuzumab. J. Clin. Virol. 2014, 59, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.E.; Magaret, A.S.; Kuntz, S.R.; Selke, S.; Huang, M.L.; Corey, L.; Casper, C.; Wald, A. Valganciclovir for the Suppression of Epstein–Barr Virus Replication. J. Infect. Dis. 2017, 216, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, R.; De Clercq, E. Role of cidofovir in the treatment of DNA virus infections, other than CMV infections, in immunocompromised patients. Curr. Opin. Investig. Drugs 2002, 3, 1561–1566. [Google Scholar] [PubMed]
- Andrei, G.; Topalis, D.; De Schutter, T.; Snoeck, R. Insights into the mechanism of action of cidofovir and other acyclic nucleoside phosphonates against polyoma- and papillomaviruses and non-viral induced neoplasia. Antiviral. Res. 2015, 114, 21–46. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Wakisaka, N.; Kondo, S.; Murono, S.; Shimizu, Y.; Nakashima, M.; Tsuji, A.; Furukawa, M. Treatment of locally recurrent Epstein–Barr virus-associated nasopharyngeal carcinoma using the anti-viral agent cidofovir. J. Med. Virol. 2008, 80, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; Sadler, R.; De Clercq, E.; Raab-Traub, N.; Pagano, J.S. The antiviral agent cidofovir [(S)-1-(3-hydroxy-2-phosphonyl-methoxypropyl)cytosine] has pronounced activity against nasopharyngeal carcinoma grown in nude mice. Cancer Res. 1998, 58, 384–388. [Google Scholar] [PubMed]
- Wakisaka, N.; Yoshizaki, T.; Raab-Traub, N.; Pagano, J.S. Ribonucleotide reductase inhibitors enhance cidofovir-induced apoptosis in EBV-positive nasopharyngeal carcinoma xenografts. Int. J. Cancer 2005, 116, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulkarim, B.; Sabri, S.; Zelenika, D.; Deutsch, E.; Frascogna, V.; Klijanienko, J.; Vainchenker, W.; Joab, I.; Bourhis, J. Antiviral agent cidofovir decreases Epstein–Barr virus (EBV) oncoproteins and enhances the radiosensitivity in EBV-related malignancies. Oncogene 2003, 22, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Afshar, K.; Rao, A.P.; Patel, V.; Forrester, K.; Ganesh, S. Use of Foscarnet Therapy for EBV Infection following Control of PTLD with Enhancement of Cellular Immunity in a Lung-Transplant Recipient. J. Transplant. 2011, 2011. [Google Scholar] [CrossRef]
- Schneider, U.; Ruhnke, M.; Delecluse, H.J.; Stein, H.; Huhn, D. Regression of Epstein–Barr virus-associated lymphoproliferative disorders in patients with acquired immunodeficiency syndrome during therapy with foscarnet. Ann. Hematol. 2000, 79, 214–216. [Google Scholar] [CrossRef]
- Biron, K.K.; Harvey, R.J.; Chamberlain, S.C.; Good, S.S.; Smith, A.A., III; Davis, M.G.; Talarico, C.L.; Miller, W.H.; Ferris, R.; Dornsife, R.E.; et al. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action. Antimicrob. Agents Chemother. 2002, 46, 2365–2372. [Google Scholar] [CrossRef]
