A Single Mutation Increases the Thermostability and Activity of Aspergillus terreus Amine Transaminase

Abstract
:1. Introduction
2. Results
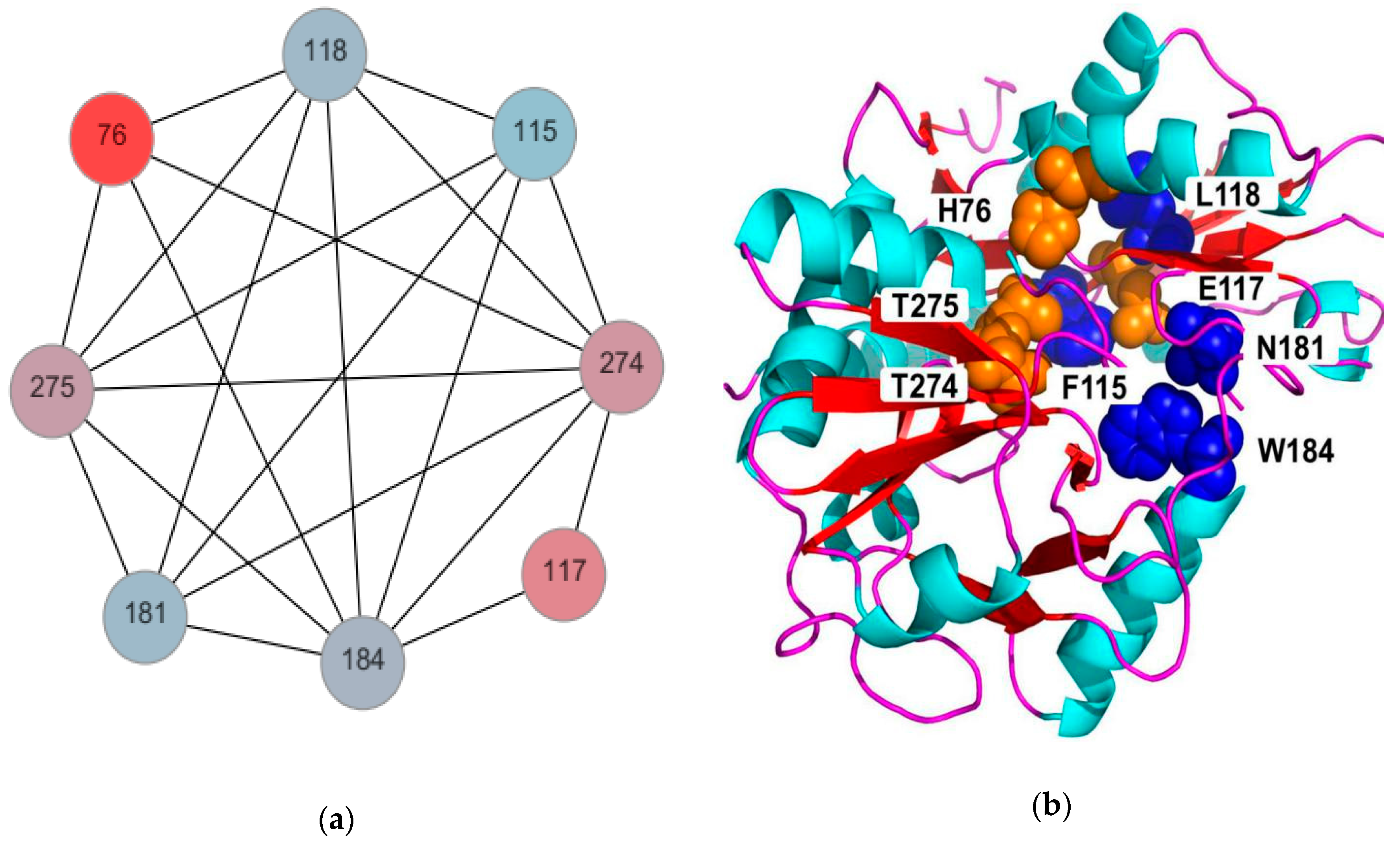
2.1. Analysis of the Mutant AT-ATAs by Alanine Scanning
2.2. Saturation Mutagenesis of Residue L118
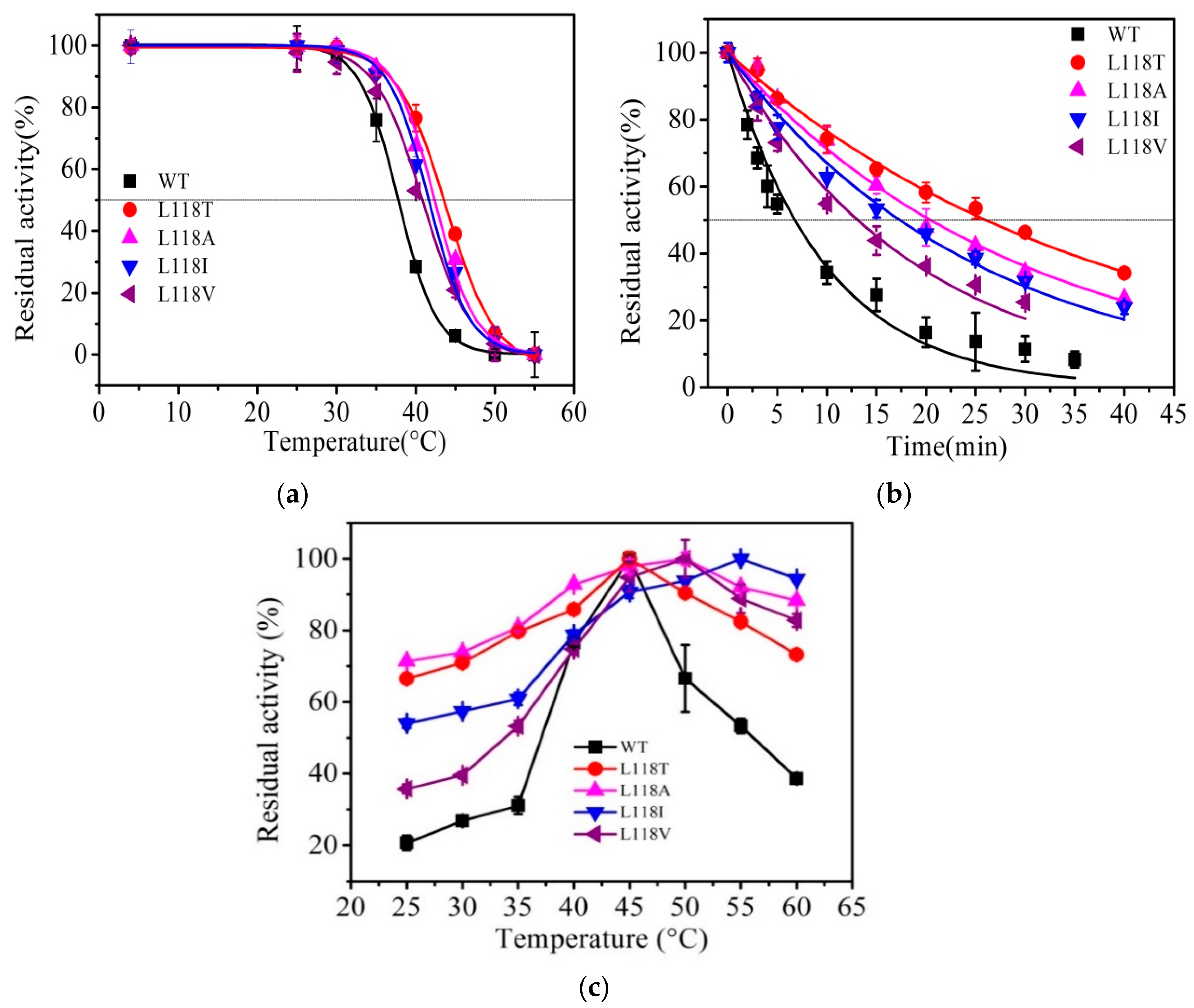
2.3. Thermal Stability of AT-ATA and its Variants
2.4. Kinetic Constants
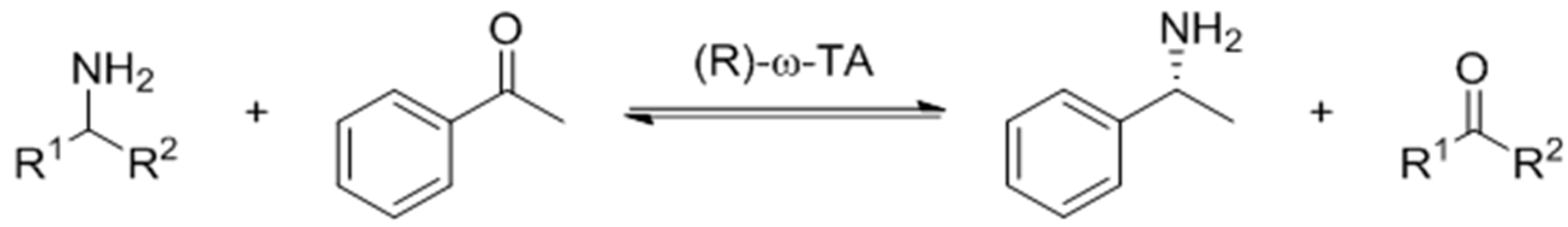
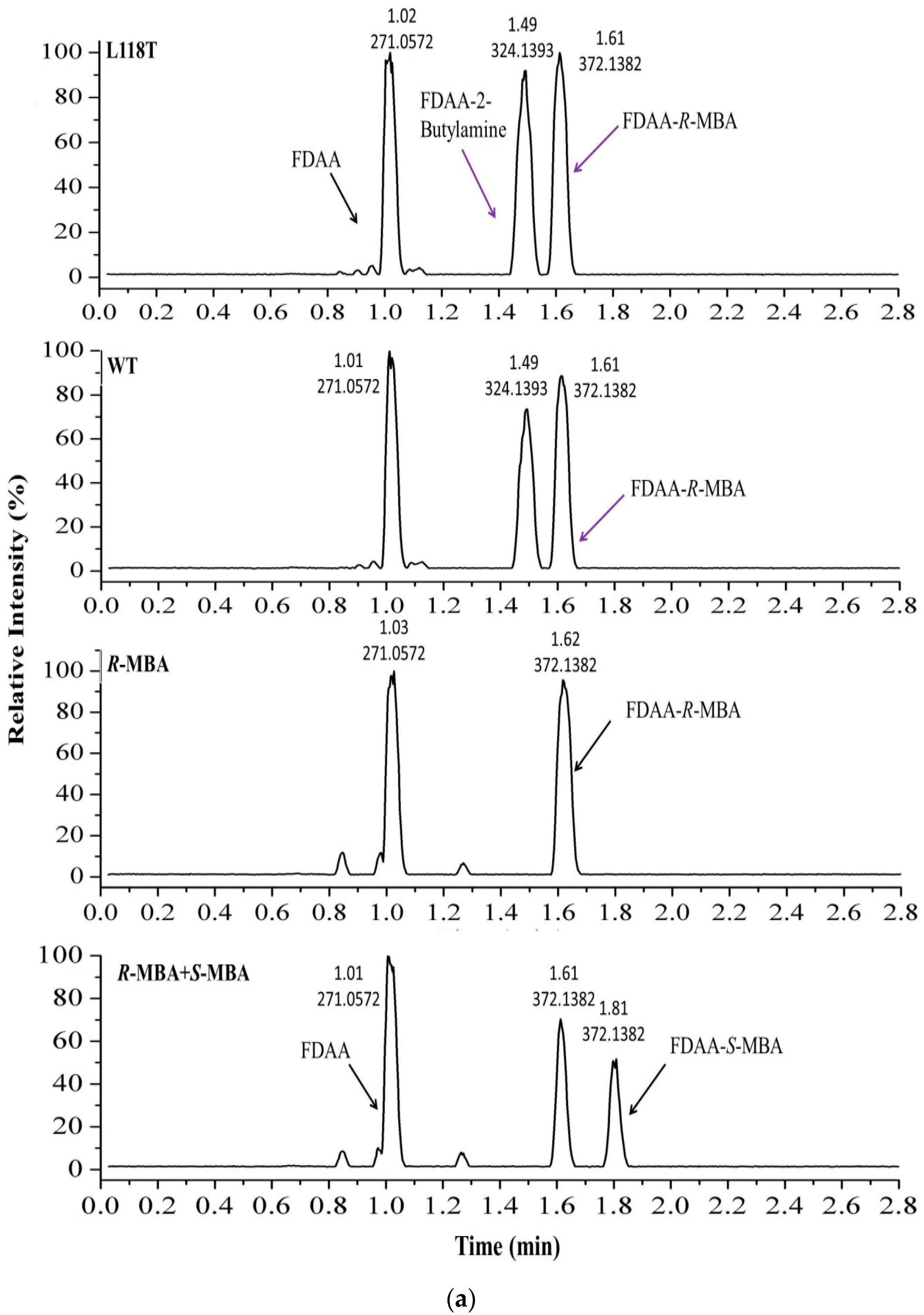
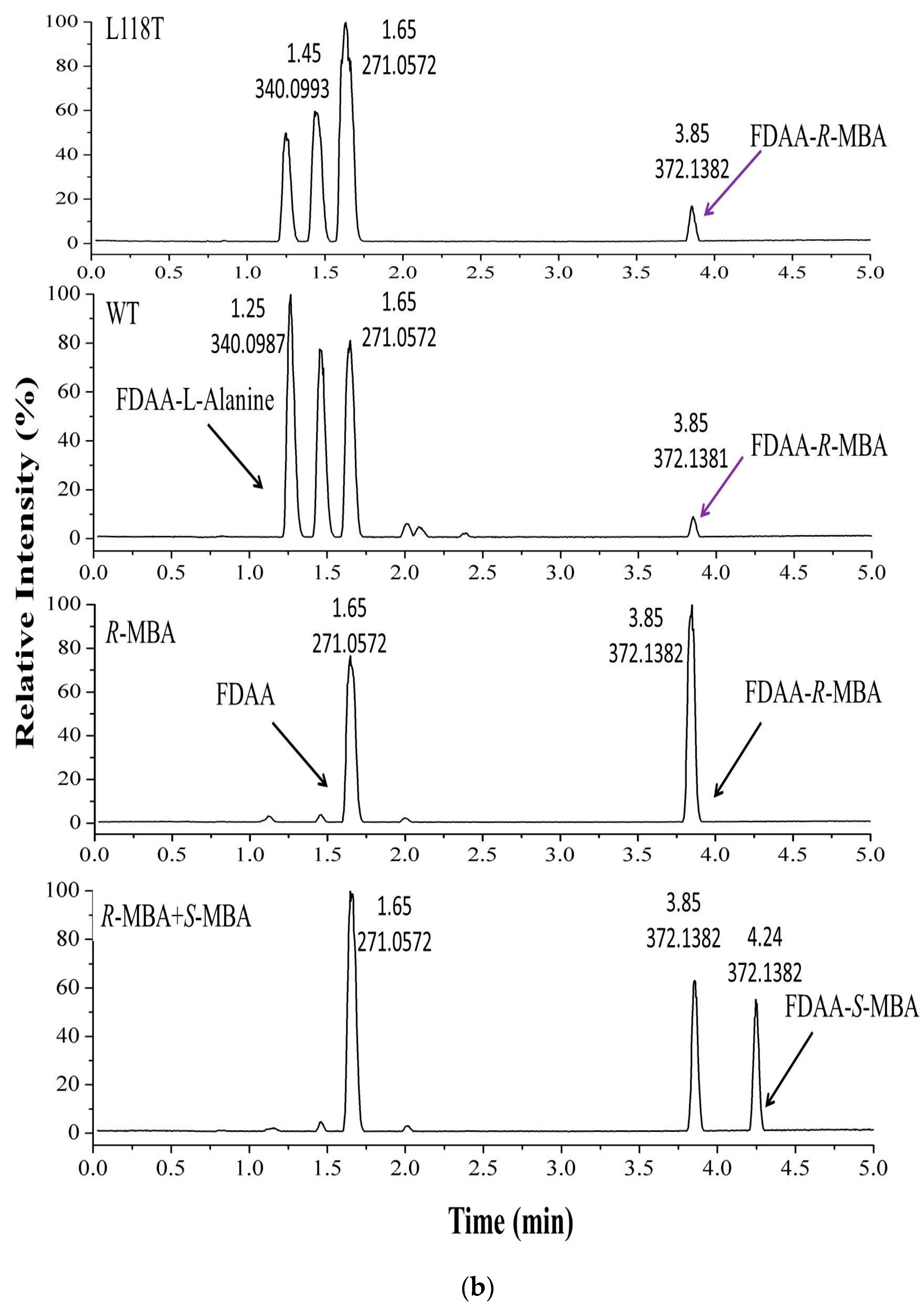
2.5. The Asymmetric Synthesis of α-MBA Catalyzed by AT-ATA and the Mutant L118T
3. Discussion
4. Materials and Methods
4.1. Molecular Biology Tools and Reagents
4.2. In Silico Analysis Procedure
4.3. Alanine Scanning of the AT-ATA Coevolution Network
4.4. Construction of the Saturation Mutagenesis Library
4.5. Library Screening
4.6. Protein Expression and Purification
4.7. Enzyme Assay and Kinetic Parameters
4.8. The Thermal Stability of AT-ATA and its Mutants
4.8.1. Kinetic Stability of AT-ATA and its Mutants
4.8.2. Thermodynamic Stability of AT-ATA and its Mutants
4.9. UPLC-MS Analysis of the Product of the Transamination Reaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferrandi, E.E.; Monti, D. Amine transaminases in chiral amines synthesis: Recent advances and challenges. World J. Microbiol. Biotechnol. 2017, 34, 13. [Google Scholar] [CrossRef]