- Zacny, V.L.; Gershburg, E.; Davis, M.G.; Biron, K.K.; Pagano, J.S. Inhibition of Epstein–Barr virus replication by a benzimidazole L-riboside: Novel antiviral mechanism of 5, 6-dichloro-2-(isopropylamino)-1-beta-L-ribofuranosyl-1H-benzimidazole. J. Virol. 1999, 73, 7271–7277. [Google Scholar]
- Prichard, M.N. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 2009, 19, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.; Bowlin, T.L. Cytomegalovirus UL97 mutations affecting cyclopropavir and ganciclovir susceptibility. Antimicrob. Agents Chemother. 2011, 55, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bender, B.J.; Kamil, J.P.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.M.; Coen, D.M. Human Cytomegalovirus UL97 Phosphorylates the Viral Nuclear Egress Complex. J. Virol. 2014, 89, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef]
- Chou, S. Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev. Med. Virol. 2008, 18, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.; Marousek, G.; Bowlin, T.L. Cyclopropavir susceptibility of cytomegalovirus DNA polymerase mutants selected after antiviral drug exposure. Antimicrob. Agents Chemother. 2012, 56, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Ercolani, R.J.; Marousek, G.; Bowlin, T.L. Cytomegalovirus UL97 kinase catalytic domain mutations that confer multidrug resistance. Antimicrob. Agents Chemother. 2013, 57, 3375–3379. [Google Scholar] [CrossRef] [PubMed]
- Winston, D.J.; Saliba, F.; Blumberg, E.; Abouljoud, M.; Garcia-Diaz, J.B.; Goss, J.A.; Clough, L.; Avery, R.; Limaye, A.P.; Ericzon, B.G.; et al. Efficacy and safety of maribavir dosed at 100 mg orally twice daily for the prevention of cytomegalovirus disease in liver transplant recipients: A randomized, double-blind, multicenter controlled trial. Am. J. Transplant. 2012, 12, 3021–3030. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Papanicolaou, G.A.; Winston, D.J.; Chemaly, R.F.; Strasfeld, L.; Young, J.A.; Rodriguez, T.; Maertens, J.; Schmitt, M.; et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: A phase 3, double-blind, placebo-controlled, randomised trial. Lancet. Infect. Dis. 2011, 11, 284–292. [Google Scholar] [CrossRef]
- Griffiths, P.; Lumley, S. Cytomegalovirus. Curr. Opin. Infect. Dis. 2014, 27, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Roy, D.; Gershburg, E.; Whitehurst, C.B.; Dittmer, D.P.; Pagano, J.S. Maribavir inhibits Epstein–Barr virus transcription in addition to viral DNA replication. J. Virol. 2009, 83, 12108–12117. [Google Scholar] [CrossRef]