- Dawood, A.W.H.; de Souza, R.O.M.A.; Bornscheuer, U.T. Asymmetric Synthesis of Chiral Halogenated Amines using Amine Transaminases. ChemCatChem. 2018, 10, 951–955. [Google Scholar] [CrossRef]
- Nugent, T.C.; El-Shazly, M. Chiral Amine Synthesis-Recent Developments and Trends for Enamide Reduction, Reductive Amination, and Imine Reduction. Adv. Synth. Catal. 2010, 352, 753–819. [Google Scholar] [CrossRef]
- Paul, C.E.; Rodríguez-Mata, M.; Busto, E.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; García-Cerrada, S.; Mendiola, J.; de Frutos, Ó.; Collado, I. Transaminases Applied to the Synthesis of High Added-Value Enantiopure Amines. Org. Process Res. Dev. 2014, 18, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Tufvesson, P.; Lima-Ramos, J.; Jensen, J.S.; Al-Haque, N.; Neto, W.; Woodley, J.M. Process considerations for the asymmetric synthesis of chiral amines using transaminases. Biotechnol. Bioeng. 2011, 108, 1479–1493. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric Synthesis of Optically Pure Pharmacologically Relevant Amines Employing ω-Transaminases. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar] [CrossRef]
- Sayer, C.; Martinez-Torres, R.J.; Richter, N.; Isupov, M.N.; Hailes, H.C.; Littlechild, J.A.; Ward, J.M. The substrate specificity, enantioselectivity and structure of the (R)-selective amine: Pyruvate transaminase from Nectria haematococca. FEBS J. 2014, 281, 2240–2253. [Google Scholar] [CrossRef]