- Murata, T.; Isomura, H.; Yamashita, Y.; Toyama, S.; Sato, Y.; Nakayama, S.; Kudoh, A.; Iwahori, S.; Kanda, T.; Tsurumi, T. Efficient production of infectious viruses requires enzymatic activity of Epstein–Barr virus protein kinase. Virology 2009, 389, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Sanders, M.K.; Law, M.; Wang, F.Z.; Xiong, J.; Dittmer, D.P.; Pagano, J.S. Maribavir inhibits Epstein–Barr virus transcription through the EBV protein kinase. J. Virol. 2013, 87, 5311–5315. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Hagemeier, S.R.; Fingeroth, J.D.; Gershburg, E.; Pagano, J.S.; Kenney, S.C. The Epstein–Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 2010, 84, 4534–4542. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.L.; Geiser, F.; Kira, T.; Gullen, E.; Cheng, Y.C.; Ptak, R.G.; Breitenbach, J.M.; Drach, J.C.; Hartline, C.B.; Kern, E.R.; et al. Synthesis and enantioselectivity of the antiviral effects of (R,Z)-,(S,Z)-methylenecyclopropane analogues of purine nucleosides and phosphoralaninate prodrugs: Influence of heterocyclic base, type of virus and host cells. Antivir. Chem. Chemother. 2000, 11, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Breitenbach, J.M.; Borysko, K.Z.; Drach, J.C.; Kern, E.R.; Gullen, E.; Cheng, Y.C.; Zemlicka, J. Synthesis and antiviral activity of (Z)- and (E)-2,2-[bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: Second-generation methylenecyclopropane analogues of nucleosides. J. Med. Chem. 2004, 47, 566–575. [Google Scholar] [CrossRef]
- Prichard, M.N.; Williams, J.D.; Komazin-Meredith, G.; Khan, A.R.; Price, N.B.; Jefferson, G.M.; Harden, E.A.; Hartline, C.B.; Peet, N.P.; Bowlin, T.L. Synthesis and antiviral activities of methylenecyclopropane analogs with 6-alkoxy and 6-alkylthio substitutions that exhibit broad-spectrum antiviral activity against human herpesviruses. Antimicrob. Agents Chemother. 2013, 57, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kern, E.R.; Drach, J.C.; Gullen, E.; Cheng, Y.C.; Zemlicka, J. Structure-activity relationships of (S,Z)-2-aminopurine methylenecyclopropane analogues of nucleosides. Variation of purine-6 substituents and activity against herpesviruses and hepatitis B virus. J. Med. Chem. 2003, 46, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Kern, E.R.; Kushner, N.L.; Hartline, C.B.; Williams-Aziz, S.L.; Harden, E.A.; Zhou, S.; Zemlicka, J.; Prichard, M.N. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother. 2005, 49, 1039–1045. [Google Scholar] [CrossRef]
- Li, C.; Quenelle, D.C.; Prichard, M.N.; Drach, J.C.; Zemlicka, J. Synthesis and antiviral activity of 6-deoxycyclopropavir, a new prodrug of cyclopropavir. Bioorg. Med. Chem. 2012, 20, 2669–2674. [Google Scholar] [CrossRef] [Green Version]
- James, S.H.; Hartline, C.B.; Harden, E.A.; Driebe, E.M.; Schupp, J.M.; Engelthaler, D.M.; Keim, P.S.; Bowlin, T.L.; Kern, E.R.; Prichard, M.N. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob. Agents Chemother. 2011, 55, 4682–4691. [Google Scholar] [CrossRef]
- Prichard, M.N.; Keith, K.A.; Quenelle, D.C.; Kern, E.R. Activity and mechanism of action of N-methanocarbathymidine against herpesvirus and orthopoxvirus infections. Antimicrob. Agents Chemother. 2006, 50, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Quenelle, D.C.; Collins, D.J.; Rice, T.L.; Rahman, A.; Glazer, R. Efficacy of orally administered low dose N-methanocarbathymidine against lethal herpes simplex virus type-2 infections of mice. Antivir. Chem. Chemother. 2011, 22, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Bravo, F.J.; Pullum, D.A.; Shen, H.; Wang, M.; Rahman, A.; Glazer, R.I.; Cardin, R.D. Efficacy of N-methanocarbathymidine against genital herpes simplex virus type 2 shedding and infection in guinea pigs. Antivir. Chem. Chemother. 2015, 24, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antiviral. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. [Google Scholar] [CrossRef]
- Wilson, J.B.; Manet, E.; Gruffat, H.; Busson, P.; Blondel, M.; Fahraeus, R. EBNA1: Oncogenic Activity, Immune Evasion and Biochemical Functions Provide Targets for Novel Therapeutic Strategies against Epstein–Barr Virus- Associated Cancers. Cancers 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xie, C.; Lung, H.L.; Lo, K.W.; Law, G.L.; Mak, N.K.; Wong, K.L. EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein–Barr virus-associated cancers. Theranostics 2018, 8, 5307–5319. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Kim, S.Y.; Noh, K.W.; Joo, E.H.; Zhao, B.; Kieff, E.; Kang, M.S. Small molecule inhibition of Epstein–Barr virus nuclear antigen-1 DNA binding activity interferes with replication and persistence of the viral genome. Antiviral. Res. 2014, 104, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianti, E.; Messick, T.E.; Lieberman, P.M.; Zauhar, R.J. Computational analysis of EBNA1 “druggability” suggests novel insights for Epstein–Barr virus inhibitor design. J. Comput. Aided Mol. Des. 2016, 30, 285–303. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M. Topoisomerase I and II activities are required for Epstein–Barr virus replication. J. Gen. Virol. 1993, 74, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Rennekamp, A.J.; Yuan, Y.; Lieberman, P.M. Topoisomerase I and RecQL1 function in Epstein–Barr virus lytic reactivation. J. Virol. 2009, 83, 8090–8098. [Google Scholar] [CrossRef]