- Huang, J.; Xie, D.F.; Feng, Y. Engineering thermostable (R)-selective amine transaminase from Aspergillus terreus through in silico design employing B-factor and folding free energy calculations. Biochem. Biophys. Res. Comm. 2017, 483, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.F.; Fang, H.; Mei, J.Q.; Gong, J.Y.; Wang, H.P.; Shen, X.Y.; Huang, J.; Mei, L.H. Improving thermostability of (R)-selective amine transaminase from Aspergillus terreus through introduction of disulfide bonds. Biotechnol. Appl. Biochem. 2018, 65, 255–262. [Google Scholar] [CrossRef]
- Xie, D.F.; Yang, J.X.; Lv, C.J.; Mei, J.Q.; Wang, H.P.; Hu, S.; Zhao, W.R.; Cao, J.R.; Tu, J.L.; Huang, J.; et al. Construction of stabilized (R)-selective amine transaminase from Aspergillus terreus by consensus mutagenesis. J. Biotechnol. 2019, 293, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Brinda, K.V.; Vishveshwara, S. A network representation of protein structures: Implications for protein stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef]
- Amitai, G.; Shemesh, A.; Sitbon, E.; Shklar, M.; Netanely, D.; Venger, I.; Pietrokovski, S. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004, 344, 1135–1146. [Google Scholar] [CrossRef]
- Lee, B.C.; Park, K.; Kim, D. Analysis of the residue-residue coevolution network and the functionally important residues in proteins. Proteins 2008, 72, 863–872. [Google Scholar] [CrossRef]
- Buslje, M.C.; Teppa, E.; Di Domenico, T.; Delfino, J.M.; Nielsen, M. Networks of high mutual information define the structural proximity of catalytic sites: Implications for catalytic residue identification. PLoS Comput. Biol. 2010, 6, e1000978. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell System Technical Journal 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Van den Bergh, T.; Tamo, G.; Nobili, A.; Tao, Y.F.; Tan, T.W.; Bornscheuer, U.T.; Kuipers, R.K.P.; Vroling, B.; de Jong, R.M.; Subramanian, K.; et al. CorNet: Assigning function to networks of co-evolving residues by automated literature mining. PLoS ONE 2017, 12, e0176427. [Google Scholar] [CrossRef]
- Dietrich, S.; Borst, N.; Schlee, S.; Schneider, D.; Janda, J.-O.; Sterner, R.; Merkl, R. Experimental assessment of the importance of amino acid positions identified by an entropy-based correlation analysis of multiplesequence alignments. Biochemistry 2012, 51, 5633–5641. [Google Scholar] [CrossRef]
- Aguilar, D.; Oliva, B.; Buslje, C.M. Mapping the mutual information network of enzymatic families in the protein structure to unveil functional features. PLoS ONE 2012, 7, e41430. [Google Scholar] [CrossRef]