- Wu, T.; Wang, Y.; Yuan, Y. Antiviral activity of topoisomerase II catalytic inhibitors against Epstein–Barr virus. Antiviral. Res. 2014, 107, 95–101. [Google Scholar] [CrossRef]
- Gonzalez-Molleda, L.; Wang, Y.; Yuan, Y. Potent antiviral activity of topoisomerase I and II inhibitors against Kaposi’s sarcoma-associated herpesvirus. Antimicrob. Agents Chemother. 2012, 56, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Widman, D.G.; Gornisiewicz, S.; Shacham, S.; Tamir, S. In vitro toxicity and efficacy of verdinexor, an exportin 1 inhibitor, on opportunistic viruses affecting immunocompromised individuals. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hayward, S.D. Potential of protein kinase inhibitors for treating herpesvirus-associated disease. Trends Microbiol. 2013, 21, 286–295. [Google Scholar] [CrossRef]
- Vigano, M.; Dengler, T.; Mattei, M.F.; Poncelet, A.; Vanhaecke, J.; Vermes, E.; Kleinloog, R.; Li, Y.; Gezahegen, Y.; Delgado, J.F.; et al. Lower incidence of cytomegalovirus infection with everolimus versus mycophenolate mofetil in de novo cardiac transplant recipients: A randomized, multicenter study. Transpl. Infect. Dis. 2010, 12, 23–30. [Google Scholar] [CrossRef]
- Hill, J.A.; Hummel, M.; Starling, R.C.; Kobashigawa, J.A.; Perrone, S.V.; Arizon, J.M.; Simonsen, S.; Abeywickrama, K.H.; Bara, C. A lower incidence of cytomegalovirus infection in de novo heart transplant recipients randomized to everolimus. Transplantation 2007, 84, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, J.; Ross, H.; Bara, C.; Delgado, J.F.; Dengler, T.; Lehmkuhl, H.B.; Wang, S.S.; Dong, G.; Witte, S.; Junge, G.; et al. Everolimus is associated with a reduced incidence of cytomegalovirus infection following de novo cardiac transplantation. Transpl. Infect. Dis. 2013, 15, 150–162. [Google Scholar] [CrossRef]
- Tan, L.; Sato, N.; Shiraki, A.; Yanagita, M.; Yoshida, Y.; Takemura, Y.; Shiraki, K. Everolimus delayed and suppressed cytomegalovirus DNA synthesis, spread of the infection, and alleviated cytomegalovirus infection. Antiviral. Res. 2019, 162, 30–38. [Google Scholar] [CrossRef]
- Lin, T.P.; Chen, S.Y.; Duh, P.D.; Chang, L.K.; Liu, Y.N. Inhibition of the Epstein–Barr virus lytic cycle by andrographolide. Biol. Pharm. Bull. 2008, 31, 2018–2023. [Google Scholar] [CrossRef]