- Oteri, F.; Nadalin, F.; Champeimont, R.; Carbone, A. BIS2Analyzer: A server for co-evolution analysis of conserved protein families. Nucleic Acids Res. 2017, 45, W307–W314. [Google Scholar] [CrossRef]
- Suplatov, D.; Sharapova, Y.; Timonina, D.; Kopylov, K.; Svedas, V. The visualCMAT: A web-server to select and interpret correlated mutations/co-evolving residues in protein families. J. Bioinform. Comput. Biol. 2018, 16, 1840005. [Google Scholar] [CrossRef]
- Janda, J.-O.; Popal, A.; Bauer, J.; Busch, M.; Klocke, M.; Spitzer, W.; Keller, J.; Merkl, R. H2rs:Deducing evolutionary and functionally important residue positions by means of an entropy and similarity based analysis of multiple sequence alignments. BMC Bioinform. 2014, 15, 118. [Google Scholar] [CrossRef]
- Tillier, E.R.M.; Biro, L.; Li, G.; Tillo, D. Codep: Maximizing co-evolutionary interdependencies to discover interacting proteins. Proteins 2006, 63, 822–831. [Google Scholar] [CrossRef]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Buslje, C.M. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, 8–14. [Google Scholar] [CrossRef]
- Łyskowski, A.; Gruber, C.; Steinkellner, G.; Schürmann, M.; Schwab, H.; Gruber, K.; Steiner, K. Crystal Structure of an (R)-Selective ω-Transaminase from Aspergillus terreus. PLoS ONE 2014, 9, 1–11. [Google Scholar] [CrossRef]
- Lin, L.; Wang, Y.; Wu, M.B.; Zhu, L.; Yang, L.R.; Lin, J.P. Enhancing the thermostability of fumarase C from Corynebacterium glutamicum via molecular modification. Enzyme Microb. Technol. 2018, 115, 45–51. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Schätzle, S.; Höhne, M.; Redestad, E.; Robins, K.; Bornscheuer, U.T. Rapid and Sensitive Kinetic Assay for Characterization of ω-Transaminases. Anal. Chem. 2009, 81, 8244–8248. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Shin, J.B.; Kim, B.G. Asymmetric synthesis of chiral amines with ω-transaminase. Biotechnol. Bioeng. 1999, 65, 206–211. [Google Scholar] [CrossRef]
- Fesko, K.; Steiner, K.; Breinbauer, R.; Schwab, H.; Schürmann, M.; Strohmeier, G.A. Investigation of one-enzyme systems in the ω-transaminase-catalyzed synthesis of chiral amines. J. Mol. Biol. Catal. B-Enzym. 2013, 96, 103–110. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | T5010 (°C) | t1/2 (min) | Tm (°C) |
|---|---|---|---|
| WT-AT | 38.5 ± 0.5 | 6.9 ± 0.6 | 41.4 ± 0.2 |
| L118A | 42.2 ± 0.2 | 20.6 ± 0.7 | 43.5 ± 0.1 |
| L118T | 43.8 ± 0.3 | 26.1 ± 0.6 | 46.4 ± 0.1 |
| L118I | 41.6 ± 0.2 | 17.4 ± 0.6 | 42.9 ± 0.1 |
| L118V | 40.7 ± 0.3 | 13.3 ± 0.6 | 42.3 ± 0.1 |
| Name | kcatpyruvate (s−1) | Kmpyruvate (mM) | kcat/Kmpyruvate (s−1·mM−1) | kcatα-MBA (s−1) | Kmα-MBA (mM) | kcat/Kmα-MBA (s−1·mM−1) |