- Uttekar, M.M.; Das, T.; Pawar, R.S.; Bhandari, B.; Menon, V.; Nutan; Gupta, S.K.; Bhat, S.V. Anti-HIV activity of semisynthetic derivatives of andrographolide and computational study of HIV-1 gp120 protein binding. Eur. J. Med. Chem. 2012, 56, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Aromdee, C.; Suebsasana, S.; Ekalaksananan, T.; Pientong, C.; Thongchai, S. Stage of action of naturally occurring andrographolides and their semisynthetic analogues against herpes simplex virus type 1 in vitro. Planta Med. 2011, 77, 915–921. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, D.; Wu, X. Biological activities and corresponding SARs of andrographolide and its derivatives. Mini. Rev. Med. Chem. 2013, 13, 298–309. [Google Scholar] [PubMed]
- Wiart, C.; Kumar, K.; Yusof, M.Y.; Hamimah, H.; Fauzi, Z.M.; Sulaiman, M. Antiviral properties of ent-labdene diterpenes of Andrographis paniculata nees, inhibitors of herpes simplex virus type 1. Phytother. Res. 2005, 19, 1069–1070. [Google Scholar] [CrossRef] [PubMed]
- Yiu, C.Y.; Chen, S.Y.; Yang, T.H.; Chang, C.J.; Yeh, D.B.; Chen, Y.J.; Lin, T.P. Inhibition of Epstein–Barr virus lytic cycle by an ethyl acetate subfraction separated from Polygonum cuspidatum root and its major component, emodin. Molecules 2014, 19, 1258–1272. [Google Scholar] [CrossRef]
- Li, D.; Zhang, N.; Cao, Y.; Zhang, W.; Su, G.; Sun, Y.; Liu, Z.; Li, F.; Liang, D.; Liu, B.; et al. Emodin ameliorates lipopolysaccharide-induced mastitis in mice by inhibiting activation of NF-kappaB and MAPKs signal pathways. Eur. J. Pharmacol. 2013, 705, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, E.; Lee, J.; Huh, S.; Hwang, C.H.; Lee, H.Y.; Kim, E.J.; Cheon, J.M.; Hyun, C.G.; Kim, Y.S.; et al. Emodin inhibits TNF alpha-induced MMP-1 expression through suppression of activator protein-1 (AP-1). Life Sci. 2006, 79, 2480–2485. [Google Scholar] [CrossRef]
- Lin, H.J.; Chao, P.D.; Huang, S.Y.; Wan, L.; Wu, C.J.; Tsai, F.J. Aloe-emodin suppressed NMDA-induced apoptosis of retinal ganglion cells through regulation of ERK phosphorylation. Phytother. Res. 2007, 21, 1007–1014. [Google Scholar] [CrossRef]
- Liu, S.; Li, H.; Chen, L.; Yang, L.; Li, L.; Tao, Y.; Li, W.; Li, Z.; Liu, H.; Tang, M.; et al. (−)-Epigallocatechin-3-gallate inhibition of Epstein–Barr virus spontaneous lytic infection involves ERK1/2 and PI3-K/Akt signaling in EBV-positive cells. Carcinogenesis 2013, 34, 627–637. [Google Scholar] [CrossRef]
- Chang, L.K.; Wei, T.T.; Chiu, Y.F.; Tung, C.P.; Chuang, J.Y.; Hung, S.K.; Li, C.; Liu, S.T. Inhibition of Epstein–Barr virus lytic cycle by (−)-epigallocatechin gallate. Biochem. Biophys. Res. Commun. 2003, 301, 1062–1068. [Google Scholar] [CrossRef]
- Cui, H.; Xu, B.; Wu, T.; Xu, J.; Yuan, Y.; Gu, Q. Potential antiviral lignans from the roots of Saururus chinensis with activity against Epstein–Barr virus lytic replication. J. Nat. Prod. 2014, 77, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.Y.; Lee, J.H.; Nam, J.B.; Hong, Y.S.; Lee, J.J. Lignans from Saururus chinensis inhibiting the transcription factor NF-kappaB. Phytochemistry 2003, 64, 765–771. [Google Scholar] [CrossRef]