|---|---|---|---|---|---|---|
| WT-AT | 0.50 ± 0.01 | 0.23 ± 0.02 | 2.22 | 0.64 ± 0.01 | 0.23 ± 0.03 | 2.82 |
| L118A | 1.48 ± 0.05 | 0.36 ± 0.05 | 4.15 | 1.46 ± 0.01 | 0.18 ± 0.01 | 7.93 |
| L118T | 1.95 ± 0.02 | 0.40 ± 0.02 | 4.85 | 1.79 ± 0.02 | 0.18 ± 0.01 | 9.98 |
| L118I | 0.82 ± 0.01 | 0.28 ± 0.02 | 2.95 | 0.82 ± 0.01 | 0.13 ± 0.01 | 6.40 |
| L118V | 0.51 ± 0.01 | 0.29 ± 0.02 | 1.76 | 0.57 ± 0.01 | 0.16 ± 0.01 | 3.57 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.-L.; Hu, S.; Lv, C.-J.; Zhao, W.-R.; Wang, H.-P.; Mei, J.-Q.; Mei, L.-H.; Huang, J. A Single Mutation Increases the Thermostability and Activity of Aspergillus terreus Amine Transaminase. Molecules 2019, 24, 1194. https://doi.org/10.3390/molecules24071194
Zhu W-L, Hu S, Lv C-J, Zhao W-R, Wang H-P, Mei J-Q, Mei L-H, Huang J. A Single Mutation Increases the Thermostability and Activity of Aspergillus terreus Amine Transaminase. Molecules. 2019; 24(7):1194. https://doi.org/10.3390/molecules24071194
Chicago/Turabian StyleZhu, Wan-Li, Sheng Hu, Chang-Jiang Lv, Wei-Rui Zhao, Hong-Peng Wang, Jia-Qi Mei, Le-He Mei, and Jun Huang. 2019. "A Single Mutation Increases the Thermostability and Activity of Aspergillus terreus Amine Transaminase" Molecules 24, no. 7: 1194. https://doi.org/10.3390/molecules24071194