- Lee, J.; Huh, M.S.; Kim, Y.C.; Hattori, M.; Otake, T. Lignan, sesquilignans and dilignans, novel HIV-1 protease and cytopathic effect inhibitors purified from the rhizomes of Saururus chinensis. Antiviral. Res. 2010, 85, 425–428. [Google Scholar] [CrossRef]
- Oh, K.S.; Choi, Y.H.; Ryu, S.Y.; Oh, B.K.; Seo, H.W.; Yon, G.H.; Kim, Y.S.; Lee, B.H. Cardiovascular effects of lignans isolated from Saururus chinensis. Planta Med. 2008, 74, 233–238. [Google Scholar] [CrossRef]
- Yu, D.; Sakurai, Y.; Chen, C.H.; Chang, F.R.; Huang, L.; Kashiwada, Y.; Lee, K.H. Anti-AIDS agents 69. Moronic acid and other triterpene derivatives as novel potent anti-HIV agents. J. Med. Chem. 2006, 49, 5462–5469. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.R.; Hsieh, Y.C.; Chang, Y.F.; Lee, K.H.; Wu, Y.C.; Chang, L.K. Inhibition of the Epstein–Barr virus lytic cycle by moronic acid. Antiviral. Res. 2010, 85, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Basnet, P.; Ohsugi, M.; Hozumi, T.; Kadota, S.; Namba, T.; Kawana, T.; Shiraki, K. Anti-herpes simplex virus activity of moronic acid purified from Rhus javanica in vitro and in vivo. J. Pharmacol. Exp. Ther. 1999, 289, 72–78. [Google Scholar]
- Tung, C.P.; Chang, F.R.; Wu, Y.C.; Chuang, D.W.; Hunyadi, A.; Liu, S.T. Inhibition of the Epstein–Barr virus lytic cycle by protoapigenone. J. Gen. Virol. 2011, 92, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Jeong, S.G.; Park, J.E.; Han, J.A.; Kang, H.R.; Lee, D.; Song, M.J. Antiviral activity of angelicin against gammaherpesviruses. Antiviral. Res. 2013, 100, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.Y.; Coumar, M.S.; Horng, J.T.; Shiao, H.Y.; Kuo, F.M.; Lee, H.L.; Chen, I.C.; Chang, C.W.; Tang, W.F.; Tseng, S.N.; et al. Anti-influenza drug discovery: Structure-activity relationship and mechanistic insight into novel angelicin derivatives. J. Med. Chem. 2010, 53, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- Miolo, G.; Tomanin, R.; De Rossi, A.; Dall’Acqua, F.; Zacchello, F.; Scarpa, M. Antiretroviral activity of furocoumarins plus UVA light detected by a replication-defective retrovirus. J. Photochem. Photobiol. 1994, 26, 241–247. [Google Scholar] [CrossRef]
- Gorres, K.L.; Daigle, D.; Mohanram, S.; Miller, G. Activation and repression of Epstein–Barr Virus and Kaposi’s sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. J. Virol. 2014, 88, 8028–8044. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.; Ho, D.N.; Tsang, C.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein–Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Harmon, P.; Gulley, M.L.; Mwansambo, C.; Kazembe, P.N.; Martinson, F.; Wokocha, C.; Kenney, S.C.; Hoffman, I.; Sigel, C.; et al. Viral response to chemotherapy in endemic burkitt lymphoma. Clin. Cancer Res. 2010, 16, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.X.; Tanhehco, Y.; Chen, J.; Foss, C.A.; Fox, J.J.; Chong, J.M.; Hobbs, R.F.; Fukayama, M.; Sgouros, G.; Kowalski, J.; et al. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nat. Med. 2008, 14, 1118–1122. [Google Scholar] [CrossRef] [Green Version]
- Countryman, J.K.; Gradoville, L.; Miller, G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein–Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol. 2008, 82, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.; Bruggeling, C.E.; Schurch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef]

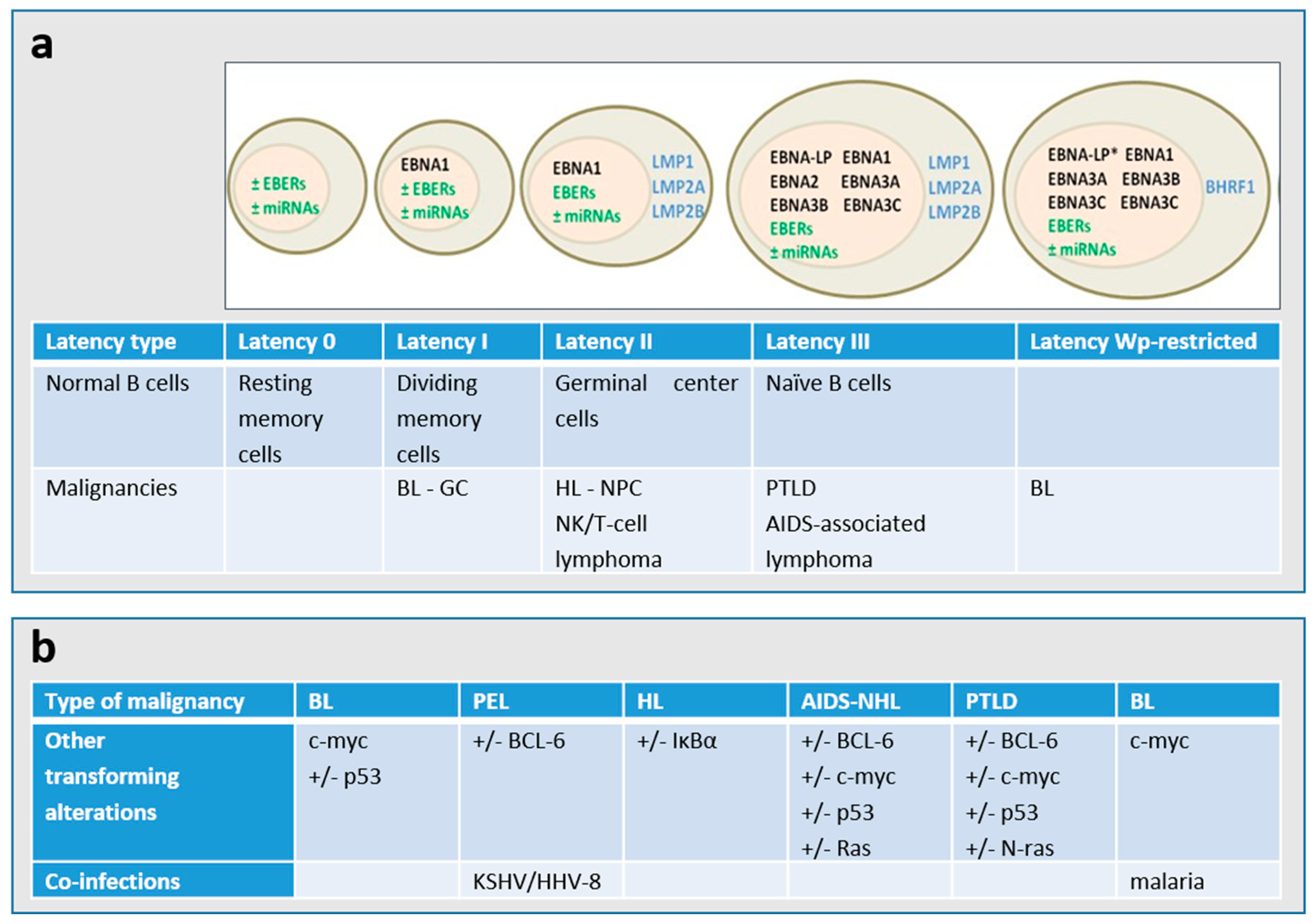

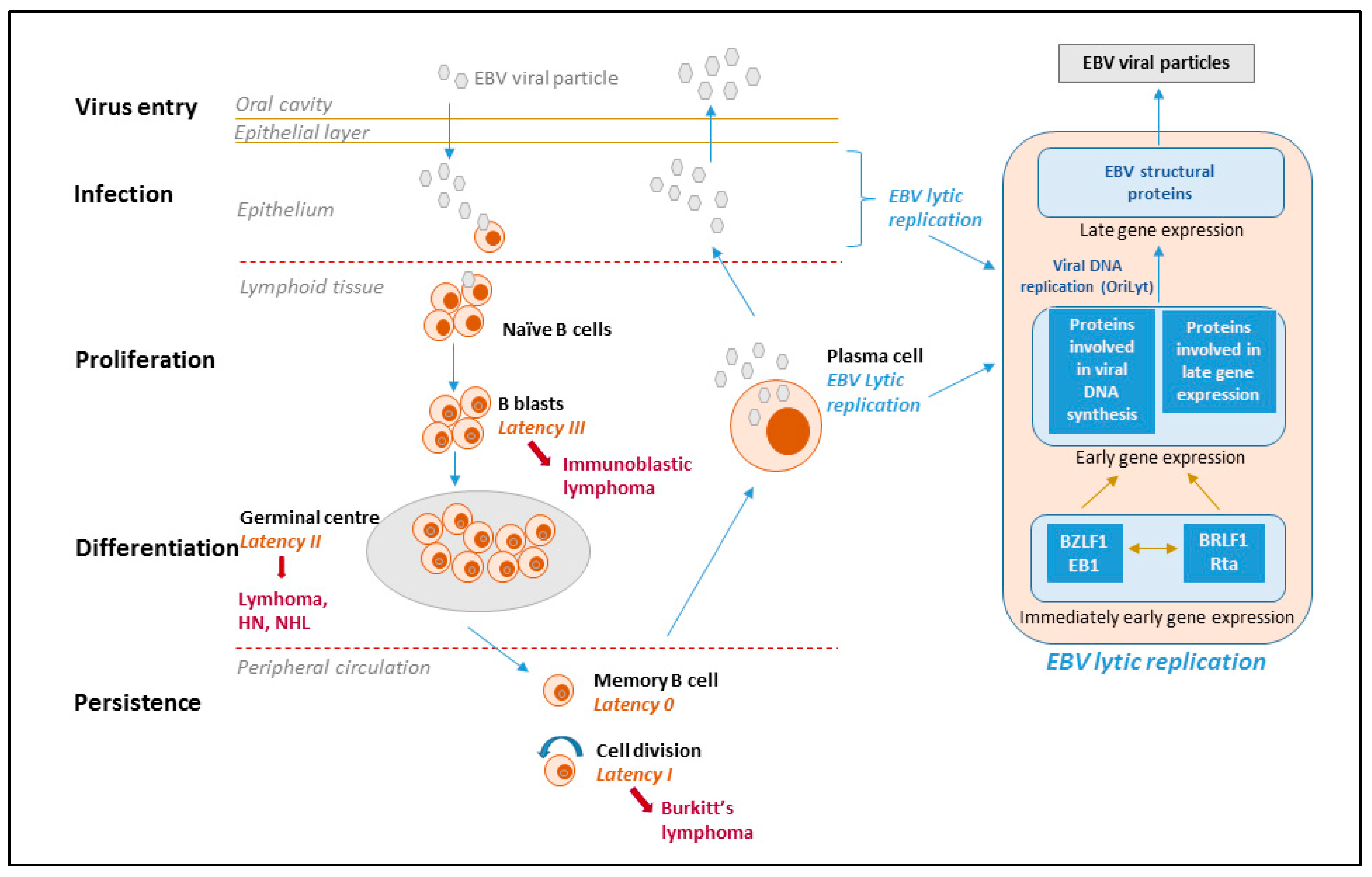{kind=link}
{kind=link}
| Immunocompetent Host | Immunocompromised Hosts | ||||
|---|---|---|---|---|---|
| Lymphoma | EBV Association | Latency Program | Lymphoma | EBV Association | Latency Program |
| BL (endemic) | 100% | I or Wp- restricted | PTLD, B-cell | >90% | III |
| BL (sporadic) | 15–85% | I | BL (HIV) | 25–35% | I |
| Classical HL | 40% | II | HL (HIV) | >80% | II |
| DLBCL associated with chronic inflammation | ~70% | II | PEL (primary effusion lymphoma) | >80% | I |
| EBV-positive DLBCL of the elderly | 100% | II | Plasmablastic lymphoma | ~70% | I or II |
| Lymphomatoid granulomatosis | 100% | II | Plasmablastic lymphoma, oral type (HIV) | 100% | I |
| Angioimmunoblastic T-cell lymphoma * | >90% | II | Primary CNS lymphoma (HIV) | 100% | III |
| Extranodal NK/T-cell lymphoma, nasal type * | 100% | II | NHLs with primary immune disorders | >90% | III |
| Aggressive NK-cell leukemia * | >90% | II | Iatrogenic immunodeficiency lymphoma | 40–50% | III |
| PTLD, NK/T-cell * | >70% | III | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrei, G.; Trompet, E.; Snoeck, R. Novel Therapeutics for Epstein–Barr Virus. Molecules 2019, 24, 997. https://doi.org/10.3390/molecules24050997
Andrei G, Trompet E, Snoeck R. Novel Therapeutics for Epstein–Barr Virus. Molecules. 2019; 24(5):997. https://doi.org/10.3390/molecules24050997
Chicago/Turabian StyleAndrei, Graciela, Erika Trompet, and Robert Snoeck. 2019. "Novel Therapeutics for Epstein–Barr Virus" Molecules 24, no. 5: 997. https://doi.org/10.3390/molecules24050997
APA StyleAndrei, G., Trompet, E., & Snoeck, R. (2019). Novel Therapeutics for Epstein–Barr Virus. Molecules, 24(5), 997. https://doi.org/10.3390/molecules24050997